Protofibrils of Amyloid-β are Important Targets of a Disease-Modifying Approach for Alzheimer’s Disease



{kind=link}
Abstract
:1. Introduction
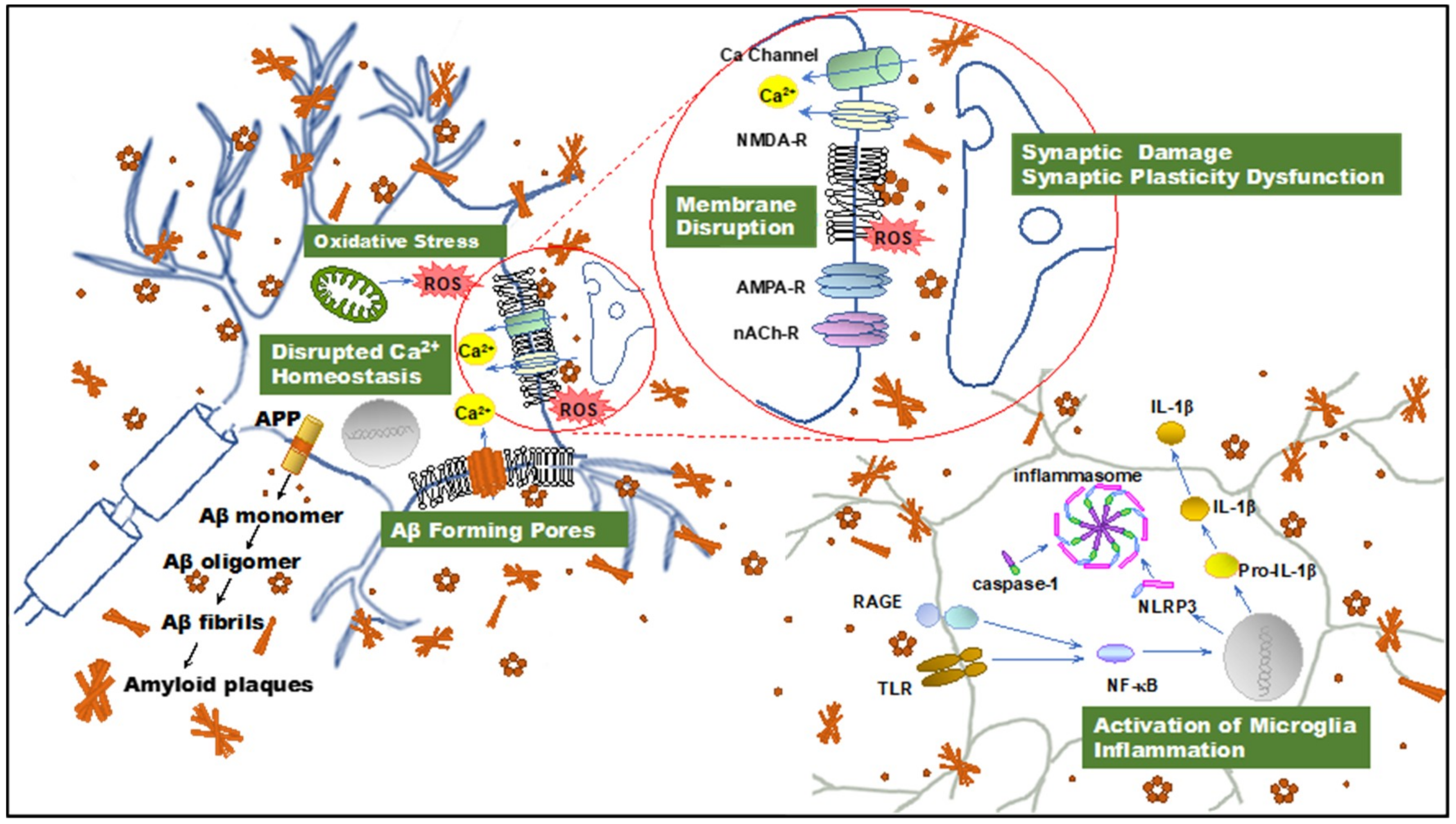
2. PFs Are Primary Toxins in AD
2.1. The Discovery of PFs and Their Role in AD Pathogenesis
2.2. PFs Are Primary Toxins in AD
2.3. Arctic Mutation Causes Aβ PF Formation
3. Therapeutic Approaches Targeting Aβ PFs
3.1. Small Molecules Inhibit the Formation of Aβ PFs
3.2. Aβ PF-Selective Antibody
3.3. Clinical Application of mAb158
4. PFs Are Present in Other Neurodegenerative Diseases
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aβ | amyloid β-protein |
| AD | Alzheimer’s disease |
| ADAS-Cog | Alzheimer’s Disease Assessment Scale cognitive subscale |
| ADCOMS | Alzheimer’s Disease Composite Score |
| AFM | atomic force microscopy |
| APP | amyloid precursor protein |
| ARIA | amyloid-related imaging abnormalities |
| αS | α-synuclein |
| CSF | cerebrospinal fluid |
| DMF | 1,2-(dimethoxymethano)fullerene |
| ELISA | enzyme-linked immunosorbent assay |
| EM | electron microscopy |
| HMW | high molecular weight |
| IL-1β | interleukin-1β |
| LDH | lactate dehydrogenase |
| LMW | low molecular weight |
| LTPs | long-term potentiation |
| MD | molecular dynamics |
| MTT | 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide metabolism |
| MyD | myeloid differentiation protein |
| NLR | Nod-like receptor |
| PFs | protofibrils |
| p-tau | phosphorylayed-tau |
| ROS | reactive oxygen species |
| SEC | size exclusion chromatography |
| SFRP1 | secreted-frizzled-related protein 1 |
| TLR | Toll-like receptor |
| TNFα | tumor necrosis factor α |
| TTR | transthyretin |
| WST | water soluble tetrazolium |
References
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K. Alzheimer’s disease as oligomeropathy. Neurochem. Int. 2018, 119, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Condron, M.M.; Teplow, D.B. Structure-neurotoxicity relationships of amyloid β-protein oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 14745–14750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, J.D.; Lansbury, P.T., Jr. Models of amyloid seeding in Alzheimer’s disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [Google Scholar] [CrossRef]
- Walsh, D.M.; Lomakin, A.; Benedek, G.B.; Condron, M.M.; Teplow, D.B. Amyloid β-protein fibrillogenesis. Detection of a protofibrillar intermediate. J. Biol. Chem. 1997, 272, 22364–22372. [Google Scholar] [CrossRef] [Green Version]
- Johansson, A.S.; Berglind-Dehlin, F.; Karlsson, G.; Edwards, K.; Gellerfors, P.; Lannfelt, L. Physiochemical characterization of the Alzheimer’s disease-related peptides Aβ 1-42Arctic and Aβ 1-42wt. FEBS J. 2006, 273, 2618–2630. [Google Scholar] [CrossRef]
