Engineering Biology to Construct Microbial Chassis for the Production of Difficult-to-Express Proteins




Abstract
:1. Introduction
2. Heterologous Expression of Biologically Functional Proteins by Bacteria
2.1. Engineered E. coli for Wide Array of Recombinant Proteins
2.2. Bacillus Subtilis as A Versatile Host with Highly Efficient Protein Secretion Systems
2.3. Lactococcus Lactis for the Expression of Recombinant Membrane Proteins
2.4. Extremophiles as Alternative Protein Expression Systems
3. Heterologous Protein Expression by Systematically Engineered Bacteria
3.1. Concept and Overview of Synthetic Minimal Genome
3.2. Applications of the Minimal Genome: from Gene Essentiality to Protein Production
3.2.1. Construction of Genome-Reduced Microbial Strains
3.2.2. Effect of Genomic Stability on Protein Production
3.2.3. Increased Availability of Cellular Resources
3.2.4. Optimization of Codon Usage
3.2.5. Changes in Translation Efficiency
4. Conclusion and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Itakura, K.; Hirose, T.; Crea, R.; Riggs, A.D.; Heyneker, H.L.; Bolivar, F.; Boyer, H.W. Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin. Science 1977, 198, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.C.; Van Frank, R.M.; Muth, W.L.; Burnett, J.P. Cytoplasmic inclusion bodies in Escherichia coli producing biosynthetic human insulin proteins. Science 1982, 215, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Carlson, R. Estimating the biotech sector’s contribution to the US economy. Nat. Biotechnol. 2016, 34, 247–255. [Google Scholar] [CrossRef]
- Sanchez-Garcia, L.; Martin, L.; Mangues, R.; Ferrer-Miralles, N.; Vazquez, E.; Villaverde, A. Recombinant pharmaceuticals from microbial cells: A 2015 update. Microb. Cell Fact. 2016, 15, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanhaiean, A.; Azghandi, M.; Razmyar, J.; Mohammadi, E.; Sekhavati, M.H. Recombinant production of a chimeric antimicrobial peptide in E. coli and assessment of its activity against some avian clinically isolated pathogens. Microb. Pathog. 2018, 122, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.P.; Ke, N.; Lobstein, J.; Peterson, C.; Szkodny, A.; Mansell, T.J.; Tuckey, C.; Riggs, P.D.; Colussi, P.A.; Noren, C.J.; et al. Efficient expression of full-length antibodies in the cytoplasm of engineered bacteria. Nat. Commun. 2015, 6, 8072. [Google Scholar] [CrossRef]
- Jaeger, K.E.; Eggert, T.; Eipper, A.; Reetz, M.T. Directed evolution and the creation of enantioselective biocatalysts. Appl. Microbiol. Biotechnol. 2001, 55, 519–530. [Google Scholar] [CrossRef]
- Derman, A.I.; Beckwith, J. Escherichia coli alkaline phosphatase fails to acquire disulfide bonds when retained in the cytoplasm. J. Bacteriol. 1991, 173, 7719–7722. [Google Scholar] [CrossRef] [Green Version]
- Kane, J.F. Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli. Curr. Opin. Biotechnol. 1995, 6, 494–500. [Google Scholar] [CrossRef]
- Vallejo, L.F.; Rinas, U. Strategies for the recovery of active proteins through refolding of bacterial inclusion body proteins. Microb. Cell Fact. 2004, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, N.; Curling, E.M. Glycosylation of recombinant proteins: Problems and prospects. Enzym. Microb. Technol. 1994, 16, 354–364. [Google Scholar] [CrossRef]
- Paddon, C.J.; Westfall, P.J.; Pitera, D.J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M.D.; Tai, A.; Main, A.; Eng, D.; et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luli, G.W.; Strohl, W.R. Comparison of growth, acetate production, and acetate inhibition of Escherichia coli strains in batch and fed-batch fermentations. Appl. Environ. Microbiol. 1990, 56, 1004–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Fu, G.; Tu, R.; Jin, Z.; Zhang, D. High-efficiency expression and secretion of human FGF21 in Bacillus subtilis by intercalation of a mini-cistron cassette and combinatorial optimization of cell regulatory components. Microb. Cell Fact. 2019, 18, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studier, F.W.; Moffatt, B.A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1986, 189, 113–130. [Google Scholar] [CrossRef]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.F.; Kushner, S.R. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 1991, 100, 195–199. [Google Scholar] [CrossRef]
- Nallamsetty, S.; Waugh, D.S. A generic protocol for the expression and purification of recombinant proteins in Escherichia coli using a combinatorial His6-maltose binding protein fusion tag. Nat. Protoc. 2007, 2, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Baneyx, F.; Mujacic, M. Recombinant protein folding and misfolding in Escherichia coli. Nat. Biotechnol. 2004, 22, 1399–1408. [Google Scholar] [CrossRef]
- Dittrich, C.R.; Vadali, R.V.; Bennett, G.N.; San, K.Y. Redistribution of metabolic fluxes in the central aerobic metabolic pathway of E. coli mutant strains with deletion of the ackA-pta and poxB pathways for the synthesis of isoamyl acetate. Biotechnol. Prog. 2005, 21, 627–631. [Google Scholar] [CrossRef]
