Dendritic Spines in Alzheimer’s Disease: How the Actin Cytoskeleton Contributes to Synaptic Failure

Abstract
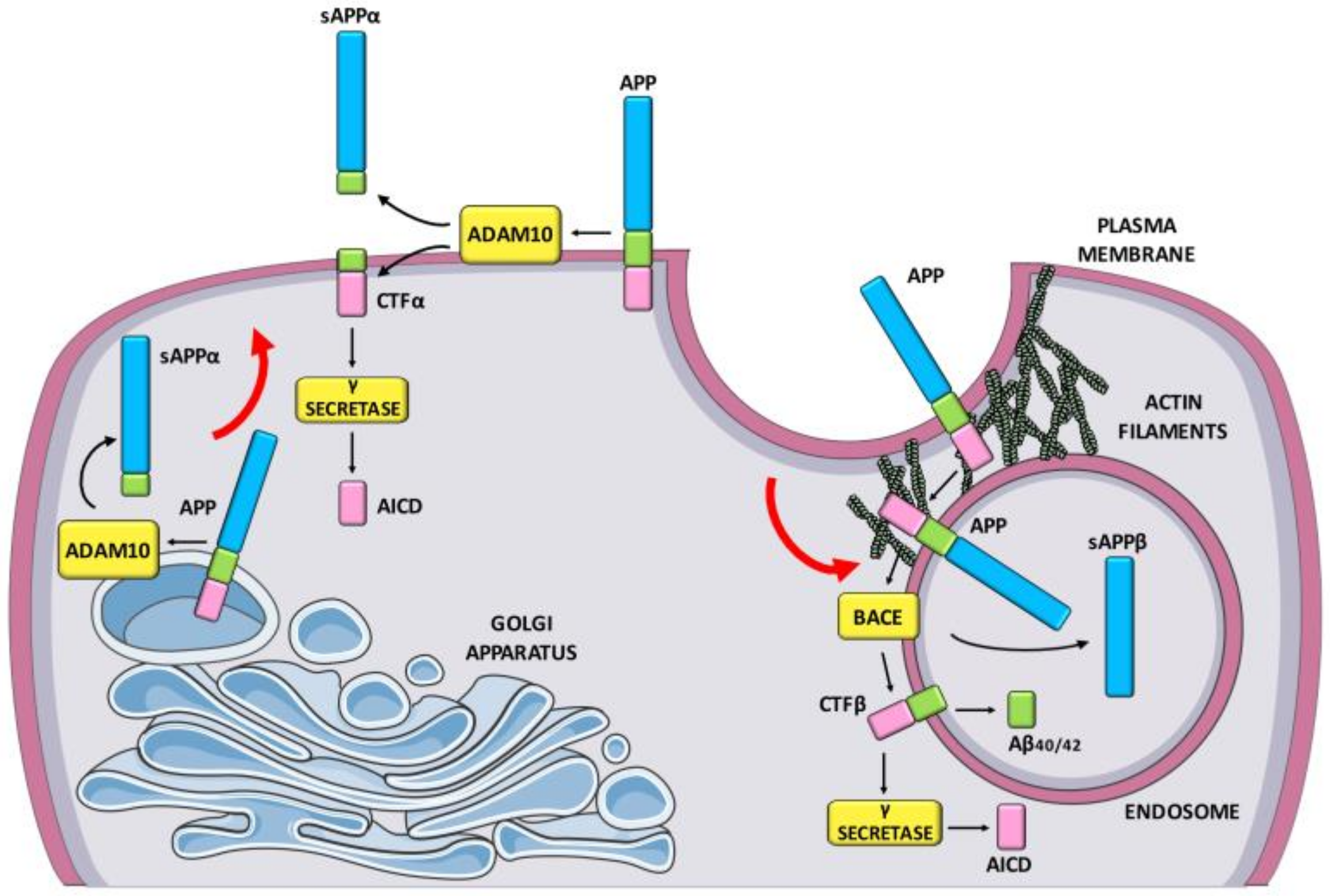
:1. Introduction
2. The Actin Cytoskeleton as the Architect of Spines
2.1. Synaptic Actin-Binding Proteins Orchestrating Actin Cytoskeleton Dynamics
2.1.1. Actin-Related Proteins-2/3
2.1.2. Profilin
2.1.3. Rho Family of GTPases
2.1.4. ADF/Cofilin
2.1.5. Cyclase-Associated Proteins
2.1.6. Epidermal Growth Factor Receptor Pathway Substrate 8
2.1.7. Myosins V and VI
2.1.8. Tropomyosins
2.1.9. Drebrin
2.1.10. Ca2+/calmodulin dependent protein kinase II β
2.1.11. α-actinin
2.2. The Actin Cytoskeleton in Spines: A Key Player of Activity-Dependent Synaptic Plasticity Events
2.2.1. Actin Cytoskeleton Remodeling to Change Spines Structure
2.2.2. Actin Cytoskeleton and Endocytosis
3. Actin Cytoskeleton Pathology in AD
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer‘s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Vermunt, L.; Sikkes, S.A.M.; van den Hout, A.; Handels, R.; Bos, I.; van der Flier, W.M.; Kern, S.; Ousset, P.-J.; Maruff, P.; Skoog, I.; et al. Alzheimer Disease Neuroimaging Initiative; AIBL Research Group; ICTUS/DSA study groups Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimers Dement. 2019, 15, 888–898. [Google Scholar] [CrossRef]
- DiLuca, M.; Olesen, J. The Cost of Brain Diseases: A Burden or a Challenge? Neuron 2014, 82, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haass, C.; Selkoe, D.J. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell 1993, 75, 1039–1042. [Google Scholar] [CrossRef]
- Das, U.; Scott, D.A.; Ganguly, A.; Koo, E.H.; Tang, Y.; Roy, S. Activity-induced convergence of APP and BACE-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron 2013, 79, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Lammich, S.; Buell, D.; Zilow, S.; Ludwig, A.-K.; Nuscher, B.; Lichtenthaler, S.F.; Prinzen, C.; Fahrenholz, F.; Haass, C. Expression of the anti-amyloidogenic secretase ADAM10 is suppressed by its 5′-untranslated region. J. Biol. Chem. 2010, 285, 15753–15760. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, P.-H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Rossner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. Embo J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.C.; Ludewig, S.; Winschel, A.; Abel, T.; Bold, C.; Salzburger, L.R.; Klein, S.; Han, K.; Weyer, S.W.; Fritz, A.-K.; et al. Distinct in vivo roles of secreted APP ectodomain variants APPsα and APPsβ in regulation of spine density, synaptic plasticity, and cognition. Embo J. 2018, 37, 116. [Google Scholar] [CrossRef] [PubMed]
- Lammich, S.; Kojro, E.; Postina, R.; Gilbert, S.; Pfeiffer, R.; Jasionowski, M.; Haass, C.; Fahrenholz, F. Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. USA 1999, 96, 3922–3927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.Z.A.; Gleeson, P.A. The trans-Golgi network is a major site for α-secretase processing of amyloid precursor protein in primary neurons. J. Biol. Chem. 2019, 294, 1618–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcello, E.; Gardoni, F.; Mauceri, D.; Romorini, S.; Jeromin, A.; Epis, R.; Borroni, B.; Cattabeni, F.; Sala, C.; Padovani, A.; et al. Synapse-associated protein-97 mediates alpha-secretase ADAM10 trafficking and promotes its activity. J. Neurosci. 2007, 27, 1682–1691. [Google Scholar] [CrossRef] [Green Version]
- Marcello, E.; Saraceno, C.; Musardo, S.; Vara, H.; de la Fuente, A.G.; Pelucchi, S.; Di Marino, D.; Borroni, B.; Tramontano, A.; Pérez-Otaño, I.; et al. Endocytosis of synaptic ADAM10 in neuronal plasticity and Alzheimer’s disease. J. Clin. Invest. 2013, 123, 2523–2538. [Google Scholar] [CrossRef]
- Saraceno, C.; Marcello, E.; Di Marino, D.; Borroni, B.; Claeysen, S.; Perroy, J.; Padovani, A.; Tramontano, A.; Gardoni, F.; Di Luca, M. SAP97-mediated ADAM10 trafficking from Golgi outposts depends on PKC phosphorylation. Cell Death Dis. 2014, 5, 1547. [Google Scholar] [CrossRef] [Green Version]
- Marcello, E.; Musardo, S.; Vandermeulen, L.; Pelucchi, S.; Gardoni, F.; Santo, N.; Antonucci, F.; Di Luca, M. Amyloid-β Oligomers Regulate ADAM10 Synaptic Localization Through Aberrant Plasticity Phenomena. Mol. Neurobiol. 2019, 27, 457. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.Z.A.; Fourriere, L.; Wang, J.; Perez, F.; Boncompain, G.; Gleeson, P.A. Distinct anterograde trafficking pathways of BACE1 and amyloid precursor protein from the TGN and the regulation of amyloid-β production. Mol. Biol. Cell 2019, 31, 27–44. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]
- Bolmont, T.; Clavaguera, F.; Meyer-Luehmann, M.; Herzig, M.C.; Radde, R.; Staufenbiel, M.; Lewis, J.; Hutton, M.; Tolnay, M.; Jucker, M. Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice. Am. J. Pathol. 2007, 171, 2012–2020. [Google Scholar] [CrossRef] [Green Version]
- Götz, J.; Chen, F.; van Dorpe, J.; Nitsch, R.M. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, D.E.; Molina-Porcel, L.; Iba, M.; Aboagye, A.K.; Paul, S.M.; Trojanowski, J.Q.; Lee, V.M.-Y. A{beta} accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am. J. Pathol. 2010, 177, 1977–1988. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pooler, A.M.; Polydoro, M.; Maury, E.A.; Nicholls, S.B.; Reddy, S.M.; Wegmann, S.; William, C.; Saqran, L.; Cagsal-Getkin, O.; Pitstick, R.; et al. Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer’s disease. Acta Neuropathol. Commun. 2015, 3, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Nguyen, L.N.; Kessels, H.W.; Hagiwara, H.; Sisodia, S.; Malinow, R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 2010, 13, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar] [CrossRef]
