Dietary Antioxidants Significantly Attenuate Hyperoxia-Induced Acute Inflammatory Lung Injury by Enhancing Macrophage Function via Reducing the Accumulation of Airway HMGB1


Abstract
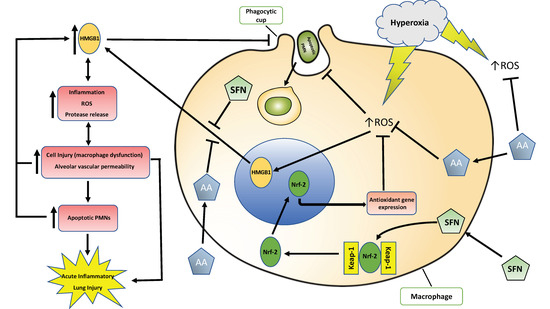
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
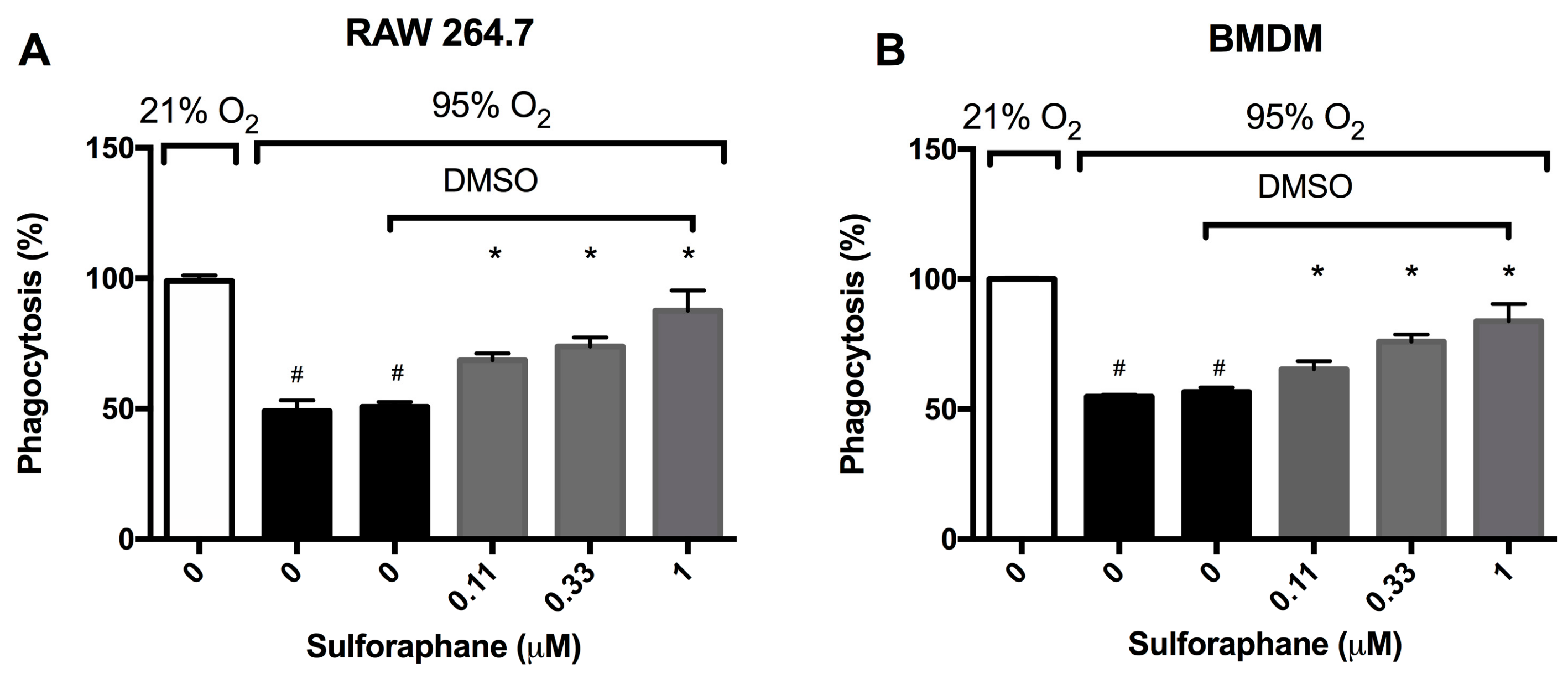
2.1. Sulforaphane Significantly Attenuates Hyperoxia-Induced Dysfunction of Macrophage Phagocytosis
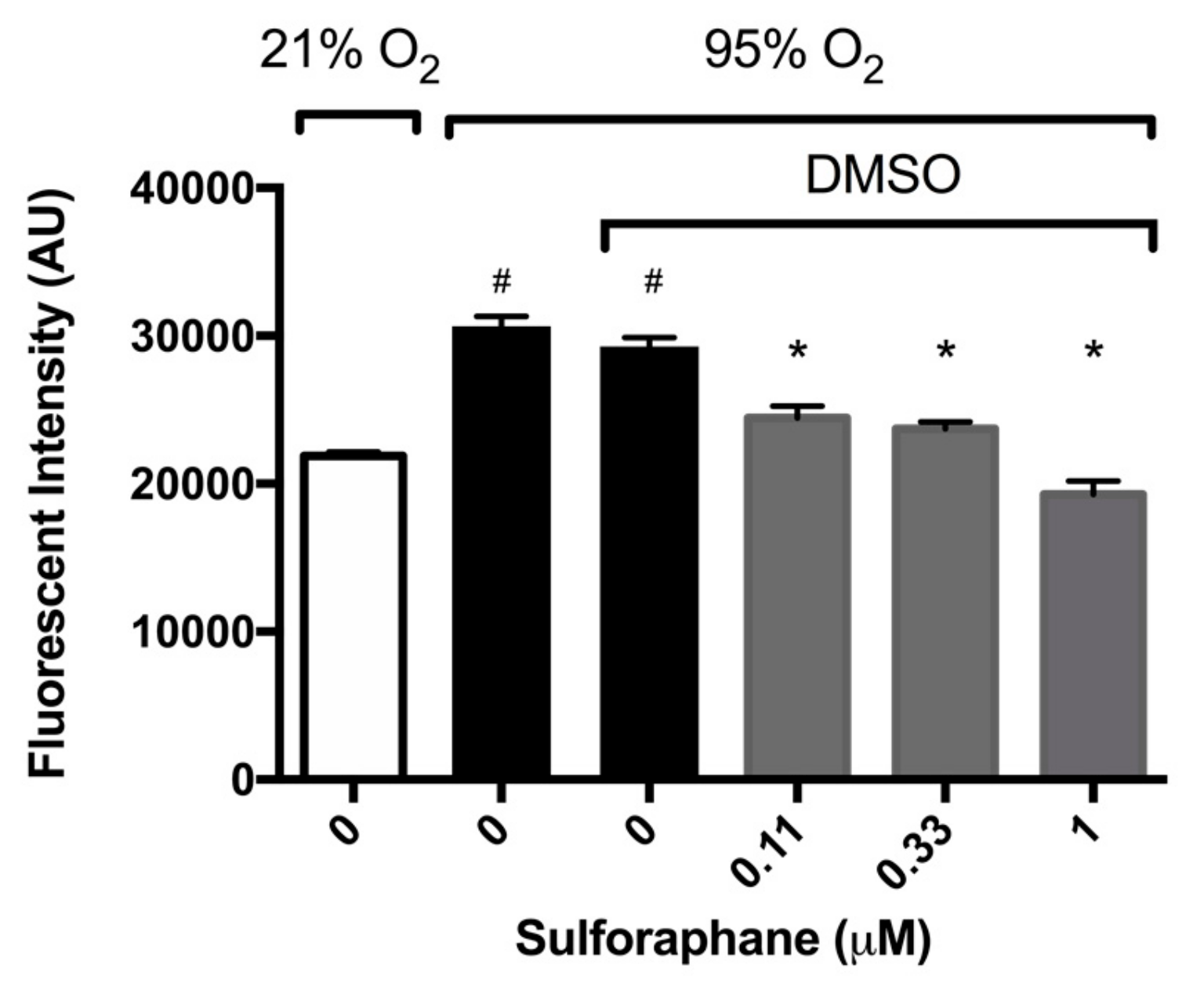
2.2. Sulforaphane Significantly Attenuates Hyperoxia-Induced Oxidative Stress
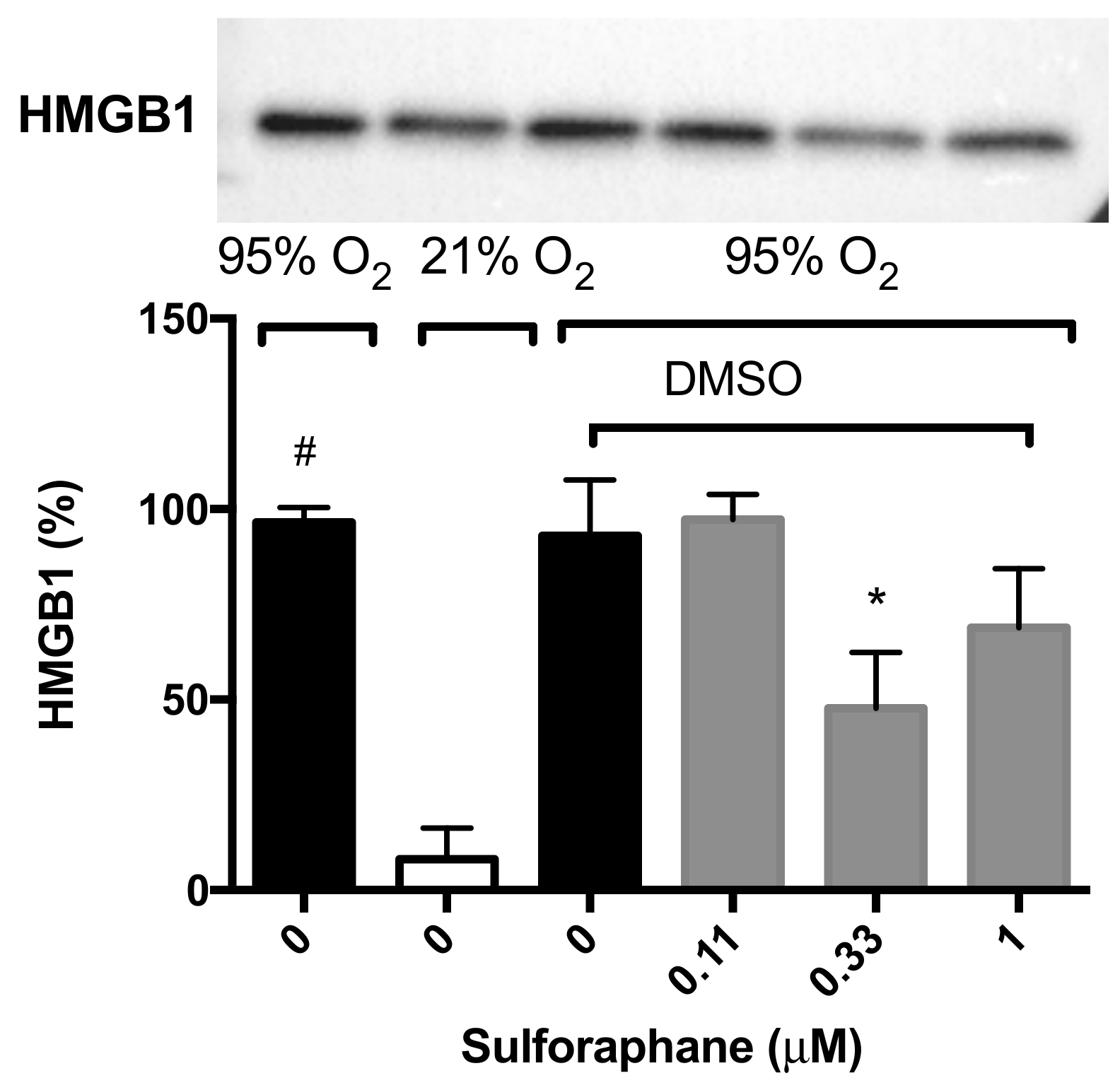
2.3. Sulforaphane Significantly Inhibits Hyperoxia-Induced Release of HMGB1 from Cultured Macrophages
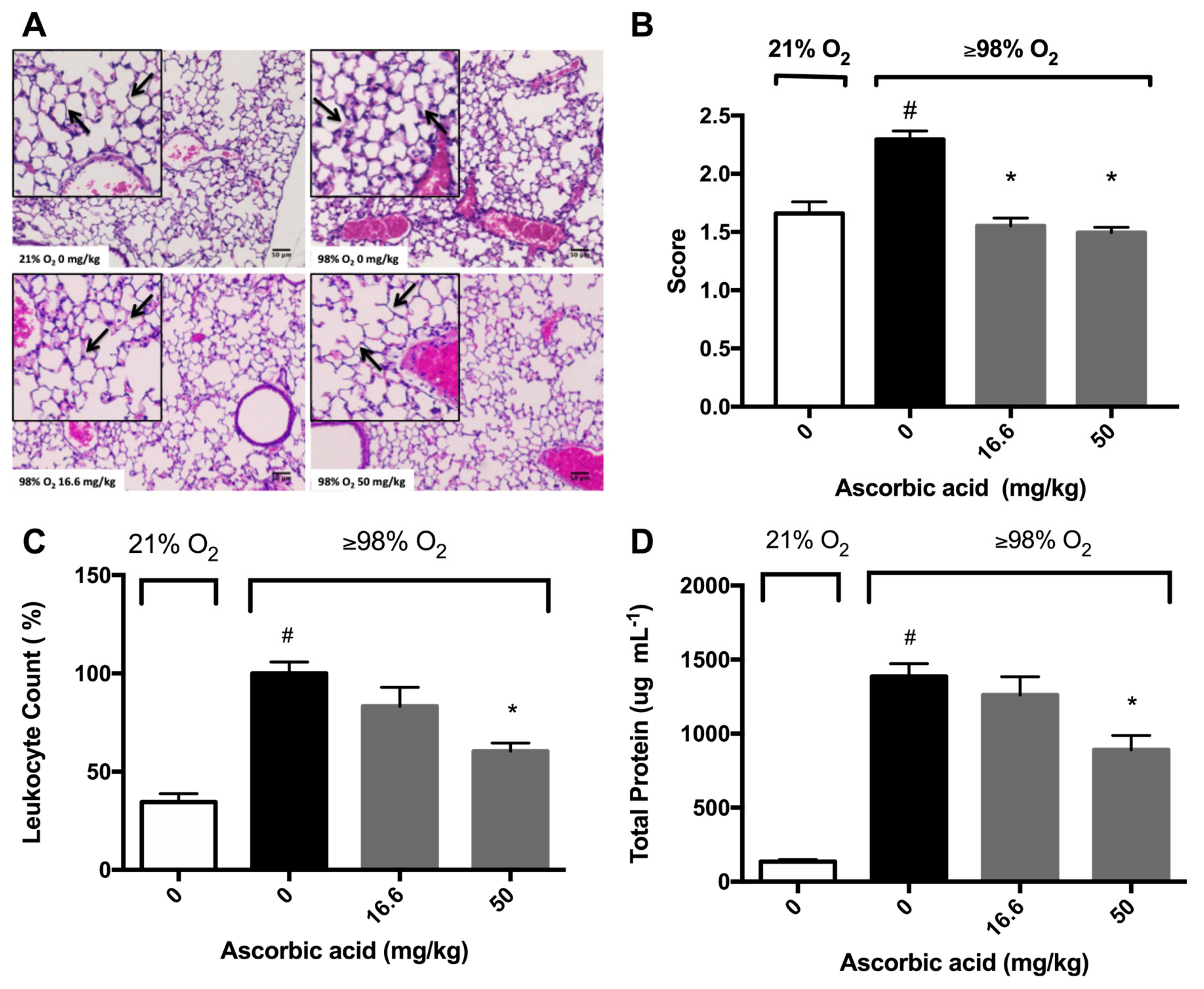
2.4. Ascorbic Acid Significantly Attenuates Hyperoxia-Induced Acute Inflammatory Lung Injury
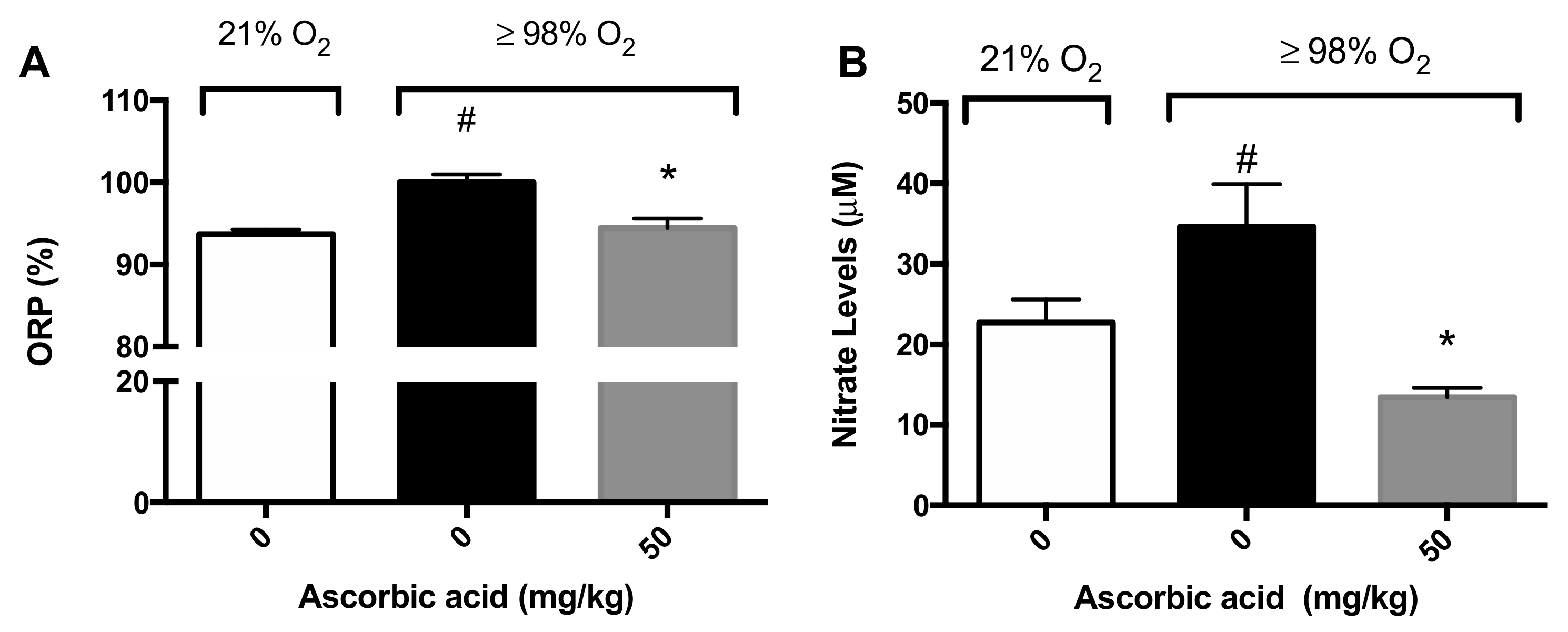
2.5. Ascorbic Acid Significantly Attenuates Hyperoxia-Induced Oxidative Stress
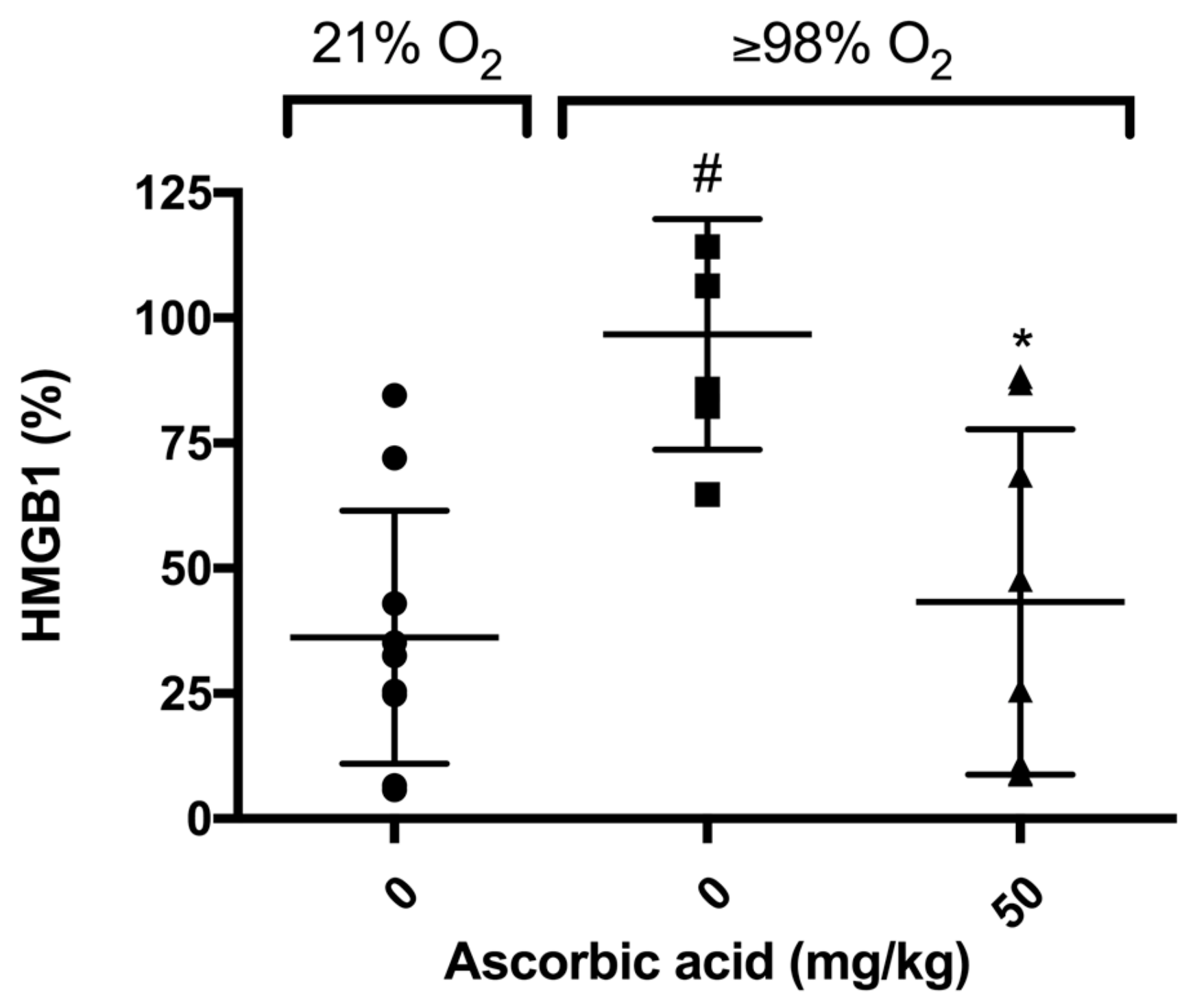
2.6. Ascorbic Acid Significantly Decreases Hyperoxia-Induced Accumulation of HMGB1 in Airways of Mice
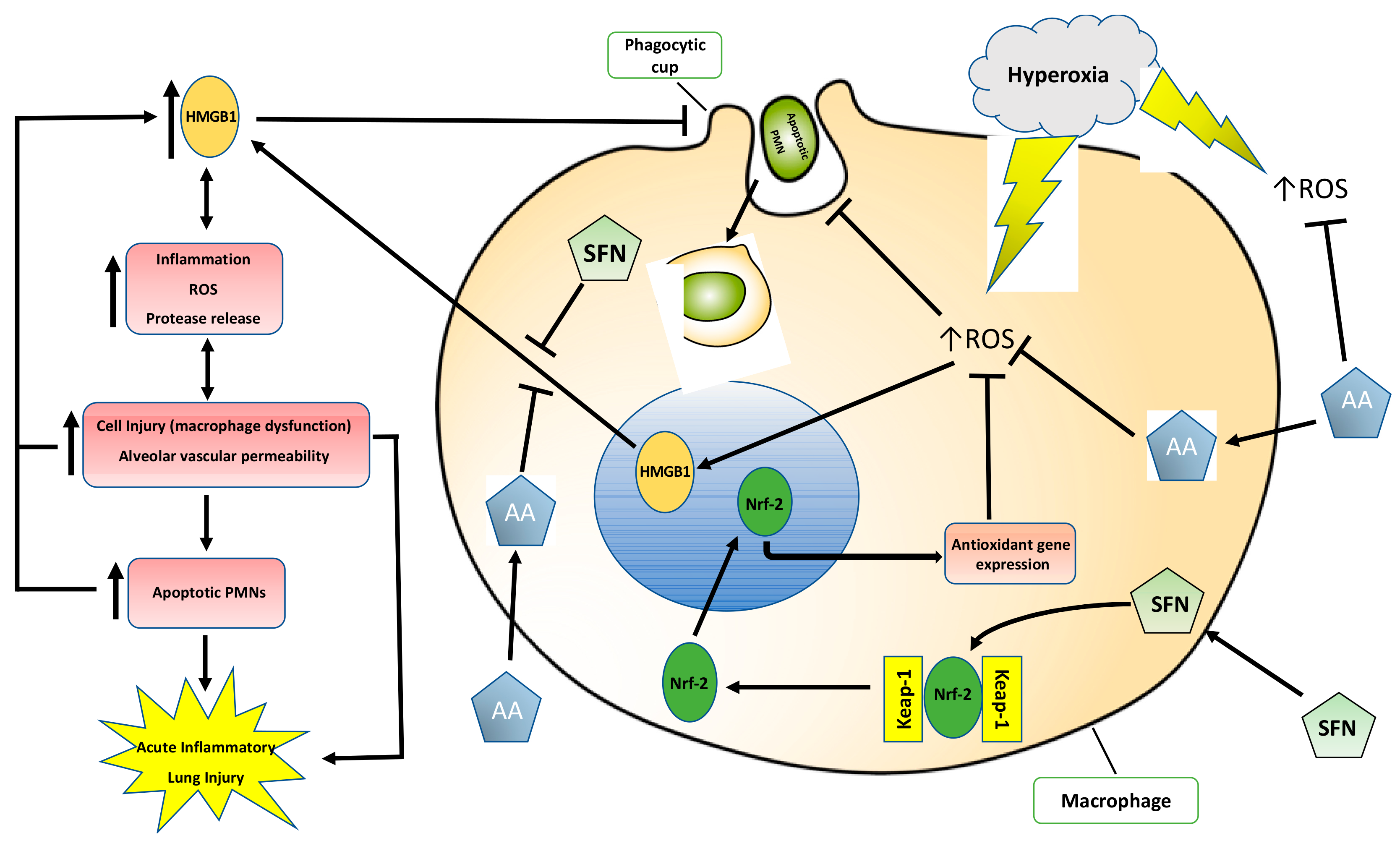
3. Discussion
3.1. Antioxidants Are Effective in Mitigating Cellular Damage and HALI by Reducing Oxidative Stress in Cultured Macrophages and Animals Exposed to Hyperoxia
3.2. Antioxidants Are Effective in Reducing the Release of HMGB1 and Accumulation of Airway Levels of HMGB1
3.3. Antioxidants Can Mitigate Hyperoxia-Impaired Macrophage Function in Phagocytosis and Reduce the Accumulation of Leukocytes in Hyperoxic Lung Tissues
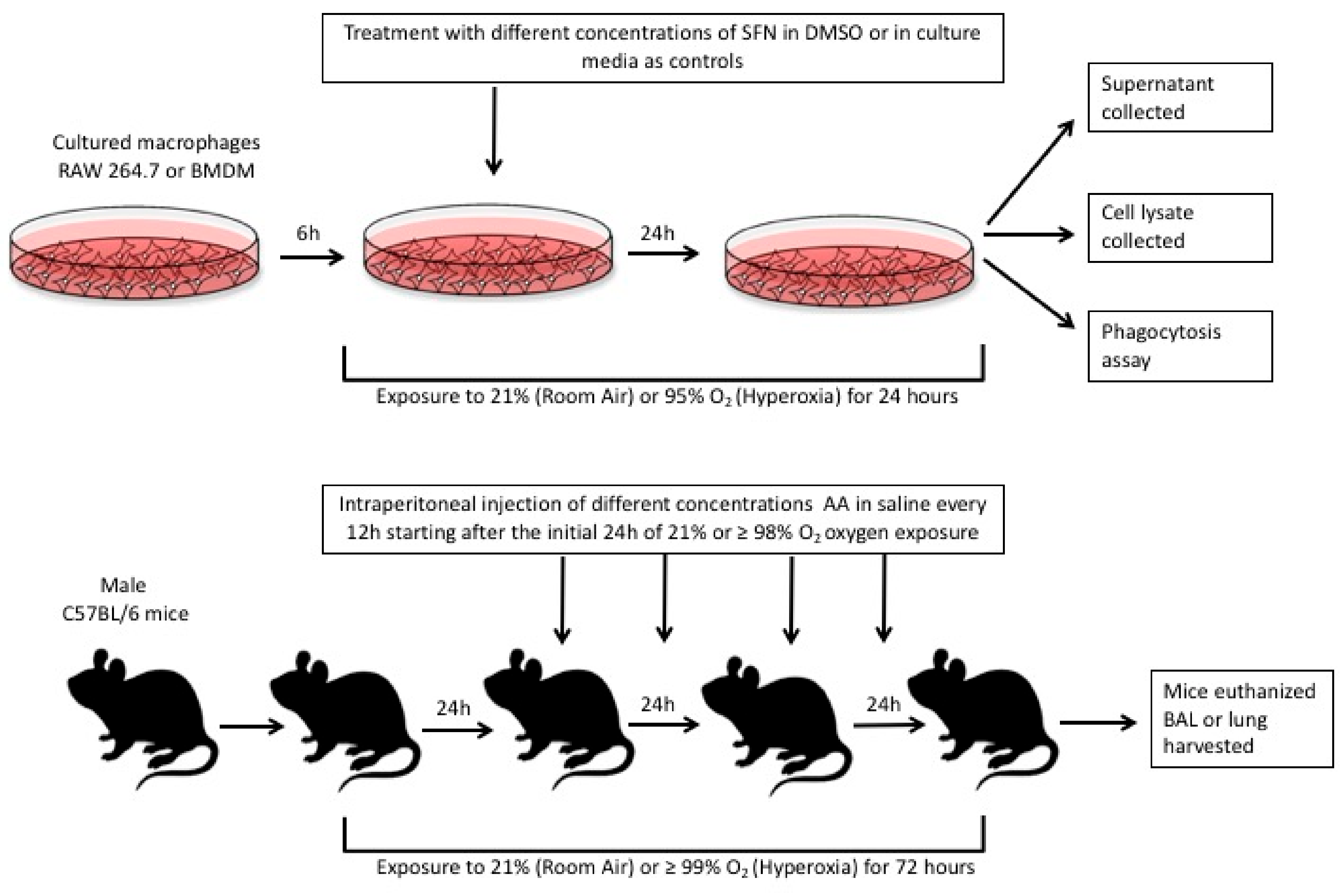
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Isolation and Culture of Bone Marrow Derived Macrophages
4.3. Phagocytosis Assay
4.4. DCFH-DA Assay
4.5. HMGB1 Release
4.6. Western Blot Analysis
4.7. Animal Studies
4.8. Lung Histopathology
4.9. Assay for Oxidative Stress
4.10. Assay for Nitrogen Oxide Species (NOx)
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA | Ascorbic Acid |
| ALI | Acute Lung Injury |
| ARDS | Acute Respiratory Distress Syndrome |
| ARE | Antioxidant Response Element |
| BALF | Bronchoalveolar Lavage Fluid |
| BMDM | Bone Marrow Derived Macrophages |
| DAMP | Damage-Associated Molecular Pattern |
| DMSO | Dimethyl Sulfoxide |
| FBS | Fetal bovine serum |
| FITC | Fluorescein Isothiocyanate |
| HALI | Hyperoxia-Induced Acute Lung Injury |
