IBTK Haploinsufficiency Affects the Tumor Microenvironment of Myc-Driven Lymphoma in E-myc Mice

 ,
,  ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
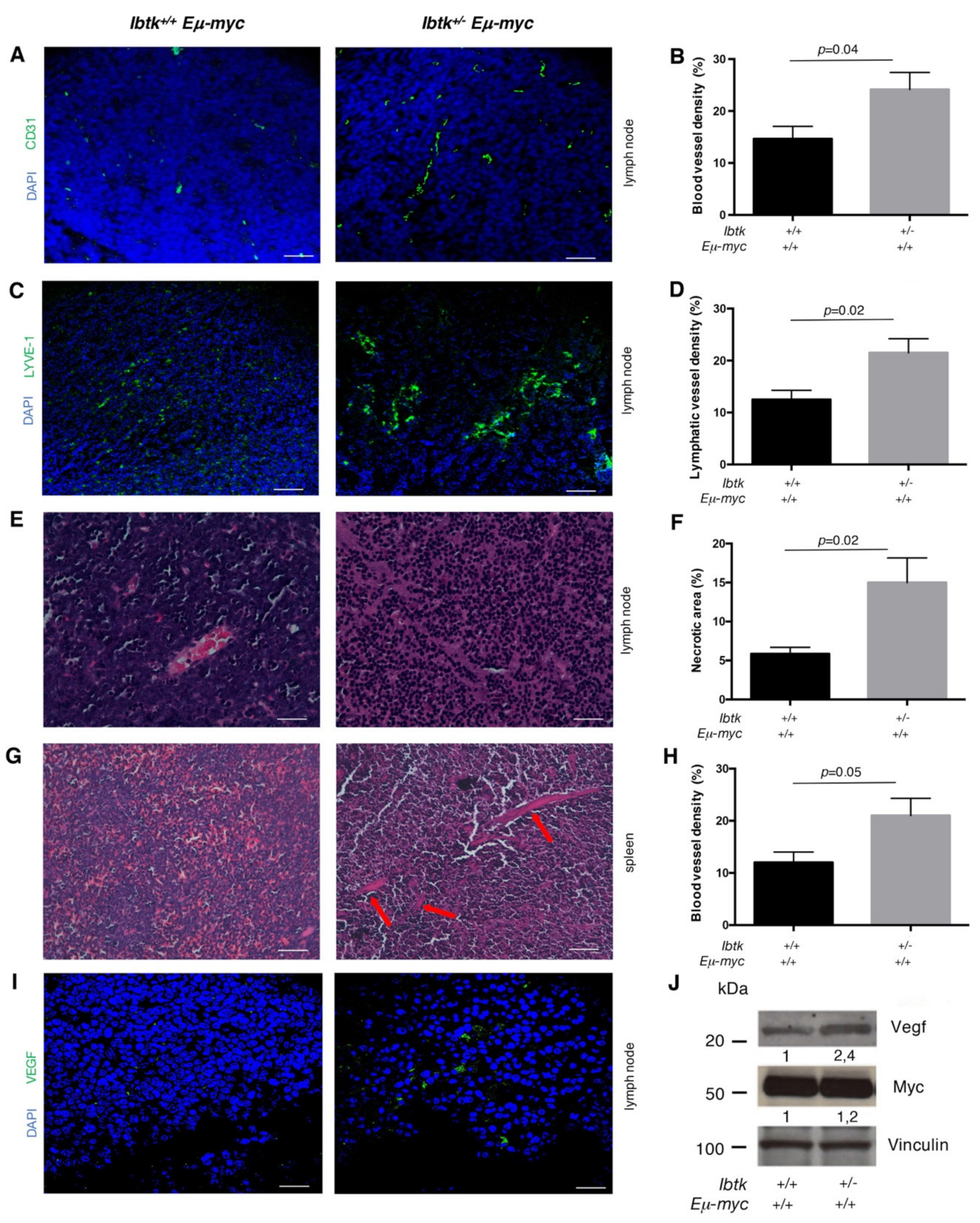
2.1. IBTK Haploinsufficiency Increases the Size and Vascularization of Spleen and Lymph Nodes of Eμ-myc Tumor Mice
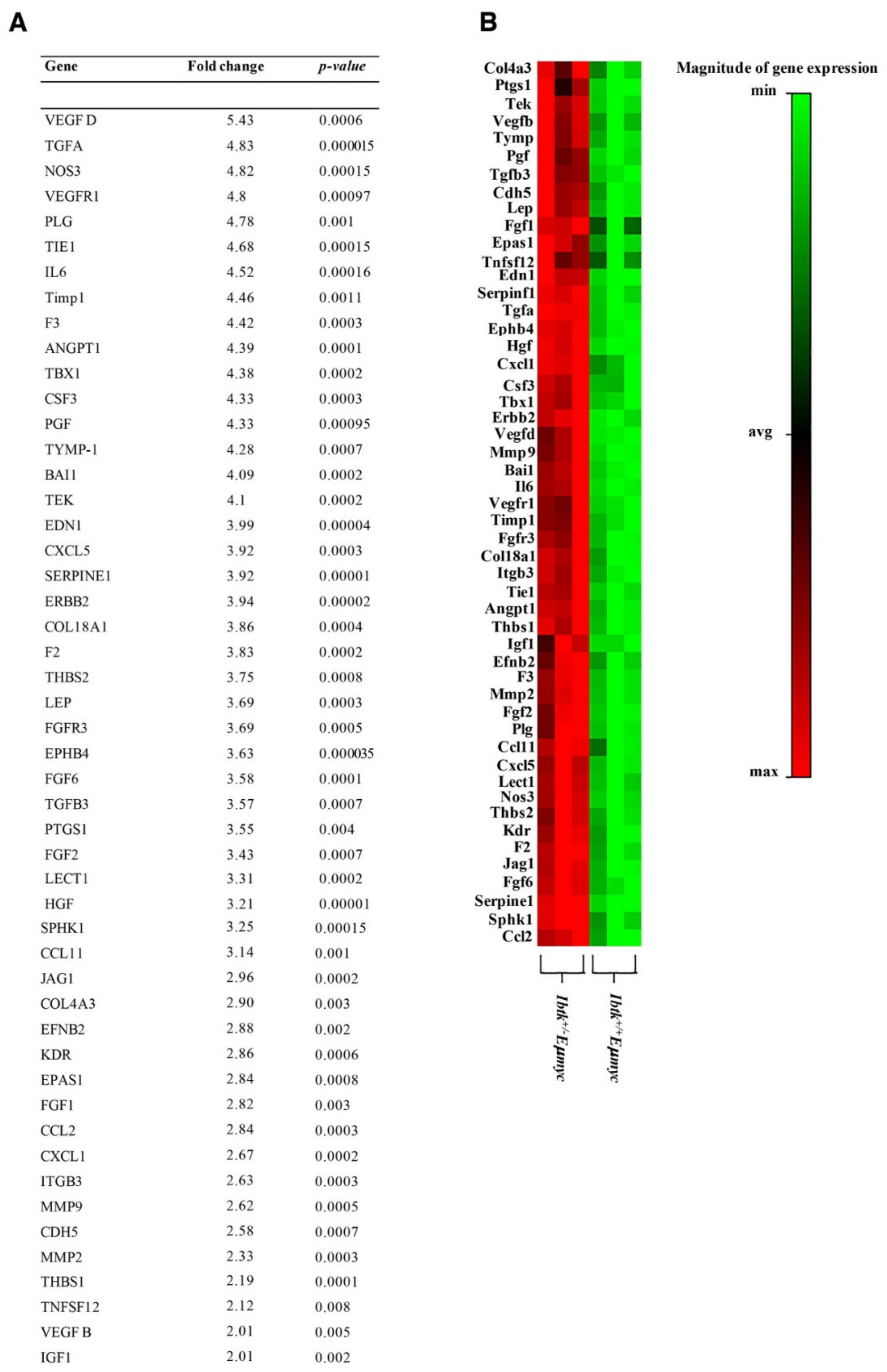
2.2. IBTK Haploinsufficiency Increases the Production of Pro-Angiogenic Factors and Cytokines in the Tumor Microenvironment of Eμ-myc Mice
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Histological Analysis
4.3. Western Blot Analysis and Cytokine Antibody Arrays
4.4. Real Time PCR, Data Normalization, and Analysis
4.5. Isolation of B Cells
4.6. Flow Cytometry
4.7. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Balkwill, F.; Mantovani, A. Cancer and inflammation: implications for pharmacology and therapeutics. Clin. Pharmacol. Ther. 2010, 87, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Vakkila, J.; Lotze, M.T. Inflammation and necrosis promote tumour growth. Nat. Rev. Immunol. 2004, 4, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef]
- Ziyad, S.; Iruela-Arispe, M.L. Molecular mechanism of tumor angiogenesis. Genes Cancer 2011, 2, 1085–1096. [Google Scholar] [CrossRef] [Green Version]
- Falcomatà, M.; Bärthel, S.; Schneider, G.; Saur, D.; Veltkamp, C. Deciphering the universe of genetic context-dependencies using mouse models of cancer. Curr. Opin. Gen. Dev. 2019, 54, 97–104. [Google Scholar] [CrossRef]
- James, Y.M. Mouse Models of Lymphoma and Lymphoid Leukemia. In Neoplastic Hematopathology: Experimental and Clinical Approaches; Jones, D., Ed.; Springer-Verlag New York, LLC: New York, NY, USA, 2010; Chapter 36; pp. 583–596. [Google Scholar]
- Frese, K.K.; Tuveson, D.A. Maximizing mouse cancer models. Nat. Rev. Cancer. 2007, 7, 645–658. [Google Scholar] [CrossRef]
- Carroll, P.A.; Freie, B.W.; Mathsyaraja, H.; Eisenman, R.N. The MYC transcription factor network: balancing metabolism, proliferation and oncogenesis. Front. Med. 2018, 12, 412–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.M.; Harris, A.W.; Pinkert, C.A.; Corcoran, L.M.; Alexander, W.S.; Cory, S.; Palmiter, R.D.; Brinster, R.L. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985, 318, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.W.; Pinkert, C.A.; Crawford, M.; Langdon, W.Y.; Brinster, R.L.; Adams, J.M. The E mu-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J. Exp. Med. 1988, 167, 363–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egle, A.; Harris, A.W.; Bath, M.L.; O’Reilly, L.; Cory, S. VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood 2004, 103, 2276–2283. [Google Scholar] [CrossRef]
- Ruddell, A.; Mezquita, P.; Brandvold, K.A.; Farr, A.; Iritani, B.M. B lymphocyte-specific c-Myc expression stimulates early and functional expansion of the vasculature and lymphatics during lymphomagenesis. Am. J. Pathol. 2003, 163, 2233–2245. [Google Scholar] [CrossRef] [Green Version]
- Baird, T.D.; Palam, L.R.; Fusakio, M.E.; Willy, J.A.; Davis, C.M.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. Selective mRNA translation during eIF2 phosphorylation induces expression of IBTKalpha. Mol. Biol. Cell 2014, 25, 1686–1697. [Google Scholar] [CrossRef]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.K.; Elledge, S.J. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Pisano, A.; Ceglia, S.; Palmieri, C.; Vecchio, E.; Fiume, G.; de Laurentiis, A.; Mimmi, S.; Falcone, C.; Iaccino, E.; Scialdone, A.; et al. CRL3IBTK regulates the tumor suppressor Pdcd4 through ubiquitylation coupled to proteasomal degradation. J. Biol. Chem. 2015, 290, 13958–13971. [Google Scholar] [CrossRef] [Green Version]
- Fiume, G.; Scialdone, A.; Rizzo, F.; De Filippo, M.R.; Laudanna, C.; Albano, F.; Golino, G.; Vecchio, E.; Pontoriero, M.; Mimmi, S.; et al. IBTK Differently Modulates Gene Expression and RNA Splicing in HeLa and K562 Cells. Int. J. Mol. Sci. 2016, 17, 1711–1848. [Google Scholar] [CrossRef] [Green Version]
- Albano, F.; Chiurazzi, F.; Mimmi, S.; Vecchio, E.; Pastore, A.; Cimmino, C.; Frieri, C.; Iaccino, E.; Pisano, A.; Golino, G.; et al. The expression of inhibitor of bruton’s tyrosine kinase gene is progressively up regulated in the clinical course of chronic lymphocytic leukaemia conferring resistance to apoptosis. Cell Death Dis. 2018, 1, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Vecchio, E.; Golino, G.; Pisano, A.; Albano, F.; Falcone, C.; Ceglia, S.; Iaccino, E.; Mimmi, S.; Fiume, G.; Giurato, G.; et al. IBTK contributes to B-cell lymphomagenesis in Eμ-myc transgenic mice conferring resistance to apoptosis. Cell Death Dis. 2019, 10, 320. [Google Scholar] [CrossRef] [PubMed]