- Bourne, J.N.; Harris, K.M. Balancing Structure and Function at Hippocampal Dendritic Spines. Annu. Rev. Neurosci. 2008, 31, 47–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sala, C.; Segal, M. Dendritic spines: The locus of structural and functional plasticity. Physiol. Rev. 2014, 94, 141–188. [Google Scholar] [CrossRef] [PubMed]
- Matus, A. Actin-based plasticity in dendritic spines. Science 2000, 290, 754–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landis, D.M.; Reese, T.S. Cytoplasmic organization in cerebellar dendritic spines. J. Cell Biol. 1983, 97, 1169–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotulainen, P.; Hoogenraad, C.C. Actin in dendritic spines: Connecting dynamics to function. J. Cell Biol. 2010, 189, 619–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, T.D. The cytoskeleton, cellular motility and the reductionist agenda. Nature 2003, 422, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Skruber, K.; Read, T.-A.; Vitriol, E.A. Reconsidering an active role for G-actin in cytoskeletal regulation. J. Cell. Sci. 2018, 131, jcs203760. [Google Scholar] [CrossRef] [Green Version]
- Dugina, V.B.; Shagieva, G.S.; Kopnin, P.B. Biological Role of Actin Isoforms in Mammalian Cells. Biochem. Mosc. 2019, 84, 583–592. [Google Scholar] [CrossRef]
- Hlushchenko, I.; Koskinen, M.; Hotulainen, P. Dendritic spine actin dynamics in neuronal maturation and synaptic plasticity. Cytoskeleton 2016, 73, 435–441. [Google Scholar] [CrossRef]
- Sheng, M.; Hoogenraad, C.C. The Postsynaptic Architecture of Excitatory Synapses: A More Quantitative View. Annu. Rev. Biochem. 2007, 76, 823–847. [Google Scholar] [CrossRef] [Green Version]
- MacGillavry, H.D.; Kerr, J.M.; Kassner, J.; Frost, N.A.; Blanpied, T.A. Shank-cortactin interactions control actin dynamics to maintain flexibility of neuronal spines and synapses. Eur. J. Neurosci. 2016, 43, 179–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matt, L.; Kim, K.; Hergarden, A.C.; Patriarchi, T.; Malik, Z.A.; Park, D.K.; Chowdhury, D.; Buonarati, O.R.; Henderson, P.B.; Gökçek Saraç, Ç.; et al. α-Actinin Anchors PSD-95 at Postsynaptic Sites. Neuron 2018, 97, 1094–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, L.A.; Goda, Y. Actin in action: The interplay between the actin cytoskeleton and synaptic efficacy. Nat. Rev. Neurosci. 2008, 9, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhong, G.; Zhuang, X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 2013, 339, 452–456. [Google Scholar] [CrossRef] [Green Version]
- Bär, J.; Kobler, O.; van Bommel, B.; Mikhaylova, M. Periodic F-actin structures shape the neck of dendritic spines. Sci. Rep. 2016, 6, 37136. [Google Scholar] [CrossRef] [PubMed]
- Bucher, M.; Fanutza, T.; Mikhaylova, M. Cytoskeletal makeup of the synapse: Shaft vs. spine. Cytoskelet 2019, cm.21583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef] [Green Version]
- Ichetovkin, I.; Grant, W.; Condeelis, J. Cofilin produces newly polymerized actin filaments that are preferred for dendritic nucleation by the Arp2/3 complex. Curr. Biol. 2002, 12, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Goley, E.D.; Ohkawa, T.; Mancuso, J.; Woodruff, J.B.; D’Alessio, J.A.; Cande, W.Z.; Volkman, L.E.; Welch, M.D. Dynamic nuclear actin assembly by Arp2/3 complex and a baculovirus WASP-like protein. Science 2006, 314, 464–467. [Google Scholar] [CrossRef] [Green Version]
- Wegner, A. Head to tail polymerization of actin. J. Mol. Biol. 1976, 108, 139–150. [Google Scholar] [CrossRef]
- Hotulainen, P.; Llano, O.; Smirnov, S.; Tanhuanpää, K.; Faix, J.; Rivera, C.; Lappalainen, P. Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J. Cell Biol. 2009, 185, 323–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeckel, A.; Ahuja, R.; Gundelfinger, E.D.; Qualmann, B.; Kessels, M.M. The actin-binding protein Abp1 controls dendritic spine morphology and is important for spine head and synapse formation. J. Neurosci. 2008, 28, 10031–10044. [Google Scholar] [CrossRef] [Green Version]
- Hering, H.; Sheng, M. Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J. Neurosci. 2003, 23, 11759–11769. [Google Scholar] [CrossRef]
- Soderling, S.H.; Guire, E.S.; Kaech, S.; White, J.; Zhang, F.; Schutz, K.; Langeberg, L.K.; Banker, G.; Raber, J.; Scott, J.D. A WAVE-1 and WRP signaling complex regulates spine density, synaptic plasticity, and memory. J. Neurosci. 2007, 27, 355–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegner, A.M.; Nebhan, C.A.; Hu, L.; Majumdar, D.; Meier, K.M.; Weaver, A.M.; Webb, D.J. N-wasp and the arp2/3 complex are critical regulators of actin in the development of dendritic spines and synapses. J. Biol. Chem. 2008, 283, 15912–15920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Sokolova, O.S.; Chung, J.; Padrickm, S.; Gelles, J.; Goode, B.L. Abp1 promotes Arp2/3 complex-dependent actin nucleation and stabilizes branch junctions by antagonizing GMF. Nat. Commun. 2018, 24, 2895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarmola, E.G.; Bubb, M.R. How depolymerization can promote polymerization: The case of actin and profilin. Bioessays 2009, 31, 1150–1160. [Google Scholar] [CrossRef]