| HMGB1 | High-Mobility Group Box-1 |
| HO-1 | Heme Oxigenase-1 |
| i.p. | Intraperitoneal |
| NOx | Nitric Oxide Species (NO2 and NO3) |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| ORP | Oxidation-reduction potential |
| PMN | Polymorphonuclear cell |
| ROS | Reactive Oxygen Species |
| SFN | Sulforaphane |
References
- Fan, E.; Brodie, D.; Slutsky, A.S. Acute Respiratory Distress Syndrome: Advances in Diagnosis and Treatment. JAMA 2018, 319, 698. [Google Scholar] [CrossRef] [PubMed]
- Dias-Freitas, F.; Metelo-Coimbra, C.; Roncon-Albuquerque, R. Molecular mechanisms underlying hyperoxia acute lung injury. Respir. Med. 2016, 119, 23–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehme, S.; Hartmann, E.K.; Tripp, T.; Thal, S.C.; David, M.; Abraham, D.; Baumgardner, J.E.; Markstaller, K.; Klein, K.U. PO2 oscillations induce lung injury and inflammation. Crit. Care Lond. Engl. 2019, 23, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, D.; Ablordeppey, E.; Wessman, B.T.; Mohr, N.M.; Trzeciak, S.; Kollef, M.H.; Roberts, B.W.; Fuller, B.M. Emergency department hyperoxia is associated with increased mortality in mechanically ventilated patients: A cohort study. Crit. Care 2018, 22, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggiu, M.; Aissaoui, N.; Nael, J.; Haw-Berlemont, C.; Herrmann, B.; Augy, J.-L.; Ortuno, S.; Vimpère, D.; Diehl, J.-L.; Bailleul, C.; et al. Hyperoxia effects on intensive care unit mortality: A retrospective pragmatic cohort study. Crit. Care 2018, 22, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Entezari, M.; Javdan, M.; Antoine, D.J.; Morrow, D.M.; Sitapara, R.A.; Patel, V.; Wang, M.; Sharma, L.; Gorasiya, S.; Zur, M.; et al. Inhibition of extracellular HMGB1 attenuates hyperoxia-induced inflammatory acute lung injury. Redox Biol. 2014, 2, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Han, C.H.; Guan, Z.B.; Zhang, P.X.; Fang, H.L.; Li, L.; Zhang, H.M.; Zhou, F.J.; Mao, Y.F.; Liu, W.W. Oxidative stress induced necroptosis activation is involved in the pathogenesis of hyperoxic acute lung injury. Biochem. Biophys. Res. Commun. 2018, 495, 2178–2183. [Google Scholar] [CrossRef]
- Pham, T.; Brochard, L.J.; Slutsky, A.S. Mechanical Ventilation: State of the Art. Mayo Clin. Proc. 2017, 92, 1382–1400. [Google Scholar] [CrossRef] [Green Version]
