Unique k-mers as Strain-Specific Barcodes for Phylogenetic Analysis and Natural Microbiome Profiling


Abstract
:1. Introduction
2. Results
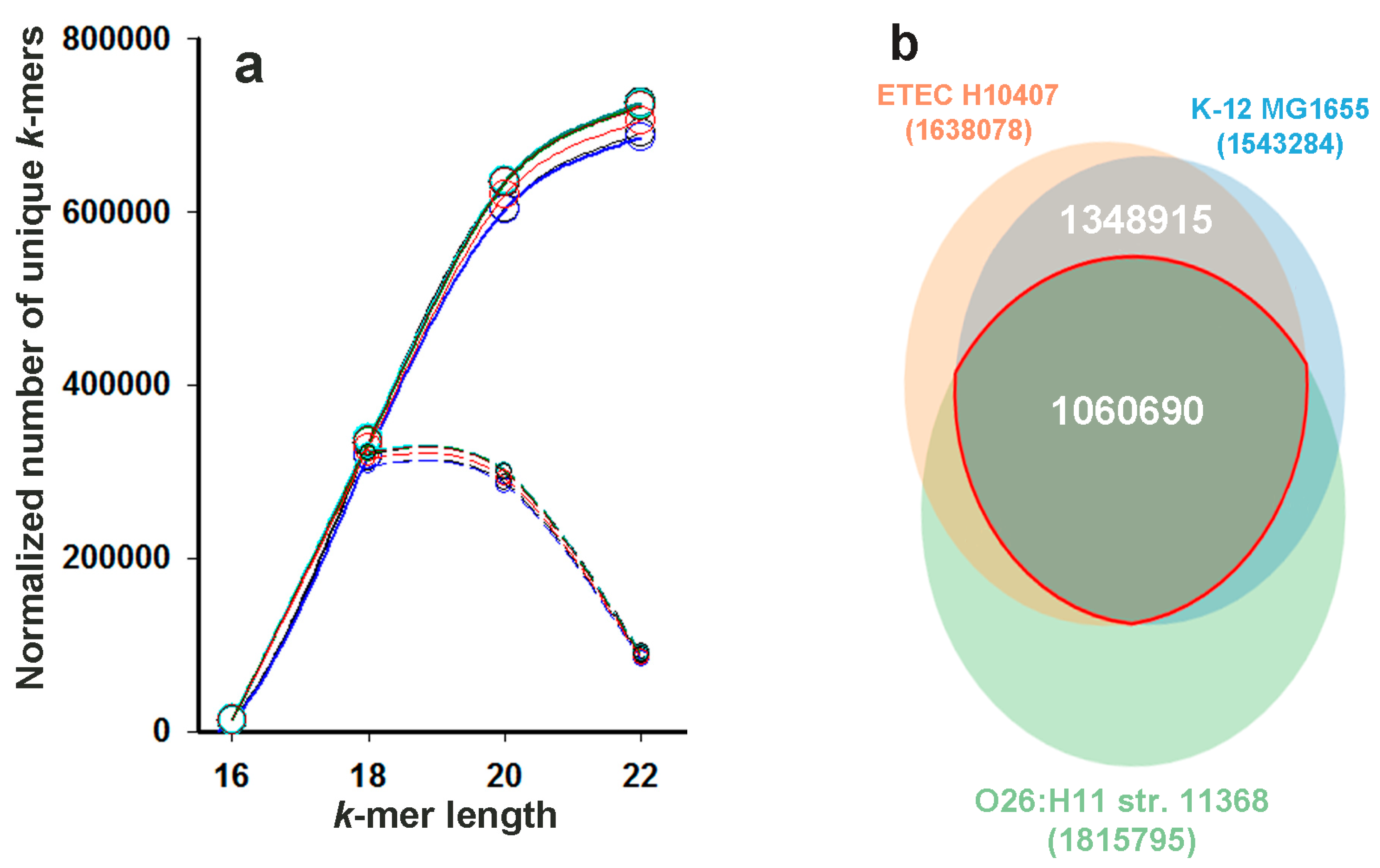
2.1. Selection of k-Values for Phylogenetic and Taxonomic Analysis
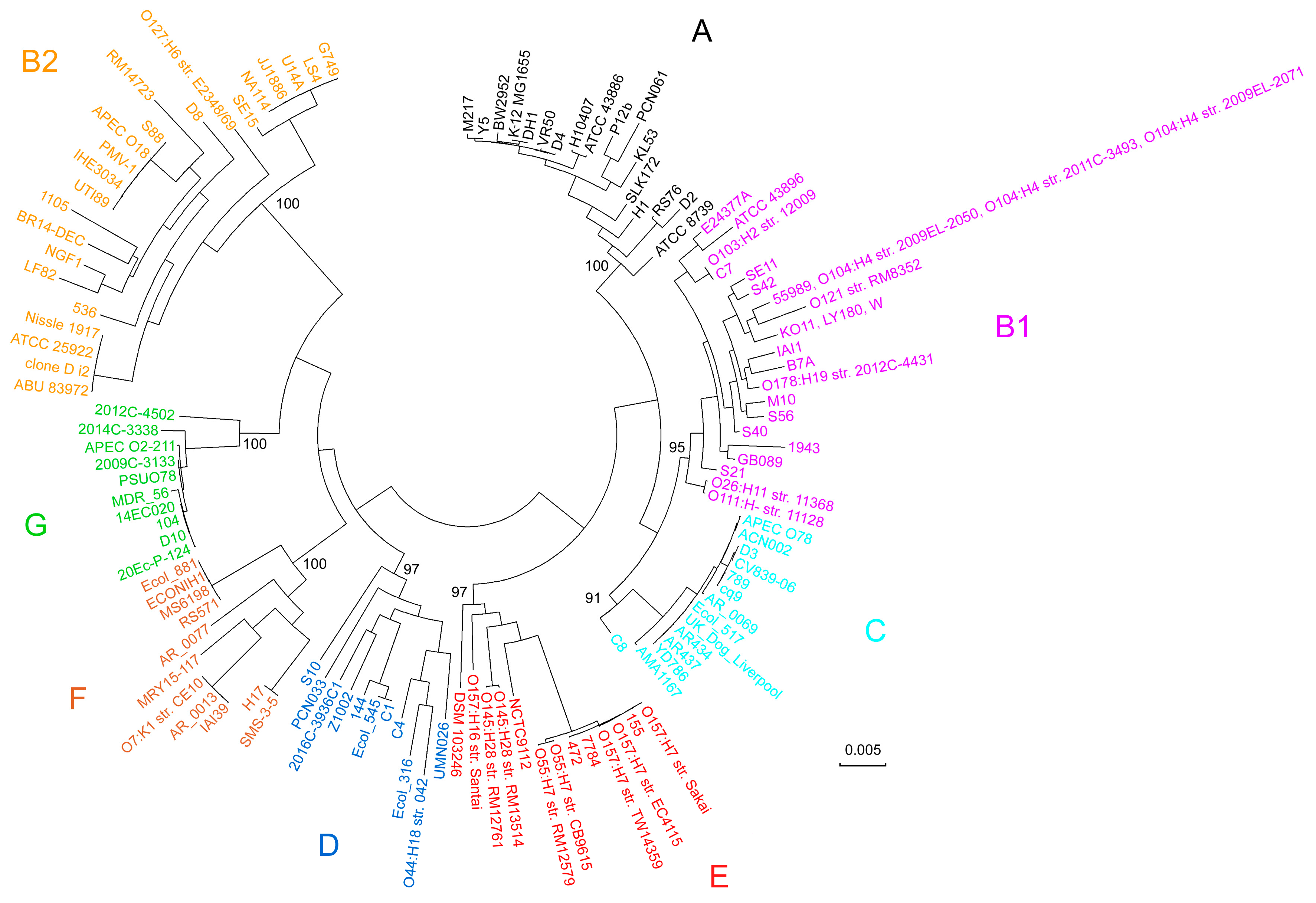
2.2. Alignment-Based Multilocus Sequence Typing Resulted in Tree with Expected Topology and Predicted New Members for E. coli Phylogroups
2.3. Phylotyping Based on Unique 18-mers and 22-mers Result in Identical Trees with the Same Topology as the Alignment-Based Approach
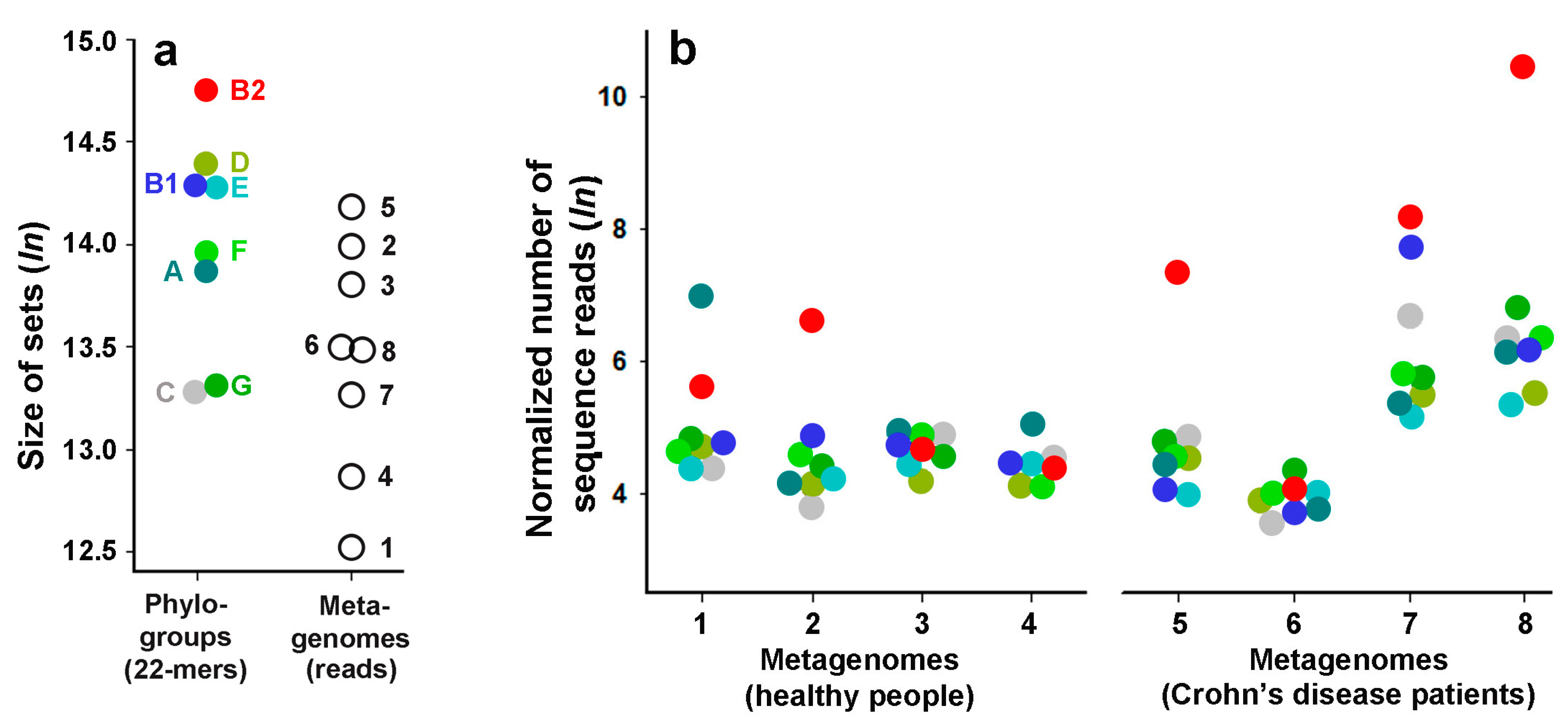
2.4. Phylogroup-Dependent Profiling of E. coli Presence in Human Intestinal Microbiomes
3. Discussion
4. Materials and Methods
4.1. Database
4.2. Outlines of UniSeq and Identification of “used” K-Mers
- a) compute(h[z], Cd(z)) for z;
- b) if z is degenerated, then continue 3);
- c) compute Id[p] that is min{(h[z’],Cd[z’]), (h[z],Cd(z)};
- if(Index[Id[p].h] is not equal to 0) then go to 3; //because such Id was already registered//
- else//save Id components of used k-mer at position p
- {Index[Id[p].h]:=1; h[nu]:=Id[p].h]; Cd[nu]:=Id[p].Cd;
- nu+=1;
- }
4.3. Detection of Unique K-Mers in the Genomes
4.4. Phylogenetic Inference
4.5. E. coli Phylogroup Taxonomy of Metagenomic Data
4.6. Ethics Statement
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetrovsky, T.; Baldrian, P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS ONE 2013, 8, e57923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, A.F.; Lindberg, M.; Jakobsson, H.; Backhed, F.; Nyren, P.; Engstrand, L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 2008, 3, e2836. [Google Scholar] [CrossRef] [PubMed]
- Glaeser, S.P.; Kampfer, P. Multilocus sequence analysis (MLSA) in prokaryotic taxonomy. Syst. Appl. Microbiol. 2015, 38, 237–245. [Google Scholar] [CrossRef]
- Bernard, G.; Chan, C.X.; Ragan, M.A. Alignment-free microbial phylogenomics under scenarios of sequence divergence, genome rearrangement and lateral genetic transfer. Sci. Rep. 2016, 6, 28970. [Google Scholar] [CrossRef] [Green Version]
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-free sequence comparison: Benefits, applications, and tools. Genome Biol. 2017, 18, 186. [Google Scholar] [CrossRef] [Green Version]
- Blaisdell, B.E. A measure of the similarity of sets of sequences not requiring sequence alignment. Proc. Natl. Acad. Sci. USA 1986, 83, 5155–5159. [Google Scholar] [CrossRef] [Green Version]
- Brendel, V.; Beckmann, J.S.; Trifonov, E.N. Linguistics of nucleotide sequences: Morphology and comparison of vocabularies. J. Biomol. Struct. Dyn. 1986, 4, 11–21. [Google Scholar] [CrossRef]