- Hartley, D.M.; Walsh, D.M.; Ye, C.P.; Diehl, T.; Vasquez, S.; Vassilev, P.M.; Teplow, D.B.; Selkoe, D.J. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J. Neurosci. 1999, 19, 8876–8884. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.M.; Hartley, D.M.; Kusumoto, Y.; Fezoui, Y.; Condron, M.M.; Lomakin, A.; Benedek, G.B.; Selkoe, D.J.; Teplow, D.B. Amyloid β-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 1999, 274, 25945–25952. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.V.; Jennings, K.H.; Jepras, R.; Neville, W.; Owen, D.E.; Hawkins, J.; Christie, G.; Davis, J.B.; George, A.; Karran, E.H.; et al. Fractionation and characterization of oligomeric, protofibrillar and fibrillar forms of β-amyloid peptide. Biochem. J. 2000, 348, 137–144. [Google Scholar] [CrossRef]
- Johansson, A.S.; Garlind, A.; Berglind-Dehlin, F.; Karlsson, G.; Edwards, K.; Gellerfors, P.; Ekholm-Pettersson, F.; Palmblad, J.; Lannfelt, L. Docosahexaenoic acid stabilizes soluble amyloid-β protofibrils and sustains amyloid-β-induced neurotoxicity in vitro. FEBS J. 2007, 274, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C.B.; Condron, M.M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D.B.; Younkin, S.G.; et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat. Neurosci. 2001, 4, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, T.; Takamura, Y.; Tsuji, M.; Watanabe-Nakayama, T.; Imamura, K.; Inoue, H.; Nakamura, S.; Inoue, T.; Kimura, A.; Yano, S.; et al. High molecular weight amyloid β1-42 oligomers induce neurotoxicity via plasma membrane damage. FASEB J. 2019, 33, 9220–9234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, S.; Moller, C.; Tegerstedt, K.; Lord, A.; Laudon, H.; Sjodahl, J.; Soderberg, L.; Spens, E.; Sahlin, C.; Waara, E.R.; et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid-β protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J. Alzheimers Dis. 2015, 43, 575–588. [Google Scholar] [CrossRef]
- Gauthier, S.; Albert, M.; Fox, N.; Goedert, M.; Kivipelto, M.; Mestre-Ferrandiz, J.; Middleton, L.T. Why has therapy development for dementia failed in the last two decades? Alzheimers Dement. 2016, 12, 60–64. [Google Scholar] [CrossRef]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frolich, L.; Jack, C.R., Jr.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer’s disease: The path to 2025. Alzheimers Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Burki, T. Alzheimer’s disease research: The future of BACE inhibitors. Lancet 2018, 391, 2486. [Google Scholar] [CrossRef]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef]
- Vandenberghe, R.; Rinne, J.O.; Boada, M.; Katayama, S.; Scheltens, P.; Vellas, B.; Tuchman, M.; Gass, A.; Fiebach, J.B.; Hill, D.; et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res. Ther. 2016, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef]