- Gore, A.; Muralidhar, M.; Espey, M.G.; Degenhardt, K.; Mantell, L.L. Hyperoxia sensing: From molecular mechanisms to significance in disease. J. Immunotoxicol. 2010, 7, 239–254. [Google Scholar] [CrossRef]
- Pagano, A.; Barazzone-Argiroffo, C. Alveolar Cell Death in Hyperoxia-Induced Lung Injury. Ann. NY Acad. Sci. 2003, 1010, 405–416. [Google Scholar] [CrossRef]
- Freeman, B.A.; Topolosky, M.K.; Crapo, J.D. Hyperoxia increases oxygen radical production in rat lung homogenates. Arch. Biochem. Biophys. 1982, 216, 477–484. [Google Scholar] [CrossRef]
- Bhandari, V. Molecular mechanisms of hyperoxia-induced acute lung injury. Front. Biosci. J. Virtual Libr. 2008, 13, 6653–6661. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, P.J.; Hickman-Davis, J.M.; Davis, I.C.; Matalon, S. Hyperoxia Impairs Antibacterial Function of Macrophages Through Effects on Actin. Am. J. Respir. Cell Mol. Biol. 2003, 28, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.M.; Entezari-Zaher, T.; Romashko, J., 3rd; Azghani, A.O.; Javdan, M.; Ulloa, L.; Miller, E.J.; Mantell, L.L. Antioxidants preserve macrophage phagocytosis of Pseudomonas aeruginosa during hyperoxia. Free Radic. Biol. Med. 2007, 42, 1338–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baleeiro, C.E.O.; Wilcoxen, S.E.; Morris, S.B.; Standiford, T.J.; Paine, R. Sublethal hyperoxia impairs pulmonary innate immunity. J. Immunol. Baltim. 2003, 171, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.S.; Sitapara, R.A.; Gore, A.; Phan, B.; Sharma, L.; Sampat, V.; Li, J.H.; Yang, H.; Chavan, S.S.; Wang, H.; et al. High Mobility Group Box–1 Mediates Hyperoxia-Induced Impairment of Pseudomonas aeruginosa Clearance and Inflammatory Lung Injury in Mice. Am. J. Respir. Cell Mol. Biol. 2013, 48, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Entezari, M.; Weiss, D.J.; Sitapara, R.; Whittaker, L.; Wargo, M.J.; Li, J.; Wang, H.; Yang, H.; Sharma, L.; Phan, B.D.; et al. Inhibition of high-mobility group box 1 protein (HMGB1) enhances bacterial clearance and protects against Pseudomonas Aeruginosa pneumonia in Cystic Fibrosis. Mol. Med. Camb. Mass 2012, 18, 477–485. [Google Scholar] [CrossRef]
- Liu, G.; Wang, J.; Park, Y.-J.; Tsuruta, Y.; Lorne, E.F.; Zhao, X.; Abraham, E. HMGB1 inhibits phagocytosis of apoptotic neutrophils through binding to phosphatidylserine. J. Immunol. Baltim. 2008, 181, 4240–4246. [Google Scholar] [CrossRef]