- Aristidou, A.A.; San, K.Y.; Bennett, G.N. Metabolic engineering of Escherichia coli to enhance recombinant protein production through acetate reduction. Biotechnol. Prog. 1995, 11, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Dragosits, M.; Mattanovich, D. Adaptive laboratory evolution-principles and applications for biotechnology. Microb. Cell Fact. 2013, 12, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liponska, A.; Ousalem, F.; Aalberts, D.P.; Hunt, J.F.; Boel, G. The new strategies to overcome challenges in protein production in bacteria. Microb. Biotechnol. 2019, 12, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Choe, D.; Cho, S.; Kim, S.C.; Cho, B.K. Minimal genome: Worthwhile or worthless efforts toward being smaller? Biotechnol. J. 2016, 11, 199–211. [Google Scholar] [CrossRef]
- Cho, B.K.; Zengler, K.; Qiu, Y.; Park, Y.S.; Knight, E.M.; Barrett, C.L.; Gao, Y.; Palsson, B.O. The transcription unit architecture of the Escherichia coli genome. Nat. Biotechnol. 2009, 27, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Nakahigashi, K.; Toya, Y.; Ishii, N.; Soga, T.; Hasegawa, M.; Watanabe, H.; Takai, Y.; Honma, M.; Mori, H.; Tomita, M. Systematic phenome analysis of Escherichia coli multiple-knockout mutants reveals hidden reactions in central carbon metabolism. Mol. Syst. Biol. 2009, 5, 306. [Google Scholar] [CrossRef] [Green Version]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Carr, P.A.; Church, G.M. Genome engineering. Nat. Biotechnol. 2009, 27, 1151–1162. [Google Scholar] [CrossRef]
- Chin, J.W. Molecular biology. Reprogramming the genetic code. Science 2012, 336, 428–429. [Google Scholar]
- Smanski, M.J.; Bhatia, S.; Zhao, D.; Park, Y.; Lauren, B.A.W.; Giannoukos, G.; Ciulla, D.; Busby, M.; Calderon, J.; Nicol, R.; et al. Functional optimization of gene clusters by combinatorial design and assembly. Nat. Biotechnol. 2014, 32, 1241–1249. [Google Scholar] [CrossRef]
- Chi, H.; Wang, X.; Shao, Y.; Qin, Y.; Deng, Z.; Wang, L.; Chen, S. Engineering and modification of microbial chassis for systems and synthetic biology. Synth. Syst. Biotechnol. 2019, 4, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids. Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saccardo, P.; Corchero, J.L.; Ferrer-Miralles, N. Tools to cope with difficult-to-express proteins. Appl. Microbiol. Biotechnol. 2016, 100, 4347–4355. [Google Scholar] [CrossRef] [PubMed]
- Li, G.W.; Oh, E.; Weissman, J.S. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature 2012, 484, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Salis, H.M.; Mirsky, E.A.; Voigt, C.A. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009, 27, 946–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, S.W.; Yang, J.S.; Cho, H.S.; Yang, J.; Kim, S.C.; Park, J.M.; Kim, S.; Jung, G.Y. Predictive combinatorial design of mRNA translation initiation regions for systematic optimization of gene expression levels. Sci. Rep. 2014, 4, 4515. [Google Scholar] [CrossRef] [Green Version]
- Miksch, G.; Bettenworth, F.; Friehs, K.; Flaschel, E.; Saalbach, A.; Twellmann, T.; Nattkemper, T.W. Libraries of synthetic stationary-phase and stress promoters as a tool for fine-tuning of expression of recombinant proteins in Escherichia coli. J. Biotechnol. 2005, 120, 25–37. [Google Scholar] [CrossRef]
- Chen, Y.J.; Liu, P.; Nielsen, A.A.; Brophy, J.A.; Clancy, K.; Peterson, T.; Voigt, C.A. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat. Methods 2013, 10, 659–664. [Google Scholar] [CrossRef]
- Zaslaver, A.; Bren, A.; Ronen, M.; Itzkovitz, S.; Kikoin, I.; Shavit, S.; Liebermeister, W.; Surette, M.G.; Alon, U. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat. Methods 2006, 3, 623–628. [Google Scholar] [CrossRef]
- Strandberg, L.; Andersson, L.; Enfors, S.O. The use of fed batch cultivation for achieving high cell densities in the production of a recombinant protein in Escherichia coli. FEMS Microbiol. Rev. 1994, 14, 53–56. [Google Scholar] [CrossRef]
- Terpe, K. Overview of bacterial expression systems for heterologous protein production: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2006, 72, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Hermjakob, H.; Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1999, 1473, 4–8. [Google Scholar] [CrossRef]
- Brown, J.L.; Roberts, W.K. Evidence that approximately eighty per cent of the soluble proteins from Ehrlich ascites cells are Nalpha-acetylated. J. Biol. Chem. 1976, 251, 1009–1014. [Google Scholar] [PubMed]