- Pevsner, P.A. l-Tuple DNA sequencing: Computer analysis. J. Biomol. Struct. Dyn. 1989, 7, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT - the blast-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kielbasa, S.M.; Wan, R.; Sato, K.; Horton, P.; Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hach, F.; Hormozdiari, F.; Alkan, C.; Hormozdiari, F.; Birol, I.; Eichler, E.E.; Sahinalp, S.C. mrsFAST: A cache-oblivious algorithm for short-read mapping. Nat. Methods 2010, 7, 576–577. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Patel, J.M.; Terrell, A. Wham: A high-throughput sequence alignment method. ACM Transact. Database Syst. 2012, 37, 28. [Google Scholar] [CrossRef]
- Batzoglou, S.; Jaffe, D.B.; Stanley, K.; Butler, J.; Gnerre, S.; Mauceli, E.; Berger, B.; Mesirov, J.P.; Lander, E.S. Arachne: A whole-genome shotgun assembler. Genome Res. 2002, 12, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [Green Version]
- Compeau, P.E.C.; Pevzner, P.A.; Tesler, G. How to apply de Bruijn graphs to genome assembly. Nature Biotechnol. 2011, 29, 987–991. [Google Scholar] [CrossRef]
- Mahadik, K.; Wright, C.; Kulkarni, M.; Bagchi, S.; Chaterji, S. Scalable genome assembly through parallel de Bruijn graph construction for multiple k-mers. Sci. Rep. 2019, 9, 14882. [Google Scholar] [CrossRef]
- Kurtz, S.; Narechania, A.; Stein, J.C.; Ware, D. A new method to compute k-mer frequencies and its application to annotate large repetitive plant genomes. BMC Genom. 2008, 9, 517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mapleson, D.; Garcia Accinelli, G.; Kettleborough, G.; Wright, J.; Clavijo, B.J. KAT: A k-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics. 2017, 33, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, K.J.; Albani, M.C.; James, G.V.; Gutjahr, C.; Hartwig, B.; Turck, F.; Paszkowski, U.; Coupland, G.; Schneeberger, K. Mutation identification by direct comparison of whole-genome sequencing data from mutant and wild-type individuals using k-mers. Nat. Biotechnol. 2013, 31, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.R.; Schatz, M.C.; Salzberg, S.L. Quake: Quality-aware detection and correction of sequencing errors. Genome Biol. 2010, 11, R116. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Dorman, K.S.; Aluru, S. Reptile: Representative tiling for short read error correction. Bioinformatics. 2010, 26, 2526–2533. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Schroder, J.; Schmidt, B. Musket: A multistage k-mer spectrum-based error corrector for Illumina sequence data. Bioinformatics. 2013, 29, 308–315. [Google Scholar] [CrossRef]