- Lin, X.; Yang, H.; Sakuragi, T.; Hu, M.; Mantell, L.L.; Hayashi, S.; Al-Abed, Y.; Tracey, K.J.; Ulloa, L.; Miller, E.J. Alpha-chemokine receptor blockade reduces high mobility group box 1 protein-induced lung inflammation and injury and improves survival in sepsis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L583–L590. [Google Scholar] [CrossRef] [Green Version]
- Abraham, E.; Arcaroli, J.; Carmody, A.; Wang, H.; Tracey, K.J. HMG-1 as a mediator of acute lung inflammation. J. Immunol. Baltim. 2000, 165, 2950–2954. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.-Y.; Jedlicka, A.E.; Reddy, S.P.M.; Kensler, T.W.; Yamamoto, M.; Zhang, L.-Y.; Kleeberger, S.R. Role of NRF2 in protection against hyperoxic lung injury in mice. Am. J. Respir. Cell Mol. Biol. 2002, 26, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-Y.; Miller-DeGraff, L.; Blankenship-Paris, T.; Wang, X.; Bell, D.A.; Lih, F.; Deterding, L.; Panduri, V.; Morgan, D.L.; Yamamoto, M.; et al. Sulforaphane enriched transcriptome of lung mitochondrial energy metabolism and provided pulmonary injury protection via Nrf2 in mice. Toxicol. Appl. Pharmacol. 2019, 364, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-Y.; Jedlicka, A.E.; Reddy, S.P.M.; Zhang, L.-Y.; Kensler, T.W.; Kleeberger, S.R. Linkage Analysis of Susceptibility to Hyperoxia: Nrf2 Is a Candidate Gene. Am. J. Respir. Cell Mol. Biol. 2002, 26, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.-H.; Chen, S.; Corpe, C.; Dutta, A.; Dutta, S.K.; et al. Vitamin C as an antioxidant: Evaluation of its role in disease prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef]
- Sorice, A.; Guerriero, E.; Capone, F.; Colonna, G.; Castello, G.; Costantini, S. Ascorbic Acid: Its Role in Immune System and Chronic Inflammation Diseases. Mini-Rev. Med. Chem. 2014, 14, 444–452. [Google Scholar] [CrossRef]
- Carr, A.; Maggini, S. Vitamin C and Immune Function. Nutrients 2017, 9, 1211. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. BBA Rev. Cancer 2012, 1826, 443–457. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.S.; Sampat, V.; Espey, M.G.; Sitapara, R.; Wang, H.; Yang, X.; Ashby, C.R.; Thomas, D.D.; Mantell, L.L. Ascorbic Acid Attenuates Hyperoxia-Compromised Host Defense against Pulmonary Bacterial Infection. Am. J. Respir. Cell Mol. Biol. 2016, 55, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Sitapara, R.A.; Antoine, D.J.; Sharma, L.; Patel, V.S.; Ashby, C.R.; Gorasiya, S.; Yang, H.; Zur, M.; Mantell, L.L. The α7 nicotinic acetylcholine receptor agonist GTS-21 improves bacterial clearance in mice by restoring hyperoxia-compromised macrophage function. Mol. Med. Camb. Mass 2014, 20, 238–247. [Google Scholar]