- Witke, W.; Sutherland, J.D.; Sharpe, A.; Arai, M.; Kwiatkowski, D.J. Profilin I is essential for cell survival and cell division in early mouse development. Proc. Natl. Acad. Sci. USA 2001, 98, 3832–3836. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Cordero, J.J.; Magalhaes, M.A.O.; Eddy, R.J.; Hodgson, L.; Condeelis, J. Functions of cofilin in cell locomotion and invasion. Nat. Rev. Mol. Cell Biol. 2013, 14, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Bamburg, J.R.; Bernstein, B.W. Actin dynamics and cofilin-actin rods in alzheimer disease. Cytoskelet 2016, 73, 477–497. [Google Scholar] [CrossRef] [Green Version]
- Hotulainen, P.; Paunola, E.; Vartiainen, M.K.; Lappalainen, P. Actin-depolymerizing factor and cofilin-1 play overlapping roles in promoting rapid F-actin depolymerization in mammalian nonmuscle cells. Mol. Biol. Cell 2005, 16, 649–664. [Google Scholar] [CrossRef] [PubMed]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schneider, C.; Stanyon, C.A.; Bernard, O.; Caroni, P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 1998, 393, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Niwa, R.; Nagata-Ohashi, K.; Takeichi, M.; Mizuno, K.; Uemura, T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 2002, 108, 233–246. [Google Scholar] [CrossRef] [Green Version]
- Okreglak, V.; Drubin, D.G. Loss of Aip1 reveals a role in maintaining the actin monomer pool and an in vivo oligomer assembly pathway. J. Cell Biol. 2010, 188, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, S. Regulation of actin filament dynamics by actin depolymerizing factor/cofilin and actin-interacting protein 1: New blades for twisted filaments. Biochemistry 2003, 42, 13363–13370. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, A.V.; Brieher, W.M. Aip1 destabilizes cofilin-saturated actin filaments by severing and accelerating monomer dissociation from ends. Curr. Biol. 2014, 24, 2749–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, S. The role of cyclase-associated protein in regulating actin filament dynamics-more than a monomer-sequestration factor. J. Cell. Sci. 2013, 126, 3249–3258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriyama, K.; Yahara, I. Human CAP1 is a key factor in the recycling of cofilin and actin for rapid actin turnover. J. Cell. Sci. 2002, 115, 1591–1601. [Google Scholar]
- Quintero-Monzon, O.; Jonasson, E.M.; Bertling, E.; Talarico, L.; Chaudhry, F.; Sihvo, M.; Lappalainen, P.; Goode, B.L. Reconstitution and dissection of the 600-kDa Srv2/CAP complex: Roles for oligomerization and cofilin-actin binding in driving actin turnover. J. Biol. Chem. 2009, 284, 10923–10934. [Google Scholar] [CrossRef] [Green Version]
- Kotila, T.; Wioland, H.; Enkavi, G.; Kogan, K.; Vattulainen, I.; Jégou, A.; Romet-Lemonne, G.; Lappalainen, P. Mechanism of synergistic actin filament pointed end depolymerization by cyclase-associated protein and cofilin. Nat. Commun. 2019, 10, 5320. [Google Scholar] [CrossRef] [Green Version]
- Shekhar, S.; Chung, J.; Kondev, J.; Gelles, J.; Goode, B.L. Synergy between Cyclase-associated protein and Cofilin accelerates actin filament depolymerization by two orders of magnitude. Nat. Commun. 2019, 10, 5319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiston, J.; Hubberstey, A.; Yu, G.; Young, D. Differential expression of CAP and CAP2 in adult rat tissues. Gene 1995, 165, 273–277. [Google Scholar] [CrossRef]
- Kumar, A.; Paeger, L.; Kosmas, K.; Kloppenburg, P.; Noegel, A.A.; Peche, V.S. Neuronal Actin Dynamics, Spine Density and Neuronal Dendritic Complexity Are Regulated by CAP2. Front. Cell Neurosci. 2016, 10, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, T.J.; Hammarlund, M. Syndecan promotes axon regeneration by stabilizing growth cone migration. Cell Rep. 2014, 8, 272–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menna, E.; Disanza, A.; Cagnoli, C.; Schenk, U.; Gelsomino, G.; Frittoli, E.; Hertzog, M.; Offenhauser, N.; Sawallisch, C.; Kreienkamp, H.-J.; et al. Eps8 regulates axonal filopodia in hippocampal neurons in response to brain-derived neurotrophic factor (BDNF). PLoS Biol. 2009, 7, e1000138. [Google Scholar] [CrossRef] [PubMed]
- Kneussel, M.; Wagner, W. Myosin motors at neuronal synapses: Drivers of membrane transport and actin dynamics. Nat. Rev. Neurosci. 2013, 14, 233–247. [Google Scholar] [CrossRef]
- Soldati, T.; Schliwa, M. Powering membrane traffic in endocytosis and recycling. Nat. Rev. Mol. Cell Biol. 2006, 7, 897–908. [Google Scholar] [CrossRef]
- Hartman, M.A.; Finan, D.; Sivaramakrishnan, S.; Spudich, J.A. Principles of unconventional myosin function and targeting. Annu. Rev. Cell Dev. Biol. 2011, 27, 133–155. [Google Scholar] [CrossRef] [Green Version]
- Gunning, P.; O’Neill, G.; Hardeman, E. Tropomyosin-based regulation of the actin cytoskeleton in time and space. Physiol. Rev. 2008, 88, 1–35. [Google Scholar] [CrossRef] [Green Version]
- Brettle, M.; Patel, S.; Fath, T. Tropomyosins in the healthy and diseased nervous system. Brain Res. Bull. 2016, 126, 311–323. [Google Scholar] [CrossRef]
- Gunning, P.W.; Hardeman, E.C.; Lappalainen, P.; Mulvihill, D.P. Tropomyosin-master regulator of actin filament function in the cytoskeleton. J. Cell. Sci. 2015, 128, 2965–2974. [Google Scholar] [CrossRef] [Green Version]
- Blanchoin, L.; Pollard, T.D.; Hitchcock-DeGregori, S.E. Inhibition of the Arp2/3 complex-nucleated actin polymerization and branch formation by tropomyosin. Curr. Biol. 2001, 11, 1300–1304. [Google Scholar] [CrossRef] [Green Version]
- Ono, S.; Ono, K. Tropomyosin inhibits ADF/cofilin-dependent actin filament dynamics. J. Cell Biol. 2002, 156, 1065–1076. [Google Scholar] [CrossRef] [Green Version]
- Faivre-Sarrailh, C.; Had, L.; Ferraz, C.; Sri Widada, J.S.; Liautard, J.P.; Rabié, A. Expression of tropomyosin genes during the development of the rat cerebellum. J. Neurochem. 1990, 55, 899–906. [Google Scholar] [CrossRef]