- Song, L.; Florea, L. Rcorrector: Efficient and accurate error correction for Illumina RNA-seq reads. Gigascience. 2015, 4, 48. [Google Scholar] [CrossRef] [Green Version]
- Drouin, A.; Giguere, S.; Deraspe, M.; Marchand, M.; Tyers, M.; Loo, V.G.; Bourgault, A.M.; Laviolette, F.; Corbeil, J. Predictive computational phenotyping and biomarker discovery using reference-free genome comparisons. BMC Genom. 2016, 17, 754. [Google Scholar] [CrossRef] [Green Version]
- Aun, E.; Brauer, A.; Kisand, V.; Tenson, T.; Remm, M.A. k-mer-based method for the identification of phenotype-associated genomic biomarkers and predicting phenotypes of sequenced bacteria. PLoS Comput. Biol. 2018, 14, e1006434. [Google Scholar] [CrossRef] [Green Version]
- Mahe, P.; Tournoud, M. Predicting bacterial resistance from whole-genome sequences using k-mers and stability selection. BMC Bioinform. 2018, 19, 383. [Google Scholar] [CrossRef] [PubMed]
- Maguire, F.; Rehman, M.A.; Carrillo, C.; Diarra, M.S.; Beiko, R.G. Identification of primary antimicrobial resistance drivers in agricultural nontyphoidal Salmonella enterica serovars by using machine learning. mSystems 2019, 4, e00211–e00219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitwieser, F.P.; Lu, J.; Salzberg, S.L. A review of methods and databases for metagenomic classification and assembly. Brief Bioinform. 2019, 20, 1125–1139. [Google Scholar] [CrossRef] [PubMed]
- Tu, Q.; He, Z.; Deng, Y.; Zhou, J. Strain/species-specific probe design for microbial identification microarrays. Appl. Environ. Microbiol. 2013, 79, 5085–5088. [Google Scholar] [CrossRef] [Green Version]
- Tu, Q.; He, Z.; Zhou, J. Strain/species identification in metagenomes using genome-specific markers. Nucleic Acids Res. 2014, 42, e67. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [Green Version]
- Breitwieser, F.P.; Baker, D.N.; Salzberg, S.L. KrakenUniq: Confident and fast metagenomics classification using unique k-mer counts. Genome Biol. 2018, 19, 198. [Google Scholar] [CrossRef] [Green Version]
- Ounit, R.; Wanamaker, S.; Close, T.J.; Lonardi, S. CLARK: Fast and accurate classification of metagenomic and genomic sequences using discriminative k-mer. BMC Genom. 2015, 16, 236. [Google Scholar] [CrossRef] [Green Version]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [Green Version]