- Abbasi, J. Promising Results in 18-Month Analysis of Alzheimer Drug Candidate. JAMA 2018, 320, 965. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.D.; Wong, S.S.; Lieber, C.M.; Lansbury, P.T. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem. Biol. 1997, 4, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Kodali, R.; Wetzel, R. Polymorphism in the intermediates and products of amyloid assembly. Curr. Opin. Struct. Biol. 2007, 17, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Nakayama, T.; Ono, K.; Itami, M.; Takahashi, R.; Teplow, D.B.; Yamada, M. High-speed atomic force microscopy reveals structural dynamics of amyloid β1-42 aggregates. Proc. Natl. Acad. Sci. USA 2016, 113, 5835–5840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimers Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasecke, F.; Miti, T.; Perez, C.; Barton, J.; Scholzel, D.; Gremer, L.; Gruning, C.S.R.; Matthews, G.; Meisl, G.; Knowles, T.P.J.; et al. Origin of metastable oligomers and their effects on amyloid fibril self-assembly. Chem. Sci. 2018, 9, 5937–5948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, Y.; Hashimoto, T.; Nomoto, H.; Hyman, B.T.; Iwatsubo, T. Role of Apolipoprotein E in β-Amyloidogenesis: Isoform-Specific Effects On Protofibril To Fibril Conversion Of Aβ In Vitro And Brain Aβ Deposition In Vivo. J. Biol. Chem. 2015, 290, 15163–15174. [Google Scholar] [CrossRef] [Green Version]
- Sollvander, S.; Nikitidou, E.; Brolin, R.; Soderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. Accumulation of amyloid-β by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol. Neurodegener. 2016, 11, 38. [Google Scholar] [CrossRef]
- Paranjape, G.S.; Gouwens, L.K.; Osborn, D.C.; Nichols, M.R. Isolated amyloid-β(1-42) protofibrils, but not isolated fibrils, are robust stimulators of microglia. ACS Chem. Neurosci. 2012, 3, 302–311. [Google Scholar] [CrossRef]
- Terrill-Usery, S.E.; Mohan, M.J.; Nichols, M.R. Amyloid-β(1-42) protofibrils stimulate a quantum of secreted IL-1β despite significant intracellular IL-1β accumulation in microglia. Biochim. Biophys. Acta 2014, 1842, 2276–2285. [Google Scholar] [CrossRef] [Green Version]
- Gouwens, L.K.; Makoni, N.J.; Rogers, V.A.; Nichols, M.R. Amyloid-β42 protofibrils are internalized by microglia more extensively than monomers. Brain Res. 2016, (Pt A), 485–495. [Google Scholar] [CrossRef] [Green Version]
- Rosini, M.; Simoni, E.; Milelli, A.; Minarini, A.; Melchiorre, C. Oxidative stress in Alzheimer’s disease: Are we connecting the dots? J. Med. Chem. 2014, 57, 2821–2831. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Scheff, S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohanka, M. Alzheimer’s disease and oxidative stress: A review. Curr. Med. Chem. 2014, 21, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Zimbron, L.F.; Luna-Munoz, J.; Mena, R.; Vazquez-Ramirez, R.; Kubli-Garfias, C.; Cribbs, D.H.; Manoutcharian, K.; Gevorkian, G. Amyloid-β peptide binds to cytochrome C oxidase subunit 1. PLoS ONE 2012, 7, e42344. [Google Scholar] [CrossRef] [Green Version]
- Bode, D.C.; Baker, M.D.; Viles, J.H. Ion Channel Formation by Amyloid-β42 Oligomers but Not Amyloid-β40 in Cellular Membranes. J. Biol. Chem. 2017, 292, 1404–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drolle, E.; Negoda, A.; Hammond, K.; Pavlov, E.; Leonenko, Z. Changes in lipid membranes may trigger amyloid toxicity in Alzheimer’s disease. PLoS ONE 2017, 12, e0182194. [Google Scholar] [CrossRef]