- Weinberger, R.; Schevzov, G.; Jeffrey, P.; Gordon, K.; Hill, M.; Gunning, P. The molecular composition of neuronal microfilaments is spatially and temporally regulated. J. Neurosci. 1996, 16, 238–252. [Google Scholar] [CrossRef] [Green Version]
- Dufour, C.; Weinberger, R.P.; Gunning, P. Tropomyosin isoform diversity and neuronal morphogenesis. Immunol. Cell Biol. 1998, 76, 424–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hook, J.; Lemckert, F.; Qin, H.; Schevzov, G.; Gunning, P. Gamma tropomyosin gene products are required for embryonic development. Mol. Cell. Biol. 2004, 24, 2318–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schevzov, G.; Bryce, N.S.; Almonte-Baldonado, R.; Joya, J.; Lin, J.J.-C.; Hardeman, E.; Weinberger, R.; Gunning, P. Specific features of neuronal size and shape are regulated by tropomyosin isoforms. Mol. Biol. Cell 2005, 16, 3425–3437. [Google Scholar] [CrossRef] [Green Version]
- Guven, K.; Gunning, P.; Fath, T. TPM3 and TPM4 gene products segregate to the postsynaptic region of central nervous system synapses. Bioarchitecture 2011, 1, 284–289. [Google Scholar] [CrossRef] [Green Version]
- Koganezawa, N.; Hanamura, K.; Sekino, Y.; Shirao, T. The role of drebrin in dendritic spines. Mol. Cell. Neurosci. 2017, 84, 85–92. [Google Scholar] [CrossRef]
- Hayashi, K.; Ishikawa, R.; Ye, L.H.; He, X.L.; Takata, K.; Kohama, K.; Shirao, T. Modulatory role of drebrin on the cytoskeleton within dendritic spines in the rat cerebral cortex. J. Neurosci. 1996, 16, 7161–7170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikati, M.A.; Grintsevich, E.E.; Reisler, E. Drebrin-induced stabilization of actin filaments. J. Biol. Chem. 2013, 288, 19926–19938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worth, D.C.; Daly, C.N.; Geraldo, S.; Oozeer, F.; Gordon-Weeks, P.R. Drebrin contains a cryptic F-actin-bundling activity regulated by Cdk5 phosphorylation. J. Cell Biol. 2013, 202, 793–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon-Weeks, P.R. The role of the drebrin/EB3/Cdk5 pathway in dendritic spine plasticity, implications for Alzheimer’s disease. Brain Res. Bull. 2016, 126, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Colbran, R.J.; Soderling, T.R. Calcium/calmodulin-dependent protein kinase II. Curr. Top. Cell. Regul. 1990, 31, 181–221. [Google Scholar]
- Lisman, J.; Schulman, H.; Cline, H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 2002, 3, 175–190. [Google Scholar] [CrossRef]
- Bennett, M.K.; Erondu, N.E.; Kennedy, M.B. Purification and characterization of a calmodulin-dependent protein kinase that is highly concentrated in brain. J. Biol. Chem. 1983, 258, 12735–12744. [Google Scholar]
- Okamoto, K.-I.; Narayanan, R.; Lee, S.H.; Murata, K.; Hayashi, Y. The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc. Natl. Acad. Sci. USA 2007, 104, 6418–6423. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Conte, I.; Carter, T.; Bayer, K.U.; Molloy, J.E. Multiple CaMKII Binding Modes to the Actin Cytoskeleton Revealed by Single-Molecule Imaging. Biophys. J. 2016, 111, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Brocke, L.; Chiang, L.W.; Wagner, P.D.; Schulman, H. Functional implications of the subunit composition of neuronal CaM kinase II. J. Biol. Chem. 1999, 274, 22713–22722. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, H.; Sasagawa, Y.; Yamamoto, H.; Bito, H.; Shirao, T. CaMKIIβ is localized in dendritic spines as both drebrin-dependent and drebrin-independent pools. J. Neurochem. 2018, 146, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Djinović-Carugo, K.; Young, P.; Gautel, M.; Saraste, M. Structure of the alpha-actinin rod: Molecular basis for cross-linking of actin filaments. Cell 1999, 98, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Hodges, J.L.; Vilchez, S.M.; Asmussen, H.; Whitmore, L.A.; Horwitz, A.R. α-Actinin-2 mediates spine morphology and assembly of the post-synaptic density in hippocampal neurons. PLoS ONE 2014, 9, e101770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalan-Sakrikar, N.; Bartlett, R.K.; Baucum, A.J.; Colbran, R.J. Substrate-selective and calcium-independent activation of CaMKII by α-actinin. J. Biol. Chem. 2012, 287, 15275–15283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinowska, M.; Chávez, A.E.; Lutzu, S.; Castillo, P.E.; Bukauskas, F.F.; Francesconi, A. Actinin-4 Governs Dendritic Spine Dynamics and Promotes Their Remodeling by Metabotropic Glutamate Receptors. J. Biol. Chem. 2015, 290, 15909–15920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtmaat, A.; Svoboda, K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci. 2009, 10, 647–658. [Google Scholar] [CrossRef]
- Renner, M.; Choquet, D.; Triller, A. Control of the postsynaptic membrane viscosity. J. Neurosci. 2009, 29, 2926–2937. [Google Scholar] [CrossRef]
- Okamoto, K.-I.; Nagai, T.; Miyawaki, A.; Hayashi, Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat. Neurosci. 2004, 7, 1104–1112. [Google Scholar] [CrossRef]
- Bosch, M.; Castro, J.; Saneyoshi, T.; Matsuno, H.; Sur, M.; Hayashi, Y. Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 2014, 82, 444–459. [Google Scholar] [CrossRef] [Green Version]
- Kasai, H.; Fukuda, M.; Watanabe, S.; Hayashi-Takagi, A.; Noguchi, J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010, 33, 121–129. [Google Scholar] [CrossRef]