- Holmes, W.M.; Mannakee, B.K.; Gutenkunst, R.N.; Serio, T.R. Loss of amino-terminal acetylation suppresses a prion phenotype by modulating global protein folding. Nat. Commun. 2014, 5, 4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacker, M.; Linton, D.; Hitchen, P.G.; Nita-Lazar, M.; Haslam, S.M.; North, S.J.; Panico, M.; Morris, H.R.; Dell, A.; Wren, B.W.; et al. N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science 2002, 298, 1790–1793. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Shinozuka, Y.; Obata, Y.; Yokoyama, K.K. Phosphorylation of two eukaryotic transcription factors, Jun dimerization protein 2 and activation transcription factor 2, in Escherichia coli by Jun N-terminal kinase 1. Anal. Biochem. 2008, 376, 115–121. [Google Scholar] [CrossRef]
- Ren, Y.; Yao, X.; Dai, H.; Li, S.; Fang, H.; Chen, H.; Zhou, C. Production of Nalpha-acetylated thymosin alpha1 in Escherichia coli. Microb. Cell Fact. 2011, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Eastwood, T.A.; Baker, K.; Brooker, H.R.; Frank, S.; Mulvihill, D.P. An enhanced recombinant amino-terminal acetylation system and novel in vivo high-throughput screen for molecules affecting alpha-synuclein oligomerisation. FEBS Lett. 2017, 591, 833–841. [Google Scholar] [CrossRef] [Green Version]
- Neumann, H.; Peak-Chew, S.Y.; Chin, J.W. Genetically encoding N(epsilon)-acetyllysine in recombinant proteins. Nat. Chem. Biol. 2008, 4, 232–234. [Google Scholar] [CrossRef]
- de Marco, A.; Deuerling, E.; Mogk, A.; Tomoyasu, T.; Bukau, B. Chaperone-based procedure to increase yields of soluble recombinant proteins produced in E. coli. BMC Biotechnol. 2007, 7, 32. [Google Scholar] [CrossRef] [Green Version]
- Bessette, P.H.; Aslund, F.; Beckwith, J.; Georgiou, G. Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc. Natl. Acad. Sci. USA 1999, 96, 13703–13708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatahet, F.; Nguyen, V.D.; Salo, K.E.; Ruddock, L.W. Disruption of reducing pathways is not essential for efficient disulfide bond formation in the cytoplasm of E. coli. Microb. Cell Fact. 2010, 9, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miroux, B.; Walker, J.E. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 1996, 260, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, S.; Lofblom, J.; Lee, C.; Hjelm, A.; Klepsch, M.; Strous, M.; Drew, D.; Slotboom, D.J.; de Gier, J.W. Optimizing membrane protein overexpression in the Escherichia coli strain Lemo21(DE3). J. Mol. Biol. 2012, 423, 648–659. [Google Scholar] [CrossRef]
- Tegel, H.; Tourle, S.; Ottosson, J.; Persson, A. Increased levels of recombinant human proteins with the Escherichia coli strain Rosetta(DE3). Protein Expr. Purif. 2010, 69, 159–167. [Google Scholar] [CrossRef]
- Wu, X.C.; Lee, W.; Tran, L.; Wong, S.L. Engineering a Bacillus subtilis expression-secretion system with a strain deficient in six extracellular proteases. J. Bacteriol. 1991, 173, 4952–4958. [Google Scholar] [CrossRef] [Green Version]
- Thwaite, J.E.; Baillie, L.W.; Carter, N.M.; Stephenson, K.; Rees, M.; Harwood, C.R.; Emmerson, P.T. Optimization of the cell wall microenvironment allows increased production of recombinant Bacillus anthracis protective antigen from B. subtilis. Appl. Environ. Microbiol. 2002, 68, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Kuipers, O.P.; Beerthuyzen, M.M.; de Ruyter, P.G.; Luesink, E.J.; de Vos, W.M. Autoregulation of nisin biosynthesis in Lactococcus lactis by signal transduction. J. Biol. Chem. 1995, 270, 27299–27304. [Google Scholar] [CrossRef] [Green Version]
- Sohlemann, P.; Soppa, J.; Oesterhelt, D.; Lohse, M.J. Expression of beta 2-adrenoceptors in halobacteria. Naunyn Schmiedebergs Arch. Pharmacol. 1997, 355, 150–160. [Google Scholar] [CrossRef]
- Lobasso, S.; Vitale, R.; Lopalco, P.; Corcelli, A. Haloferax volcanii, as a Novel Tool for Producing Mammalian Olfactory Receptors Embedded in Archaeal Lipid Bilayer. Life 2015, 5, 770–782. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Yoshida, K.; Ohshima, T. Polysaccharide-degrading thermophiles generated by heterologous gene expression in Geobacillus kaustophilus HTA426. Appl. Environ. Microbiol. 2013, 79, 5151–5158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacker, M.; Feldman, M.F.; Callewaert, N.; Kowarik, M.; Clarke, B.R.; Pohl, N.L.; Hernandez, M.; Vines, E.D.; Valvano, M.A.; Whitfield, C.; et al. Substrate specificity of bacterial oligosaccharyltransferase suggests a common transfer mechanism for the bacterial and eukaryotic systems. Proc. Natl. Acad. Sci. USA 2006, 103, 7088–7093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valderrama-Rincon, J.D.; Fisher, A.C.; Merritt, J.H.; Fan, Y.Y.; Reading, C.A.; Chhiba, K.; Heiss, C.; Azadi, P.; Aebi, M.; DeLisa, M.P. An engineered eukaryotic protein glycosylation pathway in Escherichia coli. Nat. Chem. Biol. 2012, 8, 434–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaffer, C.; Messner, P. Emerging facets of prokaryotic glycosylation. FEMS Microbiol. Rev. 2017, 41, 49–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurmond, J.M.; Hards, R.G.; Seipelt, C.T.; Leonard, A.E.; Hansson, L.; Stromqvist, M.; Bystrom, M.; Enquist, K.; Xu, B.C.; Kopchick, J.J.; et al. Expression and characterization of phosphorylated recombinant human beta-casein in Escherichia coli. Protein Expr. Purif. 1997, 10, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.G.; Meyer, J.G.; Baumgartner, J.T.; D’Souza, A.K.; Nelson, W.C.; Payne, S.H.; Kuhn, M.L.; Schilling, B.; Wolfe, A.J. Identification of Novel Protein Lysine Acetyltransferases in Escherichia coli. mBio 2018, 9, e01905-18. [Google Scholar] [CrossRef] [Green Version]