- Jiao, Z.; Chang, J.; Li, J.; Nie, D.; Cui, H.; Guo, D. Sulforaphane increases Nrf2 expression and protects alveolar epithelial cells against injury caused by cigarette smoke extract. Mol. Med. Rep. 2017, 16, 1241–1247. [Google Scholar] [CrossRef] [Green Version]
- Reddy, N.M.; Tamatam, C.R.; Reddy, S.P. Nrf2-Regulated Signaling Is Crucial for Alveolar Macrophage—Mediated Efferocytosis during Hyperoxic Lung Injury and Repair. Ann. Am. Thorac. Soc. 2015, 12, S71. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Niu, Z.; Wu, S.; Shan, S. Protective mechanism of sulforaphane in Nrf2 and anti-lung injury in ARDS rabbits. Exp. Ther. Med. 2018, 15, 4911–4915. [Google Scholar] [CrossRef]
- Szarka, R.J.; Wang, N.; Gordon, L.; Nation, P.N.; Smith, R.H. A murine model of pulmonary damage induced by lipopolysaccharide via intranasal instillation. J. Immunol. Methods 1997, 202, 49–57. [Google Scholar] [CrossRef]
- Adler, V.; Yin, Z.; Tew, K.D.; Ronai, Z. Role of redox potential and reactive oxygen species in stress signaling. Oncogene 1999, 18, 6104–6111. [Google Scholar] [CrossRef] [Green Version]
- Mach, W.J.; Thimmesch, A.R.; Pierce, J.T.; Pierce, J.D. Consequences of Hyperoxia and the Toxicity of Oxygen in the Lung. Nurs. Res. Pract. 2011, 2011, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kallet, R.H.; Matthay, M.A. Hyperoxic acute lung injury. Respir. Care 2013, 58, 123–141. [Google Scholar] [CrossRef] [Green Version]
- Freeman, B.A.; Crapo, J.D. Hyperoxia increases oxygen radical production in rat lungs and lung mitochondria. J. Biol. Chem. 1981, 256, 10986–10992. [Google Scholar]
- Wang, M.; Gorasiya, S.; Antoine, D.J.; Sitapara, R.A.; Wu, W.; Sharma, L.; Yang, H.; Ashby, C.R.; Vasudevan, D.; Zur, M.; et al. The Compromise of Macrophage Functions by Hyperoxia Is Attenuated by Ethacrynic Acid via Inhibition of NF-κB–Mediated Release of High-Mobility Group Box-1. Am. J. Respir. Cell Mol. Biol. 2015, 52, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Perez, M.; Robbins, M.E.; Revhaug, C.; Saugstad, O.D. Oxygen radical disease in the newborn, revisited: Oxidative stress and disease in the newborn period. Free Radic. Biol. Med. 2019, 142, 61–72. [Google Scholar] [CrossRef]