- Sekino, Y.; Tanaka, S.; Hanamura, K.; Yamazaki, H.; Sasagawa, Y.; Xue, Y.; Hayashi, K.; Shirao, T. Activation of N-methyl-D-aspartate receptor induces a shift of drebrin distribution: Disappearance from dendritic spines and appearance in dendritic shafts. Mol. Cell. Neurosci. 2006, 31, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Oser, M.; Condeelis, J. The cofilin activity cycle in lamellipodia and invadopodia. J. Cell. Biochem. 2009, 108, 1252–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanley, J.G. Actin-dependent mechanisms in AMPA receptor trafficking. Front. Cell Neurosci. 2014, 8, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, K.; Bosch, M.; Hayashi, Y. The roles of CaMKII and F-actin in the structural plasticity of dendritic spines: A potential molecular identity of a synaptic tag? Physiol. (Bethesda) 2009, 24, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Tada, T.; Sheng, M. Molecular mechanisms of dendritic spine morphogenesis. Curr. Opin. Neurobiol. 2006, 16, 95–101. [Google Scholar] [CrossRef]
- Allison, D.W.; Gelfand, V.I.; Spector, I.; Craig, A.M. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: Differential attachment of NMDA versus AMPA receptors. J. Neurosci. 1998, 18, 2423–2436. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.H.; Lisman, J.E. A role of actin filament in synaptic transmission and long-term potentiation. J. Neurosci. 1999, 19, 4314–4324. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Xiao, M.; Nicoll, R.A. Contribution of cytoskeleton to the internalization of AMPA receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 1261–1266. [Google Scholar] [CrossRef]
- Malinow, R.; Malenka, R.C. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 2002, 25, 103–126. [Google Scholar] [CrossRef] [Green Version]
- Choquet, D.; Triller, A. The dynamic synapse. Neuron 2013, 80, 691–703. [Google Scholar] [CrossRef] [Green Version]
- Fujii, S.; Tanaka, H.; Hirano, T. Detection and characterization of individual endocytosis of AMPA-type glutamate receptor around postsynaptic membrane. Genes Cells 2017, 22, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Rosendale, M.; Jullié, D.; Choquet, D.; Perrais, D. Spatial and Temporal Regulation of Receptor Endocytosis in Neuronal Dendrites Revealed by Imaging of Single Vesicle Formation. Cell Rep. 2017, 18, 1840–1847. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Liu, L.; Wang, Y.T.; Sheng, M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron 2002, 36, 661–674. [Google Scholar] [CrossRef] [Green Version]
- Loebrich, S.; Benoit, M.R.; Konopka, J.A.; Cottrell, J.R.; Gibson, J.; Nedivi, E. CPG2 Recruits Endophilin B2 to the Cytoskeleton for Activity-Dependent Endocytosis of Synaptic Glutamate Receptors. Curr. Biol. 2016, 26, 296–308. [Google Scholar] [CrossRef] [Green Version]
- Kakegawa, W.; Katoh, A.; Narumi, S.; Miura, E.; Motohashi, J.; Takahashi, A.; Kohda, K.; Fukazawa, Y.; Yuzaki, M.; Matsuda, S. Optogenetic Control of Synaptic AMPA Receptor Endocytosis Reveals Roles of LTD in Motor Learning. Neuron 2018, 99, 985–998. [Google Scholar] [CrossRef] [Green Version]
- Engqvist-Goldstein, A.E.Y.; Drubin, D.G. Actin assembly and endocytosis: From yeast to mammals. Annu. Rev. Cell Dev. Biol. 2003, 19, 287–332. [Google Scholar] [CrossRef] [Green Version]
- Blanpied, T.A.; Scott, D.B.; Ehlers, M.D. Dynamics and regulation of clathrin coats at specialized endocytic zones of dendrites and spines. Neuron 2002, 36, 435–449. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, F.M.; Chen, C.Y.; Knuehl, C.; Towler, M.C.; Wakeham, D.E. Biological basket weaving: Formation and function of clathrin-coated vesicles. Annu. Rev. Cell Dev. Biol. 2001, 17, 517–568. [Google Scholar] [CrossRef] [Green Version]
- Jarousse, N.; Kelly, R.B. Endocytotic mechanisms in synapses. Curr. Opin. Cell Biol. 2001, 13, 461–469. [Google Scholar] [CrossRef]
- Mooren, O.L.; Galletta, B.J.; Cooper, J.A. Roles for actin assembly in endocytosis. Annu. Rev. Biochem. 2012, 81, 661–686. [Google Scholar] [CrossRef]
- Bennett, E.M.; Chen, C.Y.; Engqvist-Goldstein, A.E.; Drubin, D.G.; Brodsky, F.M. Clathrin hub expression dissociates the actin-binding protein Hip1R from coated pits and disrupts their alignment with the actin cytoskeleton. Traffic 2001, 2, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Waelter, S.; Scherzinger, E.; Hasenbank, R.; Nordhoff, E.; Lurz, R.; Goehler, H.; Gauss, C.; Sathasivam, K.; Bates, G.P.; Lehrach, H.; et al. The huntingtin interacting protein HIP1 is a clathrin and alpha-adaptin-binding protein involved in receptor-mediated endocytosis. Hum. Mol. Genet. 2001, 10, 1807–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarar, D.; Waterman-Storer, C.M.; Schmid, S.L. A dynamic actin cytoskeleton functions at multiple stages of clathrin-mediated endocytosis. Mol. Biol. Cell 2005, 16, 964–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrais, D.; Merrifield, C.J. Dynamics of endocytic vesicle creation. Dev. Cell 2005, 9, 581–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrifield, C.J.; Perrais, D.; Zenisek, D. Coupling between clathrin-coated-pit invagination, cortactin recruitment, and membrane scission observed in live cells. Cell 2005, 121, 593–606. [Google Scholar] [CrossRef] [Green Version]
- Bryce, N.S.; Schevzov, G.; Ferguson, V.; Percival, J.M.; Lin, J.J.-C.; Matsumura, F.; Bamburg, J.R.; Jeffrey, P.L.; Hardeman, E.C.; Gunning, P.; et al. Specification of actin filament function and molecular composition by tropomyosin isoforms. Mol. Biol. Cell 2003, 14, 1002–1016. [Google Scholar] [CrossRef] [Green Version]
- Gormal, R.; Valmas, N.; Fath, T.; Meunier, F. A role for tropomyosins in activity-dependent bulk endocytosis? Mol. Cell. Neurosci. 2017, 84, 112–118. [Google Scholar] [CrossRef]