- Freitas, T.A.; Li, P.E.; Scholz, M.B.; Chain, P.S. Accurate read-based metagenome characterization using a hierarchical suite of unique signatures. Nucleic Acids Res. 2015, 43, e69. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Liao, X.; Zhu, W.; Fang, L.; Chen, X. An efficient classification algorithm for NGS data based on text similarity. Genet. Res. 2018, 100, e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Yu, Y.; Liu, J.; Elliott, C.F.; Qian, C.; Liu, J. A novel data structure to support ultra-fast taxonomic classification of metagenomic sequences with k-mer signatures. Bioinformatics 2018, 34, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Petit Iii, R.A.; Hogan, J.M.; Ezewudo, M.N.; Joseph, S.J.; Read, T.D. Fine-scale differentiation between Bacillus anthracis and Bacillus cereus group signatures in metagenome shotgun data. Peer J. 2018, 6, e5515. [Google Scholar] [CrossRef] [PubMed]
- Panyukov, V.V.; Kiselev, S.S.; Alikina, O.V.; Nazipova, N.N.; Ozoline, O.N. Short unique sequences in bacterial genomes as strain- and species-specific signatures. Math. Biol. Bioinf. 2017, 12, 547–558. [Google Scholar] [CrossRef]
- Clermont, O.; Christenson, J.K.; Denamur, E.; Gordon, D.M. The Clermont Escherichia coli phylo-typing method revisited: Improvement of specificity and detection of new phylo-groups. Environ. Microbiol. Rep. 2013, 5, 58–65. [Google Scholar] [CrossRef]
- Clermont, O.; Dixit, O.V.A.; Vangchhia, B.; Condamine, B.; Dion, S.; Bridier-Nahmias, A.; Denamur, E.; Gordon, D. Characterization and rapid identification of phylogroup G in Escherichia coli, a lineage with high virulence and antibiotic resistance potential. Environ. Microbiol. 2019, 21, 3107–3117. [Google Scholar] [CrossRef]
- Clermont, O.; Bonacorsi, S.; Bingen, E. Rapid and simple determination of the Escherichia coli phylogenetic group. Appl. Environ. Microbiol. 2000, 66, 4555–4558. [Google Scholar] [CrossRef] [Green Version]
- Escobar-Paramo, P.; Clermont, O.; Blanc-Potard, A.B.; Bui, H.; Le Bouguenec, C.; Denamur, E. A specific genetic background is required for acquisition and expression of virulence factors in Escherichia coli. Mol. Biol. Evol. 2004, 21, 1085–1094. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.R.; Owens, K.L.; Clabots, C.R.; Weissman, S.J.; Cannon, S.B. Phylogenetic relationships among clonal groups of extraintestinal pathogenic Escherichia coli as assessed by multi-locus sequence analysis. Microbes Infect. 2006, 8, 1702–1713. [Google Scholar] [CrossRef]
- Gordon, D.M.; Clermont, O.; Tolley, H.; Denamur, E. Assigning Escherichia coli strains to phylogenetic groups: Multi-locus sequence typing versus the PCR triplex method. Environ. Microbiol. 2008, 10, 2484–2496. [Google Scholar] [CrossRef]