- Amrein, K.E.; Takacs, B.; Stieger, M.; Molnos, J.; Flint, N.A.; Burn, P. Purification and characterization of recombinant human p50csk protein-tyrosine kinase from an Escherichia coli expression system overproducing the bacterial chaperones GroES and GroEL. Proc. Natl. Acad. Sci. USA 1995, 92, 1048–1052. [Google Scholar] [CrossRef] [Green Version]
- Nishihara, K.; Kanemori, M.; Yanagi, H.; Yura, T. Overexpression of trigger factor prevents aggregation of recombinant proteins in Escherichia coli. Appl. Environ. Microbiol. 2000, 66, 884–889. [Google Scholar] [CrossRef] [Green Version]
- Prinz, W.A.; Aslund, F.; Holmgren, A.; Beckwith, J. The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J. Biol. Chem. 1997, 272, 15661–15667. [Google Scholar] [CrossRef] [Green Version]
- Bosnjak, I.; Bojovic, V.; Segvic-Bubic, T.; Bielen, A. Occurrence of protein disulfide bonds in different domains of life: A comparison of proteins from the Protein Data Bank. Protein Eng. Des. Sel. 2014, 27, 65–72. [Google Scholar] [CrossRef]
- Freudl, R. Signal peptides for recombinant protein secretion in bacterial expression systems. Microb. Cell Fact. 2018, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.J.; Aslund, F.; Beckwith, J. Disulfide bond formation in the Escherichia coli cytoplasm: An in vivo role reversal for the thioredoxins. EMBO J. 1998, 17, 5543–5550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faulkner, M.J.; Veeravalli, K.; Gon, S.; Georgiou, G.; Beckwith, J. Functional plasticity of a peroxidase allows evolution of diverse disulfide-reducing pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 6735–6740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaciarz, A.; Khatri, N.K.; Velez-Suberbie, M.L.; Saaranen, M.J.; Uchida, Y.; Keshavarz-Moore, E.; Ruddock, L.W. Efficient soluble expression of disulfide bonded proteins in the cytoplasm of Escherichia coli in fed-batch fermentations on chemically defined minimal media. Microb. Cell Fact. 2017, 16, 108. [Google Scholar] [CrossRef]
- Schlegel, S.; Genevaux, P.; de Gier, J.W. De-convoluting the Genetic Adaptations of E. coli C41(DE3) in Real Time Reveals How Alleviating Protein Production Stress Improves Yields. Cell Rep. 2015, 10, 1758–1766. [Google Scholar] [CrossRef] [Green Version]
- Giacalone, M.J.; Gentile, A.M.; Lovitt, B.T.; Berkley, N.L.; Gunderson, C.W.; Surber, M.W. Toxic protein expression in Escherichia coli using a rhamnose-based tightly regulated and tunable promoter system. Biotechniques 2006, 40, 355–364. [Google Scholar] [CrossRef]
- Schallmey, M.; Singh, A.; Ward, O.P. Developments in the use of Bacillus species for industrial production. Can. J. Microbiol. 2004, 50, 1–17. [Google Scholar] [CrossRef]
- Westers, L.; Westers, H.; Quax, W.J. Bacillus subtilis as cell factory for pharmaceutical proteins: A biotechnological approach to optimize the host organism. Biochim. Biophys. Acta 2004, 1694, 299–310. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, D. Current development in genetic engineering strategies of Bacillus species. Microb. Cell Fact. 2014, 13, 63. [Google Scholar] [CrossRef]
- Boratynski, J.; Szermer-Olearnik, B. Endotoxin Removal from Escherichia coli Bacterial Lysate Using a Biphasic Liquid System. Methods Mol. Biol. 2017, 1600, 107–112. [Google Scholar]
- Simonen, M.; Palva, I. Protein secretion in Bacillus species. Microbiol. Rev. 1993, 57, 109–137. [Google Scholar] [CrossRef] [PubMed]
- Tjalsma, H.; Antelmann, H.; Jongbloed, J.D.; Braun, P.G.; Darmon, E.; Dorenbos, R.; Dubois, J.Y.; Westers, H.; Zanen, G.; Quax, W.J.; et al. Proteomics of protein secretion by Bacillus subtilis: Separating the secrets of the secretome. Microbiol. Mol. Biol. Rev. 2004, 68, 207–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, L.; Dong, Q.; Xu, Z.; Yang, S.; Zhou, J.; Freudl, R. Functional implementation of the posttranslational SecB-SecA protein-targeting pathway in Bacillus subtilis. Appl. Environ. Microbiol. 2012, 78, 651–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakeshita, H.; Kageyama, Y.; Ara, K.; Ozaki, K.; Nakamura, K. Enhanced extracellular production of heterologous proteins in Bacillus subtilis by deleting the C-terminal region of the SecA secretory machinery. Mol. Biotechnol. 2010, 46, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, L.; Fu, G.; Zhou, W.; Sun, Y.; Zheng, P.; Sun, J.; Zhang, D. A novel strategy for protein production using non-classical secretion pathway in Bacillus subtilis. Microb. Cell Fact. 2016, 15, 69. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Chen, J.; Sun, J.; Zhang, D. Multimer recognition and secretion by the non-classical secretion pathway in Bacillus subtilis. Sci. Rep. 2017, 7, 44023. [Google Scholar] [CrossRef] [Green Version]