- Klyubin, I.; Walsh, D.M.; Cullen, W.K.; Fadeeva, J.V.; Anwyl, R.; Selkoe, D.J.; Rowan, M.J. Soluble Arctic amyloid β protein inhibits hippocampal long-term potentiation in vivo. Eur. J. Neurosci. 2004, 19, 2839–2846. [Google Scholar] [CrossRef]
- Massaad, C.A.; Klann, E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid Redox Signal. 2011, 14, 2013–2054. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, E.L.; Bassi, F., Jr.; Gattaz, W.F. Inhibition of phospholipase A2 activity reduces membrane fluidity in rat hippocampus. J. Neural. Transm. (Vienna) 2005, 112, 641–647. [Google Scholar] [CrossRef]
- Alzheimer’s Association Calcium Hypothesis Workgroup. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 2017, 13, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Calcium signalling in health and disease. Biochem. Biophys. Res. Commun. 2017, 485, 5. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, E.; Prox, J.; Bernreuther, C.; Weber, S.; Schwanbeck, R.; Serneels, L.; Snellinx, A.; Craessaerts, K.; Thathiah, A.; Tesseur, I.; et al. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J. Neurosci. 2010, 30, 4833–4844. [Google Scholar] [CrossRef]
- Esteve, P.; Rueda-Carrasco, J.; Ines Mateo, M.; Martin-Bermejo, M.J.; Draffin, J.; Pereyra, G.; Sandonis, A.; Crespo, I.; Moreno, I.; Aso, E.; et al. Elevated levels of Secreted-Frizzled-Related-Protein 1 contribute to Alzheimer’s disease pathogenesis. Nat. Neurosci. 2019, 22, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Konietzko, U.; Krebs, D.C.; Nitsch, R.M. Intracellular Aβ and cognitive deficits precede β-amyloid deposition in transgenic arcAβ mice. Neurobiol. Aging 2007, 28, 1297–1306. [Google Scholar] [CrossRef]
- Lord, A.; Kalimo, H.; Eckman, C.; Zhang, X.Q.; Lannfelt, L.; Nilsson, L.N. The Arctic Alzheimer mutation facilitates early intraneuronal Aβ aggregation and senile plaque formation in transgenic mice. Neurobiol. Aging 2006, 27, 67–77. [Google Scholar] [CrossRef]
- Lord, A.; Englund, H.; Soderberg, L.; Tucker, S.; Clausen, F.; Hillered, L.; Gordon, M.; Morgan, D.; Lannfelt, L.; Pettersson, F.E.; et al. Amyloid-β protofibril levels correlate with spatial learning in Arctic Alzheimer’s disease transgenic mice. FEBS J. 2009, 276, 995–1006. [Google Scholar] [CrossRef] [PubMed]
- Sehlin, D.; Englund, H.; Simu, B.; Karlsson, M.; Ingelsson, M.; Nikolajeff, F.; Lannfelt, L.; Pettersson, F.E. Large aggregates are the major soluble Aβ species in AD brain fractionated with density gradient ultracentrifugation. PLoS ONE 2012, 7, e32014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K.; Condron, M.M.; Ho, L.; Wang, J.; Zhao, W.; Pasinetti, G.M.; Teplow, D.B. Effects of grape seed-derived polyphenols on amyloid β-protein self-assembly and cytotoxicity. J. Biol. Chem. 2008, 283, 32176–32187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ho, L.; Zhao, W.; Ono, K.; Rosensweig, C.; Chen, L.; Humala, N.; Teplow, D.B.; Pasinetti, G.M. Grape-derived polyphenolics prevent Aβ oligomerization and attenuate cognitive deterioration in a mouse model of Alzheimer’s disease. J. Neurosci. 2008, 28, 6388–6392. [Google Scholar] [CrossRef]