- Jaureguy, F.; Landraud, L.; Passet, V.; Diancourt, L.; Frapy, E.; Guigon, G.; Carbonnelle, E.; Lortholary, O.; Clermont, O.; Denamur, E.; et al. Phylogenetic and genomic diversity of human bacteremic Escherichia coli strains. BMC Genom. 2008, 9, 560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clermont, O.; Olier, M.; Hoede, C.; Diancourt, L.; Brisse, S.; Keroudean, M.; Glodt, J.; Picard, B.; Oswald, E.; Denamur, E. Animal and human pathogenic Escherichia coli strains share common genetic backgrounds. Infect. Genet. Evol. 2011, 11, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Jin, D.; Wu, S.; Yang, J.; Lan, R.; Bai, X.; Liu, S.; Meng, Q.; Yuan, X.; Zhou, J.; et al. Insights into the evolution of pathogenicity of Escherichia coli from genomic analysis of intestinal E. coli of Marmota himalayana in Qinghai-Tibet plateau of China. Emerg. Microbes Infect. 2016, 5, e122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venn Diagram Maker. Available online: https://www.meta-chart.com/venn#/data (accessed on 28 January 2020).
- Bohlin, J.; Brynildsrud, O.; Sekse, C.; Snipen, L. An evolutionary analysis of genome expansion and pathogenicity in Escherichia coli. BMC Genom. 2014, 15, 882. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zheng, H.; Yang, M.; Xu, Z.; Wang, X.; Wei, L.; Tang, B.; Liu, F.; Zhang, Y.; Ding, Y.; et al. Genome analysis and in vivo virulence of porcine extraintestinal pathogenic Escherichia coli strain PCN033. BMC Genom. 2015, 16, 717. [Google Scholar] [CrossRef] [Green Version]
- Massip, C.; Branchu, P.; Bossuet-Greif, N.; Chagneau, C.V.; Gaillard, D.; Martin, P.; Boury, M.; Secher, T.; Dubois, D.; Nougayrede, J.P.; et al. Deciphering the interplay between the genotoxic and probiotic activities of Escherichia coli Nissle 1917. PLoS Pathog. 2019, 15, e1008029. [Google Scholar] [CrossRef]
- Wagner, S.; Lupolova, N.; Gally, D.L.; Argyle, S.A. Convergence of plasmid architectures drives emergence of multi-drug resistance in a clonally diverse Escherichia coli population from a veterinary clinical care setting. Vet. Microbiol. 2017, 211, 6–14. [Google Scholar] [CrossRef]
- Zhang, Y.; Lin, K. A phylogenomic analysis of Escherichia coli / Shigella group: Implications of genomic features associated with pathogenicity and ecological adaptation. BMC Evol. Biol. 2012, 12, 174. [Google Scholar] [CrossRef] [Green Version]
- Clermont, O.; Gordon, D.; Denamur, E. Guide to the various phylogenetic classification schemes for Escherichia coli and the correspondence among schemes. Microbiology 2015, 161, 980–988. [Google Scholar] [CrossRef]
- Peris-Bondia, F.; Muraille, E.; Van Melderen, L. Complete genome sequence of the Escherichia coli PMV-1 strain, a model extraintestinal pathogenic E. coli strain used for host-pathogen interaction studies. Genome Announc. 2013, 1, e00913-13. [Google Scholar] [CrossRef] [Green Version]