- Cottrell, J.R.; Borok, E.; Horvath, T.L.; Nedivi, E. CPG2: A brain- and synapse-specific protein that regulates the endocytosis of glutamate receptors. Neuron 2004, 44, 677–690. [Google Scholar]
- Loebrich, S.; Djukic, B.; Tong, Z.J.; Cottrell, J.R.; Turrigiano, G.G.; Nedivi, E. Regulation of glutamate receptor internalization by the spine cytoskeleton is mediated by its PKA-dependent association with CPG2. Proc. Natl. Acad. Sci. USA 2013, 110, E4548–E4556. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Liang, F.; Walensky, L.D.; Huganir, R.L. Regulation of AMPA receptor GluR1 subunit surface expression by a 4. 1N-linked actin cytoskeletal association. J. Neurosci. 2000, 20, 7932–7940. [Google Scholar] [CrossRef]
- Lin, D.-T.; Makino, Y.; Sharma, K.; Hayashi, T.; Neve, R.; Takamiya, K.; Huganir, R.L. Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nat. Neurosci. 2009, 12, 879–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.H.; Chung, H.J.; Lee, H.K.; Huganir, R.L. Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc. Natl. Acad. Sci. USA 2001, 98, 11725–11730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocca, D.L.; Martin, S.; Jenkins, E.L.; Hanley, J.G. Inhibition of Arp2/3-mediated actin polymerization by PICK1 regulates neuronal morphology and AMPA receptor endocytosis. Nat. Cell Biol. 2008, 10, 259–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, T.W.; Nakagawa, T.; Licznerski, P.; Pawlak, V.; Kolleker, A.; Rozov, A.; Kim, J.; Dittgen, T.; Köhr, G.; Sheng, M.; et al. Actin/alpha-actinin-dependent transport of AMPA receptors in dendritic spines: Role of the PDZ-LIM protein RIL. J. Neurosci. 2004, 24, 8584–8594. [Google Scholar] [CrossRef] [Green Version]
- Parkinson, G.T.; Chamberlain, S.E.L.; Jaafari, N.; Turvey, M.; Mellor, J.R.; Hanley, J.G. Cortactin regulates endo-lysosomal sorting of AMPARs via direct interaction with GluA2 subunit. Sci. Rep. 2018, 8, 4155. [Google Scholar] [CrossRef]
- Correia, S.S.; Bassani, S.; Brown, T.C.; Lisé, M.-F.; Backos, D.S.; El-Husseini, A.; Passafaro, M.; Esteban, J.A. Motor protein-dependent transport of AMPA receptors into spines during long-term potentiation. Nat. Neurosci. 2008, 11, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Osterweil, E.; Wells, D.G.; Mooseker, M.S. A role for myosin VI in postsynaptic structure and glutamate receptor endocytosis. J. Cell Biol. 2005, 168, 329–338. [Google Scholar] [CrossRef]
- Sun, J.; Roy, S. The physical approximation of APP and BACE-1: A key event in alzheimer’s disease pathogenesis. Dev. Neurobiol. 2018, 78, 340–347. [Google Scholar] [CrossRef]
- Greenfield, J.P.; Tsai, J.; Gouras, G.K.; Hai, B.; Thinakaran, G.; Checler, F.; Sisodia, S.S.; Greengard, P.; Xu, H. Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc. Natl. Acad. Sci. USA 1999, 96, 742–747. [Google Scholar] [CrossRef] [Green Version]
- Das, U.; Wang, L.; Ganguly, A.; Saikia, J.M.; Wagner, S.L.; Koo, E.H.; Roy, S. Visualizing APP and BACE-1 approximation in neurons yields insight into the amyloidogenic pathway. Nat. Neurosci. 2016, 19, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Gowrishankar, S.; Wu, Y.; Ferguson, S.M. Impaired JIP3-dependent axonal lysosome transport promotes amyloid plaque pathology. J. Cell Biol. 2017, 216, 3291–3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, A.; Sisodia, S.S.; Trowbridge, I.S. Characterization of sorting signals in the beta-amyloid precursor protein cytoplasmic domain. J. Biol. Chem. 1995, 270, 3565–3573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, S.I.; Rebelo, S.; Esselmann, H.; Wiltfang, J.; Lah, J.; Lane, R.; Small, S.A.; Gandy, S.; da Cruz, E.; Silva, E.F.; et al. Retrieval of the Alzheimer’s amyloid precursor protein from the endosome to the TGN is S655 phosphorylation state-dependent and retromer-mediated. Mol. Neurodegener. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, O.M.; Reiche, J.; Schmidt, V.; Gotthardt, M.; Spoelgen, R.; Behlke, J.; von Arnim, C.A.F.; Breiderhoff, T.; Jansen, P.; Wu, X.; et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2005, 102, 13461–13466. [Google Scholar] [CrossRef] [Green Version]
- Fjorback, A.W.; Seaman, M.; Gustafsen, C.; Mehmedbasic, A.; Gokool, S.; Wu, C.; Militz, D.; Schmidt, V.; Madsen, P.; Nyengaard, J.R.; et al. Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J. Neurosci. 2012, 32, 1467–1480. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, Y.-F.; Zhang, L.; Huang, L.; Yu, P.; Zhu, H.; Deng, W.; Qin, C. Effective expression of Drebrin in hippocampus improves cognitive function and alleviates lesions of Alzheimer’s disease in APP (swe)/PS1 (ΔE9) mice. CNS Neurosci. 2017, 23, 590–604. [Google Scholar] [CrossRef]
- Harigaya, Y.; Shoji, M.; Shirao, T.; Hirai, S. Disappearance of actin-binding protein, drebrin, from hippocampal synapses in Alzheimer’s disease. J. Neurosci. Res. 1996, 43, 87–92. [Google Scholar] [CrossRef]
- Counts, S.E.; Nadeem, M.; Lad, S.P.; Wuu, J.; Mufson, E.J. Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J. Neuropathol. Exp. Neurol. 2006, 65, 592–601. [Google Scholar] [CrossRef] [Green Version]
- Hatanpää, K.; Isaacs, K.R.; Shirao, T.; Brady, D.R.; Rapoport, S.I. Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1999, 58, 637–643. [Google Scholar] [CrossRef]