- He, X.S.; Bruckner, R.; Doi, R.H. The protease genes of Bacillus subtilis. Res. Microbiol. 1991, 142, 797–803. [Google Scholar] [CrossRef]
- Westers, L.; Dijkstra, D.S.; Westers, H.; van Dijl, J.M.; Quax, W.J. Secretion of functional human interleukin-3 from Bacillus subtilis. J. Biotechnol. 2006, 123, 211–224. [Google Scholar] [CrossRef]
- Wong, S.L.; Ye, R.; Nathoo, S. Engineering and production of streptokinase in a Bacillus subtilis expression-secretion system. Appl. Environ. Microbiol. 1994, 60, 517–523. [Google Scholar] [CrossRef] [Green Version]
- Murashima, K.; Chen, C.L.; Kosugi, A.; Tamaru, Y.; Doi, R.H.; Wong, S.L. Heterologous production of Clostridium cellulovorans engB, using protease-deficient Bacillus subtilis, and preparation of active recombinant cellulosomes. J. Bacteriol. 2002, 184, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Ji, S.; Li, W.; Baloch, A.R.; Wang, M.; Li, H.; Cao, B.; Zhang, H. Efficient biosynthesis of a Cecropin A-melittin mutant in Bacillus subtilis WB700. Sci. Rep. 2017, 7, 40587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zweers, J.C.; Barak, I.; Becher, D.; Driessen, A.J.; Hecker, M.; Kontinen, V.P.; Saller, M.J.; Vavrova, L.; van Dijl, J.M. Towards the development of Bacillus subtilis as a cell factory for membrane proteins and protein complexes. Microb. Cell Fact. 2008, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steidler, L.; Hans, W.; Schotte, L.; Neirynck, S.; Obermeier, F.; Falk, W.; Fiers, W.; Remaut, E. Treatment of murine colitis by Lactococcus lactis secreting interleukin-10. Science 2000, 289, 1352–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mierau, I.; Kleerebezem, M. 10 years of the nisin-controlled gene expression system (NICE) in Lactococcus lactis. Appl. Microbiol. Biotechnol. 2005, 68, 705–717. [Google Scholar] [CrossRef]
- Boumaiza, M.; Colarusso, A.; Parrilli, E.; Garcia-Fruitos, E.; Casillo, A.; Aris, A.; Corsaro, M.M.; Picone, D.; Leone, S.; Tutino, M.L. Getting value from the waste: Recombinant production of a sweet protein by Lactococcus lactis grown on cheese whey. Microb. Cell Fact. 2018, 17, 126. [Google Scholar] [CrossRef] [Green Version]
- Mierau, I.; Olieman, K.; Mond, J.; Smid, E.J. Optimization of the Lactococcus lactis nisin-controlled gene expression system NICE for industrial applications. Microb. Cell Fact. 2005, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Steen, A.; Wiederhold, E.; Gandhi, T.; Breitling, R.; Slotboom, D.J. Physiological adaptation of the bacterium Lactococcus lactis in response to the production of human CFTR. Mol. Cell Proteom. 2011, 10, M000052MCP200. [Google Scholar] [CrossRef]
- Singh, S.K.; Tiendrebeogo, R.W.; Chourasia, B.K.; Kana, I.H.; Singh, S.; Theisen, M. Lactococcus lactis provides an efficient platform for production of disulfide-rich recombinant proteins from Plasmodium falciparum. Microb. Cell Fact. 2018, 17, 55. [Google Scholar] [CrossRef]
- Cano-Garrido, O.; Rueda, F.L.; Sanchez-Garcia, L.; Ruiz-Avila, L.; Bosser, R.; Villaverde, A.; Garcia-Fruitos, E. Expanding the recombinant protein quality in Lactococcus lactis. Microb. Cell Fact. 2014, 13, 167. [Google Scholar] [CrossRef] [Green Version]
- Geertsma, E.R.; Poolman, B. High-throughput cloning and expression in recalcitrant bacteria. Nat. Methods 2007, 4, 705–707. [Google Scholar] [CrossRef] [Green Version]
- Ventosa, A.; Nieto, J.J.; Oren, A. Biology of moderately halophilic aerobic bacteria. Microbiol. Mol. Biol. Rev. 1998, 62, 504–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakawa, T.; Timasheff, S.N. The stabilization of proteins by osmolytes. Biophys. J. 1985, 47, 411–414. [Google Scholar] [CrossRef]
- Tokunaga, H.; Arakawa, T.; Tokunaga, M. Novel soluble expression technologies derived from unique properties of halophilic proteins. Appl. Microbiol. Biotechnol. 2010, 88, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Nagayoshi, C.; Ishibashi, M.; Tokunaga, M. Purification and characterization of human brain serine racemase expressed in moderately halophilic bacteria. Protein Pept. Lett. 2009, 16, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Zeldes, B.M.; Keller, M.W.; Loder, A.J.; Straub, C.T.; Adams, M.W.; Kelly, R.M. Extremely thermophilic microorganisms as metabolic engineering platforms for production of fuels and industrial chemicals. Front. Microbiol. 2015, 6, 1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cava, F.; Hidalgo, A.; Berenguer, J. Thermus thermophilus as biological model. Extremophiles 2009, 13, 213–231. [Google Scholar] [CrossRef]