- Geddes, R.D.; Wang, X.; Yomano, L.P.; Miller, E.N.; Zheng, H.; Shanmugam, K.T.; Ingram, L.O. Polyamine transporters and polyamines increase furfural tolerance during xylose fermentation with ethanologenic Escherichia coli strain LY180. Appl. Environ. Microbiol. 2014, 80, 5955–5964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forde, B.M.; Ben Zakour, N.L.; Stanton-Cook, M.; Phan, M.D.; Totsika, M.; Peters, K.M.; Chan, K.G.; Schembri, M.A.; Upton, M.; Beatson, S.A. The complete genome sequence of Escherichia coli EC958: A high quality reference sequence for the globally disseminated multidrug resistant E. coli O25b:H4-ST131 clone. PLoS ONE 2014, 9, e104400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forde, B.M.; Roberts, L.W.; Phan, M.D.; Peters, K.M.; Fleming, B.A.; Russell, C.W.; Lenherr, S.M.; Myers, J.B.; Barker, A.P.; Fisher, M.A.; et al. Population dynamics of an Escherichia coli ST131 lineage during recurrent urinary tract infection. Nat. Commun. 2019, 10, 3643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beatson, S.A.; Ben Zakour, N.L.; Totsika, M.; Forde, B.M.; Watts, R.E.; Mabbett, A.N.; Szubert, J.M.; Sarkar, S.; Phan, M.D.; Peters, K.M.; et al. Molecular analysis of asymptomatic bacteriuria Escherichia coli strain VR50 reveals adaptation to the urinary tract by gene acquisition. Infect. Immun. 2015, 83, 1749–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, T.J.; Danzeisen, J.L.; Youmans, B.; Case, K.; Llop, K.; Munoz-Aguayo, J.; Flores-Figueroa, C.; Aziz, M.; Stoesser, N.; Sokurenko, E.; et al. Separate F-type plasmids have shaped the evolution of the H30 subclone of Escherichia coli sequence type 131. MSphere 2016, 1, e00121-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugawara, Y.; Akeda, Y.; Sakamoto, N.; Takeuchi, D.; Motooka, D.; Nakamura, S.; Hagiya, H.; Yamamoto, N.; Nishi, I.; Yoshida, H.; et al. Genetic characterization of blaNDM-harboring plasmids in carbapenem-resistant Escherichia coli from Myanmar. PLoS ONE 2017, 12, e0184720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, M.Q.; Pham, A. Complete genome sequences of two atypical enteropathogenic Escherichia coli O145 environmental strains. Genome Announc. 2018, 6, e00418-18. [Google Scholar] [CrossRef] [Green Version]