- Mendoza-Naranjo, A.; Gonzalez-Billault, C.; Maccioni, R.B. Abeta1-42 stimulates actin polymerization in hippocampal neurons through Rac1 and Cdc42 Rho GTPases. J. Cell. Sci. 2007, 120, 279–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borin, M.; Saraceno, C.; Catania, M.; Lorenzetto, E.; Pontelli, V.; Paterlini, A.; Fostinelli, S.; Avesani, A.; Di Fede, G.; Zanusso, G.; et al. Rac1 activation links tau hyperphosphorylation and Aβ dysmetabolism in Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galloway, P.G.; Mulvihill, P.; Siedlak, S.; Mijares, M.; Kawai, M.; Padget, H.; Kim, R.; Perry, G. Immunochemical demonstration of tropomyosin in the neurofibrillary pathology of Alzheimer’s disease. Am. J. Pathol. 1990, 137, 291–300. [Google Scholar] [PubMed]
- Feuillette, S.; Deramecourt, V.; Laquerriere, A.; Duyckaerts, C.; Delisle, M.-B.; Maurage, C.-A.; Blum, D.; Buée, L.; Frébourg, T.; Campion, D.; et al. Filamin-A and Myosin VI colocalize with fibrillary Tau protein in Alzheimer’s disease and FTDP-17 brains. Brain Res. 2010, 1345, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Hirano, A. Hirano bodies and related neuronal inclusions. Neuropathol. Appl. Neurobiol. 1994, 20, 3–11. [Google Scholar] [CrossRef]
- Mitake, S.; Ojika, K.; Hirano, A. Hirano bodies and Alzheimer’s disease. Kaohsiung J. Med. Sci. 1997, 13, 10–18. [Google Scholar]
- Galloway, P.G.; Perry, G.; Gambetti, P. Hirano body filaments contain actin and actin-associated proteins. J. Neuropathol. Exp. Neurol. 1987, 46, 185–199. [Google Scholar] [CrossRef]
- Maciver, S.K.; Harrington, C.R. Two actin binding proteins, actin depolymerizing factor and cofilin, are associated with Hirano bodies. Neuroreport 1995, 6, 1985–1988. [Google Scholar] [CrossRef]
- Castaño, E.M.; Maarouf, C.L.; Wu, T.; Leal, M.C.; Whiteside, C.M.; Lue, L.-F.; Kokjohn, T.A.; Sabbagh, M.N.; Beach, T.G.; Roher, A.E. Alzheimer disease periventricular white matter lesions exhibit specific proteomic profile alterations. Neurochem. Int. 2013, 62, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Owen, J.B.; Di Domenico, F.; Sultana, R.; Perluigi, M.; Cini, C.; Pierce, W.M.; Butterfield, D.A. Proteomics-determined differences in the concanavalin-A-fractionated proteome of hippocampus and inferior parietal lobule in subjects with Alzheimer’s disease and mild cognitive impairment: Implications for progression of AD. J. Proteome Res. 2009, 8, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Shahid-Salles, S.; Sherling, D.; Fechheimer, N.; Iyer, N.; Wells, L.; Fechheimer, M.; Furukawa, R. De novo actin polymerization is required for model Hirano body formation in Dictyostelium. Biol. Open 2016, 5, 807–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minamide, L.S.; Striegl, A.M.; Boyle, J.A.; Meberg, P.J.; Bamburg, J.R. Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat. Cell Biol. 2000, 2, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Rahman, T.; Davies, D.S.; Tannenberg, R.K.; Fok, S.; Shepherd, C.; Dodd, P.R.; Cullen, K.M.; Goldsbury, C. Cofilin rods and aggregates concur with tau pathology and the development of Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 1443–1460. [Google Scholar] [CrossRef] [PubMed]
- Maloney, M.T.; Minamide, L.S.; Kinley, A.W.; Boyle, J.A.; Bamburg, J.R. Beta-secretase-cleaved amyloid precursor protein accumulates at actin inclusions induced in neurons by stress or amyloid beta: A feedforward mechanism for Alzheimer’s disease. J. Neurosci. 2005, 25, 11313–11321. [Google Scholar] [CrossRef]
- Cichon, J.; Sun, C.; Chen, B.; Jiang, M.; Chen, X.A.; Sun, Y.; Wang, Y.; Chen, G. Cofilin aggregation blocks intracellular trafficking and induces synaptic loss in hippocampal neurons. J. Biol. Chem. 2012, 287, 3919–3929. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, B.W.; Shaw, A.E.; Minamide, L.S.; Pak, C.W.; Bamburg, J.R. Incorporation of cofilin into rods depends on disulfide intermolecular bonds: Implications for actin regulation and neurodegenerative disease. J. Neurosci. 2012, 32, 6670–6681. [Google Scholar] [CrossRef]
- Chen, B.; Wang, Y. Cofilin rod formation in neurons impairs neuronal structure and function. Cns Neurol. Disord. Drug Targets 2015, 14, 554–560. [Google Scholar] [CrossRef]
- Kang, D.E.; Woo, J.A. Cofilin, a Master Node Regulating Cytoskeletal Pathogenesis in Alzheimer’s Disease. J. Alzheimers Dis. 2019, 278, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, B.W.; Bamburg, J.R. Actin-ATP hydrolysis is a major energy drain for neurons. J. Neurosci. 2003, 23, 1–6. [Google Scholar] [CrossRef]
- Bernstein, B.W.; Chen, H.; Boyle, J.A.; Bamburg, J.R. Formation of actin-ADF/cofilin rods transiently retards decline of mitochondrial potential and ATP in stressed neurons. Am. J. Physiol. Cell Physiol. 2006, 291, C828–C839. [Google Scholar] [CrossRef] [Green Version]
- Barone, E.; Mosser, S.; Fraering, P.C. Inactivation of brain Cofilin-1 by age, Alzheimer’s disease and γ-secretase. Biochim. Biophys. Acta 2014, 1842, 2500–2509. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.; Vidal, G.S.; Djurisic, M.; William, C.M.; Birnbaum, M.E.; Garcia, K.C.; Hyman, B.T.; Shatz, C.J. Human LilrB2 is a β-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science 2013, 341, 1399–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, J.A.; Jung, A.R.; Lakshmana, M.K.; Bedrossian, A.; Lim, Y.; Bu, J.H.; Park, S.A.; Koo, E.H.; Mook-Jung, I.; Kang, D.E. Pivotal role of the RanBP9-cofilin pathway in Aβ-induced apoptosis and neurodegeneration. Cell Death Differ. 2012, 19, 1413–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshmana, M.K.; Chung, J.Y.; Wickramarachchi, S.; Tak, E.; Bianchi, E.; Koo, E.H.; Kang, D.E. A fragment of the scaffolding protein RanBP9 is increased in Alzheimer’s disease brains and strongly potentiates amyloid-beta peptide generation. Faseb J. 2010, 24, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rush, T.; Martinez-Hernandez, J.; Dollmeyer, M.; Frandemiche, M.L.; Borel, E.; Boisseau, S.; Jacquier-Sarlin, M.; Buisson, A. Synaptotoxicity in Alzheimer’s Disease Involved a Dysregulation of Actin Cytoskeleton Dynamics through Cofilin 1 Phosphorylation. J. Neurosci. 2018, 38, 10349–10361. [Google Scholar] [CrossRef] [Green Version]