- Velez, A.M.; Horta, A.C.; da Silva, A.J.; Iemma, M.R.; Giordano Rde, L.; Zangirolami, T.C. Enhanced production of recombinant thermo-stable lipase in Escherichia coli at high induction temperature. Protein Expr. Purif. 2013, 90, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, H.P.; Sperling-Petersen, H.U.; Mortensen, K.K. Production of recombinant thermostable proteins expressed in Escherichia coli: Completion of protein synthesis is the bottleneck. J. Chromatogr. B Anal. Technol. Biomed Life Sci. 2003, 786, 207–214. [Google Scholar] [CrossRef]
- Albers, S.V.; Jonuscheit, M.; Dinkelaker, S.; Urich, T.; Kletzin, A.; Tampe, R.; Driessen, A.J.; Schleper, C. Production of recombinant and tagged proteins in the hyperthermophilic archaeon Sulfolobus solfataricus. Appl. Environ. Microbiol. 2006, 72, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Drejer, E.B.; Hakvag, S.; Irla, M.; Brautaset, T. Genetic Tools and Techniques for Recombinant Expression in Thermophilic Bacillaceae. Microorganisms. 2018, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Khalil, A.S.; Collins, J.J. Synthetic biology: Applications come of age. Nat. Rev. Genet. 2010, 11, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Vickers, C.E.; Blank, L.M.; Kromer, J.O. Grand challenge commentary: Chassis cells for industrial biochemical production. Nat. Chem. Biol. 2010, 6, 875–877. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, T.; Kadoya, R.; Endo, K.; Tohata, M.; Sawada, K.; Liu, S.; Ozawa, T.; Kodama, T.; Kakeshita, H.; Kageyama, Y.; et al. Enhanced recombinant protein productivity by genome reduction in Bacillus subtilis. DNA Res. 2008, 15, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, Q.T.; Yu, P.; Wang, J.; Li, Z.Y.; Chen, X.A.; Mao, X.M.; Li, Y.Q. Rational construction of genome-reduced and high-efficient industrial Streptomyces chassis based on multiple comparative genomic approaches. Microb. Cell Fact. 2019, 18, 16. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Benders, G.A.; Andrews-Pfannkoch, C.; Denisova, E.A.; Baden-Tillson, H.; Zaveri, J.; Stockwell, T.B.; Brownley, A.; Thomas, D.W.; Algire, M.A.; et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 2008, 319, 1215–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchison, C.A.; Chuang, R.Y.; Noskov, V.N.; Assad-Garcia, N.; Deerinck, T.J.; Ellisman, M.H.; Gill, J.; Kannan, K.; Karas, B.J.; Ma, L.; et al. Design and synthesis of a minimal bacterial genome. Science 2016, 351, 6253. [Google Scholar] [CrossRef] [Green Version]
- Fredens, J.; Wang, K.; de la Torre, D.; Funke, L.F.H.; Robertson, W.E.; Christova, Y.; Chia, T.; Schmied, W.H.; Dunkelmann, D.L.; Beranek, V.; et al. Total synthesis of Escherichia coli with a recoded genome. Nature 2019, 569, 514–518. [Google Scholar] [CrossRef]
- Wang, K.; Fredens, J.; Brunner, S.F.; Kim, S.H.; Chia, T.; Chin, J.W. Defining synonymous codon compression schemes by genome recoding. Nature 2016, 539, 59–64. [Google Scholar] [CrossRef]
- Wang, K.; Sachdeva, A.; Cox, D.J.; Wilf, N.M.; Lang, K.; Wallace, S.; Mehl, R.A.; Chin, J.W. Optimized orthogonal translation of unnatural amino acids enables spontaneous protein double-labelling and FRET. Nat. Chem. 2014, 6, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [Green Version]
- Abremski, K.; Hoess, R.; Sternberg, N. Studies on the properties of P1 site-specific recombination: Evidence for topologically unlinked products following recombination. Cell 1983, 32, 1301–1311. [Google Scholar] [CrossRef]
- Broach, J.R.; Guarascio, V.R.; Jayaram, M. Recombination within the yeast plasmid 2mu circle is site-specific. Cell 1982, 29, 227–234. [Google Scholar] [CrossRef]
- Hashimoto, M.; Ichimura, T.; Mizoguchi, H.; Tanaka, K.; Fujimitsu, K.; Keyamura, K.; Ote, T.; Yamakawa, T.; Yamazaki, Y.; Mori, H.; et al. Cell size and nucleoid organization of engineered Escherichia coli cells with a reduced genome. Mol. Microbiol. 2005, 55, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Posfai, G.; Plunkett, G.; Feher, T.; Frisch, D.; Keil, G.M.; Umenhoffer, K.; Kolisnychenko, V.; Stahl, B.; Sharma, S.S.; de Arruda, M.; et al. Emergent properties of reduced-genome Escherichia coli. Science 2006, 312, 1044–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizoguchi, H.; Sawano, Y.; Kato, J.; Mori, H. Superpositioning of deletions promotes growth of Escherichia coli with a reduced genome. DNA Res. 2008, 15, 277–284. [Google Scholar] [CrossRef]
- Yu, B.J.; Sung, B.H.; Koob, M.D.; Lee, C.H.; Lee, J.H.; Lee, W.S.; Kim, M.S.; Kim, S.C. Minimization of the Escherichia coli genome using a Tn5-targeted Cre/loxP excision system. Nat. Biotechnol. 2002, 20, 1018–1023. [Google Scholar] [CrossRef]
- Park, M.K.; Lee, S.H.; Yang, K.S.; Jung, S.C.; Lee, J.H.; Kim, S.C. Enhancing recombinant protein production with an Escherichia coli host strain lacking insertion sequences. Appl. Microbiol. Biotechnol. 2014, 98, 6701–6713. [Google Scholar] [CrossRef]