- Michalak, E.M.; Jansen, E.S.; Happo, L.; Cragg, M.S.; Tai, L.; Smyth, G.K.; Strasser, A.; Adams, J.M.; Scott., C.L. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ. 2009, 16, 684–696. [Google Scholar] [CrossRef]
- Frenzel, A.; Loven, J.; Henriksson, M.A. Targeting MYC-regulated miRNAs to combat cancer. Genes Cancer 2010, 1, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.L.; Jin, G.; Cao, R.; Zhang, S.; Cao, Y.; Zhou, Z. MT1-MMP sheds LYVE-1 on lymphatic endothelial cells and suppresses VEGF-C production to inhibit lymphangiogenesis. Nat. Commun. 2016, 7, 10824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, M.; Xin, X.; Lala, P.K. A practical and sensitive method of quantitating lymphangiogenesis in vivo. Lab Invest. 2013, 93, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Kakinuma, T.; Hwang, S.T. Chemokines, chemokine receptors, and cancer metastasis. J. Leukoc. Biol. 2006, 79, 639–651. [Google Scholar] [CrossRef]
- Mira, E.; Lacalle, R.A.; Buesa, J.M.; de Buitrago, G.G.; Jimenez-Baranda, S.; Gómez-Moutón, C.; Martínez-A, C.; Mañes, S. Secreted MMP9 promotes angiogenesis more efficiently than constitutive active MMP9 bound to the tumor cell surface. J. Cell Sci. 2004, 1, 1847–1857. [Google Scholar] [CrossRef] [Green Version]
- Rundhaug, J.E. Matrix metalloproteinases and angiogenesis. J. Cell Mol. Med. 2005, 9, 267–285. [Google Scholar] [CrossRef]
- Zhang, J.; Patel, L.; Pienta, K.J. Targeting chemokine (C-C motif) ligand 2 (CCL2) as an example of translation of cancer molecular biology to the clinic. Prog. Mol. Biol. Transl. Sci. 2010, 95, 31–53. [Google Scholar]
- Erreni, M.; Mantovani, A.; Allavena, P. Tumor-associated Macrophages (TAM) and Inflammation in Colorectal Cancer. Cancer Microenviron. 2011, 4, 141–154. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 22, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, T.; Kato, T.; Kimura, T.; Miyakawa, R.; Tanaka, S.; Fujii, A.; Yamamoto, K.; Kameoka, S.; Hamano, K.; Kawakami, M.; et al. Clinicopathologic study of angiogenesis in Japanese patients with breast cancer. World J. Surg. 1997, 21, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Craig, M.J.; Loberg, R.D. CCL2 (Monocyte Chemoattractant Protein-1) in cancer bone metastases. Cancer Metastasis Rev. 2006, 25, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Happerfield, L.C.; Bobrow, L.G.; Millis, R.R. Angiogenesis and inflammation in ductal carcinoma in situ of the breast. J. Pathol. 1997, 181, 200–206. [Google Scholar] [CrossRef]