- Rust, M.B.; Gurniak, C.B.; Renner, M.; Vara, H.; Morando, L.; Görlich, A.; Sassoè-Pognetto, M.; Banchaabouchi, M.A.; Giustetto, M.; Triller, A.; et al. Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. Embo J. 2010, 29, 1889–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opazo, P.; Choquet, D. A three-step model for the synaptic recruitment of AMPA receptors. Mol. Cell. Neurosci. 2011, 46, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Lee, C.W.; Fan, Y.; Komlos, D.; Tang, X.; Sun, C.; Yu, K.; Hartzell, H.C.; Chen, G.; Bamburg, J.R.; et al. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat. Neurosci 2010, 13, 1208–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, J.-A.A.; Liu, T.; Fang, C.C.; Cazzaro, S.; Kee, T.; LePochat, P.; Yrigoin, K.; Penn, C.; Zhao, X.; Wang, X.; et al. Activated cofilin exacerbates tau pathology by impairing tau-mediated microtubule dynamics. Commun. Biol. 2019, 2, 112. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, L.; Rust, M.B.; Culmsee, C. Actin(g) on mitochondria—a role for cofilin1 in neuronal cell death pathways. Biol. Chem. 2019, 400, 1089–1097. [Google Scholar] [CrossRef]
- Rehklau, K.; Hoffmann, L.; Gurniak, C.B.; Ott, M.; Witke, W.; Scorrano, L.; Culmsee, C.; Rust, M.B. Cofilin1-dependent actin dynamics control DRP1-mediated mitochondrial fission. Cell Death Dis. 2017, 8, e3063. [Google Scholar] [CrossRef] [PubMed]
- Klamt, F.; Zdanov, S.; Levine, R.L.; Pariser, A.; Zhang, Y.; Zhang, B.; Yu, L.-R.; Veenstra, T.D.; Shacter, E. Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nat. Cell Biol. 2009, 11, 1241–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zhou, G.-L.; Vedantam, S.; Li, P.; Field, J. Mitochondrial shuttling of CAP1 promotes actin- and cofilin-dependent apoptosis. J. Cell. Sci. 2008, 121, 2913–2920. [Google Scholar] [CrossRef] [Green Version]
- Chua, B.T.; Volbracht, C.; Tan, K.O.; Li, R.; Yu, V.C.; Li, P. Mitochondrial translocation of cofilin is an early step in apoptosis induction. Nat. Cell Biol. 2003, 5, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Roh, S.-E.; Woo, J.A.; Lakshmana, M.K.; Uhlar, C.; Ankala, V.; Boggess, T.; Liu, T.; Hong, Y.-H.; Mook-Jung, I.; Kim, S.J.; et al. Mitochondrial dysfunction and calcium deregulation by the RanBP9-cofilin pathway. Faseb J. 2013, 27, 4776–4789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Woo, J.-A.A.; Yan, Y.; LePochat, P.; Bukhari, M.Z.; Kang, D.E. Dual role of cofilin in APP trafficking and amyloid-β clearance. Faseb J. 2019, 33, 14234–14247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; Vanleeuwen, J.-E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Penzes, P.; Vanleeuwen, J.-E. Impaired regulation of synaptic actin cytoskeleton in Alzheimer’s disease. Brain Res. Rev. 2011, 67, 184–192. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Protein | Physiological Function | Role in AD |
|---|---|---|
| Arp2/3 | Actin filaments nucleator: major actin-binding protein with polymerizing and filament branching activities | Required for Hirano body formation in Dictyostelium |
| Profilin | Responsible for the ADP to ATP nucleotide exchange on actin | Required for model Hirano body formation in Dictyostelium |
| Rho family of GTPases | Intermediaries between external signals and internal actin organization | - Increased activity of Rac1/Cdc42 Rho GTPases upon fibrillar Aβ exposure - Rac1 is increased in plasma samples of AD patients and abnormally activated in AD mice at early stages |
| ADF/cofilin | Promote the actin turnover | - Identified in the Hirano bodies and main component of actin rods -Alterations of its activation in in vitro and in vivo AD models |
| Myosins V and VI | Actin-based motor proteins | Myosin VI colocalizes with tau protein accumulated in neurons of AD patients |
| Drebrin | Bundles actin filaments by crosslinking | Decreased in the hippocampal dendritic spines of AD patients and in the hippocampus of AD mice |
| α-actinin | Form dimers that crosslink actin filaments | Component of the Hirano bodies |
| Tropomyosins | Form head-to-tail polymers to stabilize the actin filament and recruit myosin | Component of neurofibrillary pathology and Hirano bodies |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelucchi, S.; Stringhi, R.; Marcello, E. Dendritic Spines in Alzheimer’s Disease: How the Actin Cytoskeleton Contributes to Synaptic Failure. Int. J. Mol. Sci. 2020, 21, 908. https://doi.org/10.3390/ijms21030908
Pelucchi S, Stringhi R, Marcello E. Dendritic Spines in Alzheimer’s Disease: How the Actin Cytoskeleton Contributes to Synaptic Failure. International Journal of Molecular Sciences. 2020; 21(3):908. https://doi.org/10.3390/ijms21030908
Chicago/Turabian StylePelucchi, Silvia, Ramona Stringhi, and Elena Marcello. 2020. "Dendritic Spines in Alzheimer’s Disease: How the Actin Cytoskeleton Contributes to Synaptic Failure" International Journal of Molecular Sciences 21, no. 3: 908. https://doi.org/10.3390/ijms21030908
APA StylePelucchi, S., Stringhi, R., & Marcello, E. (2020). Dendritic Spines in Alzheimer’s Disease: How the Actin Cytoskeleton Contributes to Synaptic Failure. International Journal of Molecular Sciences, 21(3), 908. https://doi.org/10.3390/ijms21030908