- Jin, Y.; Sun, Y.; Lei, J.; Wei, G. Dihydrochalcone molecules destabilize Alzheimer’s amyloid-β protofibrils through binding to the protofibril cavity. Phys. Chem. Chem. Phys. 2018, 20, 17208–17217. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xi, W.; Luo, Y.; Cao, S.; Wei, G. Interactions of a water-soluble fullerene derivative with amyloid-β protofibrils: Dynamics, binding mechanism, and the resulting salt-bridge disruption. J. Phys. Chem. B 2014, 118, 6733–6741. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.M.; Gu, R.X.; Wang, Y.J.; Pi, Y.L.; Zhang, Y.H.; Xu, Q.; Wei, D.Q. Destabilization of Alzheimer’s Aβ42 Protofibrils with a Novel Drug Candidate wgx-50 by Molecular Dynamics Simulations. J. Phys. Chem. B 2015, 119, 11196–11202. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.K.; Shuaib, S.; Goyal, D.; Goyal, B. Insights into the inhibitory mechanism of a resveratrol and clioquinol hybrid against Aβ42 aggregation and protofibril destabilization: A molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2019, 37, 3183–3197. [Google Scholar] [CrossRef] [PubMed]
- Shuaib, S.; Narang, S.S.; Goyal, D.; Goyal, B. Computational design and evaluation of β-sheet breaker peptides for destabilizing Alzheimer’s amyloid-β42 protofibrils. J. Cell Biochem. 2019, 120, 17935–17950. [Google Scholar]
- Englund, H.; Sehlin, D.; Johansson, A.S.; Nilsson, L.N.; Gellerfors, P.; Paulie, S.; Lannfelt, L.; Pettersson, F.E. Sensitive ELISA detection of amyloid-β protofibrils in biological samples. J. Neurochem. 2007, 103, 334–345. [Google Scholar] [CrossRef]
- Sehlin, D.; Hedlund, M.; Lord, A.; Englund, H.; Gellerfors, P.; Paulie, S.; Lannfelt, L.; Pettersson, F.E. Heavy-chain complementarity-determining regions determine conformation selectivity of anti-Aβantibodies. Neurodegener. Dis. 2011, 8, 117–123. [Google Scholar] [CrossRef]
- Lannfelt, L.; Moller, C.; Basun, H.; Osswald, G.; Sehlin, D.; Satlin, A.; Logovinsky, V.; Gellerfors, P. Perspectives on future Alzheimer therapies: Amyloid-β protofibrils - a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Lord, A.; Gumucio, A.; Englund, H.; Sehlin, D.; Sundquist, V.S.; Soderberg, L.; Moller, C.; Gellerfors, P.; Lannfelt, L.; Pettersson, F.E.; et al. An amyloid-β protofibril-selective antibody prevents amyloid formation in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2009, 36, 425–434. [Google Scholar] [CrossRef]
- Sollvander, S.; Nikitidou, E.; Gallasch, L.; Zysk, M.; Soderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. The Aβ protofibril selective antibody mAb158 prevents accumulation of Aβ in astrocytes and rescues neurons from Aβ-induced cell death. J. Neuroinflammation 2018, 15, 98. [Google Scholar] [CrossRef]