- Rasko, D.A.; Del Canto, F.; Luo, Q.; Fleckenstein, J.M.; Vidal, R.; Hazen, T.H. Comparative genomic analysis and molecular examination of the diversity of enterotoxigenic Escherichia coli isolates from Chile. PLoS Negl. Trop. Dis. 2019, 13, e0007828. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Sorensen, T. A method of establishing groups of equal amplitude in plant sociology based on similarity of species content. Kongelige Danske Videnskabernes Selskab. Biol. krifter. 1948, 4, 1–34. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef]
- Costea, P.I.; Coelho, L.P.; Sunagawa, S.; Munch, R.; Huerta-Cepas, J.; Forslund, K.; Hildebrand, F.; Kushugulova, A.; Zeller, G.; Bork, P. Subspecies in the global human gut microbiome. Mol. Syst. Biol. 2017, 13, 960. [Google Scholar] [CrossRef] [Green Version]
- Buchholz, U.; Bernard, H.; Werber, D.; Bohmer, M.M.; Remschmidt, C.; Wilking, H.; Delere, Y.; an der Heiden, M.; Adlhoch, C.; Dreesman, J.; et al. German outbreak of Escherichia coli O104:H4 associated with sprouts. N. Engl. J. Med. 2011, 365, 1763–1770. [Google Scholar] [CrossRef]
- Frank, C.; Werber, D.; Cramer, J.P.; Askar, M.; Faber, M.; an der Heiden, M.; Bernard, H.; Fruth, A.; Prager, R.; Spode, A.; et al. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N. Engl. J. Med. 2011, 365, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.M.; O’Brien, C.L.; Pavli, P. Escherichia coli diversity in the lower intestinal tract of humans. Environ. Microbiol. Rep. 2015, 7, 642–648. [Google Scholar] [CrossRef]
- Cormen, T.H.; Stein, C.; Rivest, R.L.; Leiserson, C.E. Introduction to Algorithms, 2nd ed.; McGraw-Hill Higher Education: Cambridge, MA, USA, 2001; pp. 824–825. [Google Scholar]
- Qi, W.; Lacher, D.W.; Bumbaugh, A.C.; Hyma, K.E.; Quellette, L.M.; Large, T.M.; Tarr, C.L.; Whittam, T.S. EcMLST: An online database for multi locus sequence typing of pathogenic Escherichia coli. Comput Syst Bioinformatics Conf. 2004, 520–521. [Google Scholar] [CrossRef]
- Wirth, T.; Falush, D.; Lan, R.; Colles, F.; Mensa, P.; Wieler, L.H.; Karch, H.; Reeves, P.R.; Maiden, M.C.J.; Ochman, H.; et al. Sex and virulence in Escherichia coli: An evolutionary perspective. Mol. Microbiol. 2006, 60, 1136–1151. [Google Scholar] [CrossRef] [Green Version]
- Lescat, M.; Hoede, C.; Clermont, O.; Garry, L.; Darlu, P.; Tuffery, P.; Denamur, E.; Picard, B. aes, the gene encoding the esterase B in Escherichia coli, is a powerful phylogenetic marker of the species. BMC Microbiol. 2009, 9, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, G. Estimating the dimension of a model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Tavare, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Mathem. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Gu, X.; Fu, Y.X.; Li, W.H. Maximum likelihood estimation of the heterogeneity of substitution rate among nucleotide sites. Mol. Biol. Evol. 1995, 12, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [Green Version]