- Komatsu, M.; Uchiyama, T.; Omura, S.; Cane, D.E.; Ikeda, H. Genome-minimized Streptomyces host for the heterologous expression of secondary metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 2646–2651. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Escribano, J.P.; Bibb, M.J. Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb. Biotechnol. 2011, 4, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Jing, X.; Xie, P.; Chen, W.; Wang, T.; Xia, H.; Qin, Z. Sequential deletion of all the polyketide synthase and nonribosomal peptide synthetase biosynthetic gene clusters and a 900-kb subtelomeric sequence of the linear chromosome of Streptomyces coelicolor. FEMS Microbiol. Lett. 2012, 333, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Thanapipatsiri, A.; Claesen, J.; Gomez-Escribano, J.P.; Bibb, M.; Thamchaipenet, A. A Streptomyces coelicolor host for the heterologous expression of Type III polyketide synthase genes. Microb. Cell Fact. 2015, 14, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myronovskyi, M.; Rosenkranzer, B.; Nadmid, S.; Pujic, P.; Normand, P.; Luzhetskyy, A. Generation of a cluster-free Streptomyces albus chassis strains for improved heterologous expression of secondary metabolite clusters. Metab. Eng. 2018, 49, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xiao, L.P.; Zhou, Y.J.; Deng, K.H.; Tan, G.Y.; Han, Y.C.; Liu, X.H.; Deng, Z.X.; Liu, T.G. Development of Streptomyces sp. FR-008 as an emerging chassis. Synth. Syst. Biotechnol. 2016, 1, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manabe, K.; Kageyama, Y.; Morimoto, T.; Shimizu, E.; Takahashi, H.; Kanaya, S.; Ara, K.; Ozaki, K.; Ogasawara, N. Improved production of secreted heterologous enzyme in Bacillus subtilis strain MGB874 via modification of glutamate metabolism and growth conditions. Microb. Cell Fact. 2013, 12, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ara, K.; Ozaki, K.; Nakamura, K.; Yamane, K.; Sekiguchi, J.; Ogasawara, N. Bacillus minimum genome factory: Effective utilization of microbial genome information. Biotechnol. Appl. Biochem. 2007, 46, 169–178. [Google Scholar] [PubMed]
- Westers, H.; Dorenbos, R.; van Dijl, J.M.; Kabel, J.; Flanagan, T.; Devine, K.M.; Jude, F.; Seror, S.J.; Beekman, A.C.; Darmon, E.; et al. Genome engineering reveals large dispensable regions in Bacillus subtilis. Mol. Biol. Evolut. 2003, 20, 2076–2090. [Google Scholar] [CrossRef] [Green Version]
- Reuss, D.R.; Altenbuchner, J.; Mader, U.; Rath, H.; Ischebeck, T.; Sappa, P.K.; Thurmer, A.; Guerin, C.; Nicolas, P.; Steil, L.; et al. Large-scale reduction of the Bacillus subtilis genome: Consequences for the transcriptional network, resource allocation, and metabolism. Genome Res. 2017, 27, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Koo, B.M.; Kritikos, G.; Farelli, J.D.; Todor, H.; Tong, K.; Kimsey, H.; Wapinski, I.; Galardini, M.; Cabal, A.; Peters, J.M.; et al. Construction and Analysis of Two Genome-Scale Deletion Libraries for Bacillus subtilis. Cell Syst. 2017, 4, 291–305. [Google Scholar] [CrossRef]
- Zhu, D.; Fu, Y.; Liu, F.; Xu, H.; Saris, P.E.; Qiao, M. Enhanced heterologous protein productivity by genome reduction in Lactococcus lactis NZ9000. Microb. Cell Fact. 2017, 16, 1. [Google Scholar] [CrossRef] [Green Version]
- Leprince, A.; de Lorenzo, V.; Voller, P.; van Passel, M.W.; Martins dos Santos, V.A. Random and cyclical deletion of large DNA segments in the genome of Pseudomonas putida. Environ. Microbiol. 2012, 14, 1444–1453. [Google Scholar] [CrossRef] [Green Version]
- Lieder, S.; Nikel, P.I.; de Lorenzo, V.; Takors, R. Genome reduction boosts heterologous gene expression in Pseudomonas putida. Microb. Cell Fact. 2015, 14, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgart, M.; Unthan, S.; Ruckert, C.; Sivalingam, J.; Grunberger, A.; Kalinowski, J.; Bott, M.; Noack, S.; Frunzke, J. Construction of a prophage-free variant of Corynebacterium glutamicum ATCC 13032 for use as a platform strain for basic research and industrial biotechnology. Appl. Environ. Microbiol. 2013, 79, 6006–6015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giga-Hama, Y.; Tohda, H.; Takegawa, K.; Kumagai, H. Schizosaccharomyces pombe minimum genome factory. Biotechnol. Appl. Biochem. 2007, 46, 147–155. [Google Scholar] [PubMed]
- Kolisnychenko, V.; Plunkett, G.; Herring, C.D.; Feher, T.; Posfai, J.; Blattner, F.R.; Posfai, G. Engineering a reduced Escherichia coli genome. Genome Res. 2002, 12, 640–647. [Google Scholar] [CrossRef] [Green Version]
- Aguilar Suarez, R.; Stulke, J.; van Dijl, J.M. Less Is More: Toward a Genome-Reduced Bacillus Cell Factory for Difficult Proteins. ACS Synth. Biol. 2019, 8, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Csorgo, B.; Feher, T.; Timar, E.; Blattner, F.R.; Posfai, G. Low-mutation-rate, reduced-genome Escherichia coli: An improved host for faithful maintenance of engineered genetic constructs. Microb. Cell Fact. 2012, 11, 11. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Sung, B.H.; Kim, M.S.; Blattner, F.R.; Yoon, B.H.; Kim, J.H.; Kim, S.C. Metabolic engineering of a reduced-genome strain of Escherichia coli for L-threonine production. Microb. Cell Fact. 2009, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Choe, D.; Lee, J.H.; Yoo, M.; Hwang, S.; Sung, B.H.; Cho, S.; Palsson, B.; Kim, S.C.; Cho, B.K. Adaptive laboratory evolution of a genome-reduced Escherichia coli. Nat. Commun. 2019, 10, 935. [Google Scholar] [CrossRef]