- Syvanen, S.; Hultqvist, G.; Gustavsson, T.; Gumucio, A.; Laudon, H.; Soderberg, L.; Ingelsson, M.; Lannfelt, L.; Sehlin, D. Efficient clearance of Aβ protofibrils in AβPP-transgenic mice treated with a brain-penetrating bifunctional antibody. Alzheimers Res. Ther. 2018, 10, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logovinsky, V.; Satlin, A.; Lai, R.; Swanson, C.; Kaplow, J.; Osswald, G.; Basun, H.; Lannfelt, L. Safety and tolerability of BAN2401--a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res. Ther. 2016, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aducanumab. Available online: https://www.alzforum.org/therapeutics/aducanumab (accessed on 25 October 2019).
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Sengupta, U.; Castillo-Carranza, D.; Gerson, J.E.; Guerrero-Munoz, M.; Troncoso, J.C.; Jackson, G.R.; Kayed, R. The formation of tau pore-like structures is prevalent and cell specific: Possible implications for the disease phenotypes. Acta Neuropathol. Commun. 2014, 2, 56. [Google Scholar] [CrossRef] [Green Version]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T., Jr. Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 2002, 322, 1089–1102. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, G.A.P.; Silva, J.L. Alpha-synuclein stepwise aggregation reveals features of an early onset mutation in Parkinson’s disease. Commun. Biol. 2019, 2, 374. [Google Scholar] [CrossRef] [Green Version]
- Groenning, M.; Campos, R.I.; Hirschberg, D.; Hammarstrom, P.; Vestergaard, B. Considerably Unfolded Transthyretin Monomers Preceed and Exchange with Dynamically Structured Amyloid Protofibrils. Sci. Rep. 2015, 5, 11443. [Google Scholar] [CrossRef] [Green Version]
- Beasley, M.; Stonebraker, A.R.; Hasan, I.; Kapp, K.L.; Liang, B.J.; Agarwal, G.; Groover, S.; Sedighi, F.; Legleiter, J. Lipid Membranes Influence the Ability of Small Molecules To Inhibit Huntingtin Fibrillization. Biochemistry 2019, 58, 4361–4373. [Google Scholar] [CrossRef]
- Gallagher-Jones, M.; Glynn, C.; Boyer, D.R.; Martynowycz, M.W.; Hernandez, E.; Miao, J.; Zee, C.T.; Novikova, I.V.; Goldschmidt, L.; McFarlane, H.T.; et al. Sub-angstrom cryo-EM structure of a prion protofibril reveals a polar clasp. Nat. Struct. Mol. Biol. 2018, 25, 131–134. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ono, K.; Tsuji, M. Protofibrils of Amyloid-β are Important Targets of a Disease-Modifying Approach for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 952. https://doi.org/10.3390/ijms21030952
Ono K, Tsuji M. Protofibrils of Amyloid-β are Important Targets of a Disease-Modifying Approach for Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(3):952. https://doi.org/10.3390/ijms21030952
Chicago/Turabian StyleOno, Kenjiro, and Mayumi Tsuji. 2020. "Protofibrils of Amyloid-β are Important Targets of a Disease-Modifying Approach for Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 3: 952. https://doi.org/10.3390/ijms21030952
APA StyleOno, K., & Tsuji, M. (2020). Protofibrils of Amyloid-β are Important Targets of a Disease-Modifying Approach for Alzheimer’s Disease. International Journal of Molecular Sciences, 21(3), 952. https://doi.org/10.3390/ijms21030952