- Galaxy server. Available online: https://usegalaxy.org (accessed on 28 January 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylogroup | Number of Strains | Range in Size Variation for Sets of Marker 22-mers in Individual Genomes | Size of Core Sets | Size of Cumulative Sets | |
|---|---|---|---|---|---|
| Maximal | Minimal | ||||
| A | 17 | 143,024 | 24,726 | 232 | 1,055,426 |
| B1 | 25 | 161,117 | 72,365 | 143 | 1,600,260 |
| B2 | 23 | 515,073 | 379,072 | 29,343 | 2,539,510 |
| C | 14 | 148,829 | 56,030 | 8444 | 586,272 |
| D | 11 | 368,049 | 243,470 | 1298 | 1,778,210 |
| E | 13 | 463,307 | 292,542 | 10,213 | 1,582,445 |
| F | 11 | 355,845 | 248,277 | 20,640 | 1,159,521 |
| G | 10 | 235,711 | 146,632 | 51,125 | 599,863 |
| SRA ID of Metagenome (N) | A | B1 | B2 | C | D | E | F | G |
|---|---|---|---|---|---|---|---|---|
| Samples of healthy individuals | ||||||||
| SRX187518 (1) | 17 | 15 | 26 | 5 | 12 | 14 | 8 | 0 |
| SRX187521 (2) | 9382 | 223 | 5060 | 33 | 559 | 110 | 182 | 39 |
| SRX187522 (3) | 31 | 191 | 11,566 | 17 | 34 | 273 | 83 | 15 |
| SRX187523 (4) | 29 | 29 | 62 | 10 | 21 | 22 | 17 | 6 |
| Samples of Crohn’s Disease Patients | ||||||||
| SRX187524 (5) | 105 | 307 | 36,698 | 64 | 212 | 54 | 279 | 305 |
| SRX187525 (6) | 10 | 28 | 81 | 6 | 22 | 24 | 23 | 15 |
| SRX187526 (7) | 211 | 11388 | 38,389 | 3435 | 774 | 223 | 1147 | 420 |
| SRX187527 (8) | 944 | 4418 | 462,763 | 1292 | 1019 | 1104 | 1713 | 2024 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panyukov, V.V.; Kiselev, S.S.; Ozoline, O.N. Unique k-mers as Strain-Specific Barcodes for Phylogenetic Analysis and Natural Microbiome Profiling. Int. J. Mol. Sci. 2020, 21, 944. https://doi.org/10.3390/ijms21030944
Panyukov VV, Kiselev SS, Ozoline ON. Unique k-mers as Strain-Specific Barcodes for Phylogenetic Analysis and Natural Microbiome Profiling. International Journal of Molecular Sciences. 2020; 21(3):944. https://doi.org/10.3390/ijms21030944
Chicago/Turabian StylePanyukov, Valery V., Sergey S. Kiselev, and Olga N. Ozoline. 2020. "Unique k-mers as Strain-Specific Barcodes for Phylogenetic Analysis and Natural Microbiome Profiling" International Journal of Molecular Sciences 21, no. 3: 944. https://doi.org/10.3390/ijms21030944
APA StylePanyukov, V. V., Kiselev, S. S., & Ozoline, O. N. (2020). Unique k-mers as Strain-Specific Barcodes for Phylogenetic Analysis and Natural Microbiome Profiling. International Journal of Molecular Sciences, 21(3), 944. https://doi.org/10.3390/ijms21030944