- Allavena, P.; Germano, G.; Marchesi, F.; Mantovani, A. Chemokines in cancer related inflammation. Exp. Cell Res. 2011, 10, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Aref, S.; Mabed, M.; Zalata, K.; Sakrana, M.; El Askalany, H. The interplay between c-Myc oncogene expression and circulating vascular endothelial growth factor (sVEGF), its antagonist receptor, soluble Flt-1 in diffuse large B cell lymphoma (DLBCL): relationship to patient outcome. Leuk. Lymphoma 2004, 45, 499–506. [Google Scholar] [CrossRef]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front. Physiol. 2014, 5, 5–75. [Google Scholar] [CrossRef] [Green Version]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Iaccino, E.; Mimmi, S.; Dattilo, V.; Marino, F.; Candeloro, P.; Di Loria, A.; Marimpietri, D.; Pisano, A.; Albano, F.; Vecchio, E.; et al. Monitoring multiple myeloma by idiotype-specific peptide binders of tumor-derived exosomes. Mol. Cancer. 2017, 16, 159. [Google Scholar] [CrossRef]
- Lin, C.J.; Nasr, Z.; Premsrirut, P.K.; Porco, J.A., Jr.; Hippo, Y.; Lowe, S.W.; Pelletier, J. Targeting synthetic lethal interactions between Myc and the eIF4F complex impedes tumorigenesis. Cell. Rep. 2012, 1, 325–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Li, P.; Liu, C.; Ren, Y.; Tang, X.; Wang, K.; He, J. Sinomenine hydrochloride inhibits breast cancer metastasis by attenuating inflammation-related epithelial-mesenchymal transition and cancer stemness. Oncotarget 2017, 8, 13560–13574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, Y.; Zheng, L.; Liu, M.; Chen, C.D.; Jiang, H. UTX is an escape from X-inactivation tumor-suppressor in B cell lymphoma. Nat. Comm. 2018, 9, 2720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontoriero, M.; Fiume, G.; Vecchio, E.; de Laurentiis, A.; Albano, F.; Iaccino, E.; Mimmi, S.; Pisano, A.; Agosti, V.; Giovannone, E.; et al. Activation of NF-κB in B cell receptor signaling through Bruton’s tyrosine kinase-dependent phosphorylation of IκB-α. J. Mol. Med. 2019, 97, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Weidner, N.; Semple, J.P.; Welch, W.R.; Folkman, J. Tumor angiogenesis and metastasis—correlation in invasive breast carcinoma. N. Engl. J. Med. 1991, 324, 1–8. [Google Scholar] [CrossRef]