- Brozmanova, M.; Hanacek, J. Hyperoxia-induced regulation of cough reflex and its effect after antioxidant supplementation. Respir. Physiol. Neurobiol. 2018, 257, 75–81. [Google Scholar] [CrossRef]
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. BBA Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Stress-sensing mechanisms and the physiological roles of the Keap1–Nrf2 system during cellular stress. J. Biol. Chem. 2017, 292, 16817–16824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Furusawa, Y.; et al. Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol. Cell. Biol. 2015, 36, 271–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Gao, L.; Su, S.; Sargsyan, D.; Wu, R.; Raskin, I.; Kong, A.-N. Moringa Isothiocyanate Activates Nrf2: Potential Role in Diabetic Nephropathy. AAPS J. 2019, 21, 31. [Google Scholar] [CrossRef]
- La Marca, M.; Beffy, P.; Della Croce, C.; Gervasi, P.G.; Iori, R.; Puccinelli, E.; Longo, V. Structural influence of isothiocyanates on expression of cytochrome P450, phase II enzymes, and activation of Nrf2 in primary rat hepatocytes. Food Chem. Toxicol. 2012, 50, 2822–2830. [Google Scholar] [CrossRef] [Green Version]
- Katsuyama, Y.; Tsuboi, T.; Taira, N.; Yoshioka, M.; Masaki, H. 3-O-Laurylglyceryl ascorbate activates the intracellular antioxidant system through the contribution of PPAR-γ and Nrf2. J. Dermatol. Sci. 2016, 82, 189–196. [Google Scholar] [CrossRef]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef]
- Wang, M.; Gauthier, A.; Daley, L.; Dial, K.; Wu, J.; Woo, J.; Lin, M.; Ashby, C.; Mantell, L.L. The Role of HMGB1, a Nuclear Damage-Associated Molecular Pattern Molecule, in the Pathogenesis of Lung Diseases. Antioxid. Redox Signal. 2019, 31, 954–993. [Google Scholar] [CrossRef]
- Van Zoelen, M.A.; Ishizaka, A.; Wolthuls, E.K.; Choi, G.; van der Poll, T.; Schultz, M.J. Pulmonary levels of high-mobility group box 1 during mechanical ventilation and ventilator-associated pneumonia. Shock 2008, 29, 441–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, H.; Matsuda, T.; Hashimoto, S.; Amaya, F.; Kitamura, Y.; Tanaka, M.; Kobayashi, A.; Maruyama, I.; Yamada, S.; Hasegawa, N.; et al. Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am. J. Respir. Crit. Care Med. 2004, 170, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ni, S.-S.; Liu, H. Pollutional haze and COPD: Etiology, epidemiology, pathogenesis, pathology, biological markers and therapy. J. Thorac. Dis. 2016, 8, E20–E30. [Google Scholar] [PubMed]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 1998, 101, 890–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voll, R.E.; Herrmann, M.; Roth, E.A.; Stach, C.; Kalden, J.R.; Girkontaite, I. Immunosuppressive effects of apoptotic cells. Nature 1997, 390, 350–351. [Google Scholar] [CrossRef]
- Haslett, C. Granulocyte Apoptosis and Its Role in the Resolution and Control of Lung Inflammation. Am. J. Respir. Crit. Care Med. 1999, 160, S5–S11. [Google Scholar] [CrossRef]
- Friggeri, A.; Yang, Y.; Banerjee, S.; Park, Y.-J.; Liu, G.; Abraham, E. HMGB1 inhibits macrophage activity in efferocytosis through binding to the αvβ3-integrin. Am. J. Physiol. 2010, 299, C1267–C1276. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Friggeri, A.; Liu, G.; Abraham, E. The C-terminal acidic tail is responsible for the inhibitory effects of HMGB1 on efferocytosis. J. Leukoc. Biol. 2010, 88, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; de Freitas, A.; Friggeri, A.; Zmijewski, J.W.; Liu, G.; Abraham, E. Intracellular HMGB1 negatively regulates efferocytosis. J. Immunol. Baltim. 2011, 187, 4686–4694. [Google Scholar] [CrossRef] [Green Version]
- Folkard, D.L.; Melchini, A.; Traka, M.H.; Al-Bakheit, A.; Saha, S.; Mulholland, F.; Watson, A.; Mithen, R.F. Suppression of LPS-induced transcription and cytokine secretion by the dietary isothiocyanate sulforaphane. Mol. Nutr. Food Res. 2014, 58, 2286–2296. [Google Scholar] [CrossRef]
- Weischenfeldt, J.; Porse, B. Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc. 2008, 2008, pdb.prot5080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husari, A.; Khayat, A.; Bitar, H.; Hashem, Y.; Rizkallah, A.; Zaatari, G.; El Sabban, M. Antioxidant activity of pomegranate juice reduces acute lung injury secondary to hyperoxia in an animal model. BMC Res. Notes 2014, 7, 664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.K.; Park, S.U. Current potential health benefits of sulforaphane. EXCLI J. 2016, 15, 571–577. [Google Scholar] [PubMed]