- Fiume, G.; Scialdone, A.; Albano, F.; Rossi, A.; Tuccillo, F.M.; Rea, D.; Palmieri, C.; Caiazzo, E.; Cicala, C.; Bellevicine, C.; et al. Impairment of T cell development and acute inflammatory response in HIV-1 Tat transgenic mice. Sci. Rep. 2015, 5, 13864. [Google Scholar] [CrossRef] [Green Version]
- Schiavone, M.; Fiume, G.; Caivano, A.; de Laurentiis, A.; Falcone, C.; Masci, F.F.; Iaccino, E.; Mimmi, S.; Palmieri, C.; Pisano, A.; et al. Design and characterization of a peptide mimotope of the HIV-1 gp120 bridging sheet. Int. J. Mol. Sci. 2012, 13, 5674–5699. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Pre-B cell | Pre-B/B cell | B cell |
|---|---|---|---|
| lymphoma | lymphoma | lymphoma | |
| Ibtk+/+ Eμ-myc (n = 20) | 12 (60%) | 1 (5%) | 7 (35%) |
| Ibtk+/- Eμ-myc (n = 24) | 23 (96%) | 0 (0%) | 1 (4%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vecchio, E.; Fiume, G.; Mignogna, C.; Iaccino, E.; Mimmi, S.; Maisano, D.; Trapasso, F.; Quinto, I. IBTK Haploinsufficiency Affects the Tumor Microenvironment of Myc-Driven Lymphoma in E-myc Mice. Int. J. Mol. Sci. 2020, 21, 885. https://doi.org/10.3390/ijms21030885
Vecchio E, Fiume G, Mignogna C, Iaccino E, Mimmi S, Maisano D, Trapasso F, Quinto I. IBTK Haploinsufficiency Affects the Tumor Microenvironment of Myc-Driven Lymphoma in E-myc Mice. International Journal of Molecular Sciences. 2020; 21(3):885. https://doi.org/10.3390/ijms21030885
Chicago/Turabian StyleVecchio, Eleonora, Giuseppe Fiume, Chiara Mignogna, Enrico Iaccino, Selena Mimmi, Domenico Maisano, Francesco Trapasso, and Ileana Quinto. 2020. "IBTK Haploinsufficiency Affects the Tumor Microenvironment of Myc-Driven Lymphoma in E-myc Mice" International Journal of Molecular Sciences 21, no. 3: 885. https://doi.org/10.3390/ijms21030885
APA StyleVecchio, E., Fiume, G., Mignogna, C., Iaccino, E., Mimmi, S., Maisano, D., Trapasso, F., & Quinto, I. (2020). IBTK Haploinsufficiency Affects the Tumor Microenvironment of Myc-Driven Lymphoma in E-myc Mice. International Journal of Molecular Sciences, 21(3), 885. https://doi.org/10.3390/ijms21030885