- Egner, P.A.; Chen, J.G.; Wang, J.B.; Wu, Y.; Sun, Y.; Lu, J.H.; Zhu, J.; Zhang, Y.H.; Chen, Y.S.; Friesen, M.D.; et al. Bioavailability of Sulforaphane from Two Broccoli Sprout Beverages: Results of a Short-term, Cross-over Clinical Trial in Qidong, China. Cancer Prev. Res. 2011, 4, 384–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, A.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marik, P.E.; Khangoora, V.; Rivera, R.; Hooper, M.H.; Catravas, J. Hydrocortisone, Vitamin C, and Thiamine for the Treatment of Severe Sepsis and Septic Shock. Chest 2017, 151, 1229–1238. [Google Scholar] [CrossRef]
- Ohno, S.; Ohno, Y.; Suzuki, N.; Soma, G.-I.; Inoue, M. High-dose vitamin C (ascorbic acid) therapy in the treatment of patients with advanced cancer. Anticancer Res. 2009, 29, 809–815. [Google Scholar]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, V.; Dial, K.; Wu, J.; Gauthier, A.G.; Wu, W.; Lin, M.; Espey, M.G.; Thomas, D.D.; Ashby, C.R., Jr.; Mantell, L.L. Dietary Antioxidants Significantly Attenuate Hyperoxia-Induced Acute Inflammatory Lung Injury by Enhancing Macrophage Function via Reducing the Accumulation of Airway HMGB1. Int. J. Mol. Sci. 2020, 21, 977. https://doi.org/10.3390/ijms21030977
Patel V, Dial K, Wu J, Gauthier AG, Wu W, Lin M, Espey MG, Thomas DD, Ashby CR Jr., Mantell LL. Dietary Antioxidants Significantly Attenuate Hyperoxia-Induced Acute Inflammatory Lung Injury by Enhancing Macrophage Function via Reducing the Accumulation of Airway HMGB1. International Journal of Molecular Sciences. 2020; 21(3):977. https://doi.org/10.3390/ijms21030977
Chicago/Turabian StylePatel, Vivek, Katelyn Dial, Jiaqi Wu, Alex G. Gauthier, Wenjun Wu, Mosi Lin, Michael G. Espey, Douglas D. Thomas, Charles R. Ashby, Jr., and Lin L. Mantell. 2020. "Dietary Antioxidants Significantly Attenuate Hyperoxia-Induced Acute Inflammatory Lung Injury by Enhancing Macrophage Function via Reducing the Accumulation of Airway HMGB1" International Journal of Molecular Sciences 21, no. 3: 977. https://doi.org/10.3390/ijms21030977
APA StylePatel, V., Dial, K., Wu, J., Gauthier, A. G., Wu, W., Lin, M., Espey, M. G., Thomas, D. D., Ashby, C. R., Jr., & Mantell, L. L. (2020). Dietary Antioxidants Significantly Attenuate Hyperoxia-Induced Acute Inflammatory Lung Injury by Enhancing Macrophage Function via Reducing the Accumulation of Airway HMGB1. International Journal of Molecular Sciences, 21(3), 977. https://doi.org/10.3390/ijms21030977