- Hirokawa, Y.; Kawano, H.; Tanaka-Masuda, K.; Nakamura, N.; Nakagawa, A.; Ito, M.; Mori, H.; Oshima, T.; Ogasawara, N. Genetic manipulations restored the growth fitness of reduced-genome Escherichia coli. J. Biosci. Bioeng. 2013, 116, 52–58. [Google Scholar] [CrossRef]
- Mushegian, A.R.; Koonin, E.V. A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc. Natl. Acad. Sci. USA 1996, 93, 10268–10273. [Google Scholar] [CrossRef] [Green Version]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Ehrlich, S.D.; Albertini, A.; Amati, G.; Andersen, K.K.; Arnaud, M.; Asai, K.; Ashikaga, S.; Aymerich, S.; Bessieres, P.; et al. Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. USA 2003, 100, 4678–4683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Opijnen, T.; Bodi, K.L.; Camilli, A. Tn-seq: High-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 2009, 6, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Colavin, A.; Shi, H.; Czarny, T.L.; Larson, M.H.; Wong, S.; Hawkins, J.S.; Lu, C.H.S.; Koo, B.M.; Marta, E.; et al. A Comprehensive, CRISPR-based Functional Analysis of Essential Genes in Bacteria. Cell 2016, 165, 1493–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlomi, T.; Eisenberg, Y.; Sharan, R.; Ruppin, E. A genome-scale computational study of the interplay between transcriptional regulation and metabolism. Mol. Syst. Biol. 2007, 3, 101. [Google Scholar] [CrossRef]
- Rees, J.; Chalkley, O.; Landon, S.; Purcell, O.; Marucci, L.; Grierson, C. Designing genomes using whole-cell models. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Rugbjerg, P.; Myling-Petersen, N.; Porse, A.; Sarup-Lytzen, K.; Sommer, M.O.A. Diverse genetic error modes constrain large-scale bio-based production. Nat. Commun. 2018, 9, 787. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, F.; Rinas, U. Stress induced by recombinant protein production in Escherichia coli. Adv. Biochem. Eng. Biotechnol. 2004, 89, 73–92. [Google Scholar]
- Borkowski, O.; Bricio, C.; Murgiano, M.; Rothschild-Mancinelli, B.; Stan, G.B.; Ellis, T. Cell-free prediction of protein expression costs for growing cells. Nat. Commun. 2018, 9, 1457. [Google Scholar] [CrossRef] [Green Version]
- Thommen, M.; Holtkamp, W.; Rodnina, M.V. Co-translational protein folding: Progress and methods. Curr. Opin. Struct. Biol. 2017, 42, 83–89. [Google Scholar] [CrossRef]
- Kudla, G.; Murray, A.W.; Tollervey, D.; Plotkin, J.B. Coding-sequence determinants of gene expression in Escherichia coli. Science 2009, 324, 255–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavner, Y.; Kotlar, D. Codon bias as a factor in regulating expression via translation rate in the human genome. Gene 2005, 345, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Collart, M.A.; Weiss, B. Ribosome pausing, a dangerous necessity for co-translational events. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.P.; Mattocks, J.; Green, P.S.; Moffatt, F.; Kilby, P.M. Determination and control of low-level amino acid misincorporation in human thioredoxin protein produced in a recombinant Escherichia coli production system. Biotechnol. Bioeng. 2012, 109, 1987–1995. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, C.; Govindarajan, S.; Minshull, J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004, 22, 346–353. [Google Scholar] [CrossRef]
- Chung, B.K.; Lee, D.Y. Computational codon optimization of synthetic gene for protein expression. BMC Syst. Biol. 2012, 6, 134. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Lin, J.; Cen, P. 5-Aminolevulinate production with recombinant Escherichia coli using a rare codon optimizer host strain. Appl. Microbiol. Biotechnol. 2007, 75, 777–782. [Google Scholar] [CrossRef]
- Ostrov, N.; Landon, M.; Guell, M.; Kuznetsov, G.; Teramoto, J.; Cervantes, N.; Zhou, M.; Singh, K.; Napolitano, M.G.; Moosburner, M.; et al. Design, synthesis, and testing toward a 57-codon genome. Science 2016, 353, 819–822. [Google Scholar] [CrossRef] [Green Version]
- McManus, C.J.; May, G.E.; Spealman, P.; Shteyman, A. Ribosome profiling reveals post-transcriptional buffering of divergent gene expression in yeast. Genome Res. 2014, 24, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.; Kim, J.N.; Kim, M.W.; Bucca, G.; Cho, S.; Yoon, Y.J.; Kim, B.G.; Roe, J.H.; Kim, S.C.; Smith, C.P.; et al. The dynamic transcriptional and translational landscape of the model antibiotic producer Streptomyces coelicolor A3(2). Nat. Commun. 2016, 7, 11605. [Google Scholar] [CrossRef] [Green Version]
- Ebrahim, A.; Brunk, E.; Tan, J.; O’Brien, E.J.; Kim, D.; Szubin, R.; Lerman, J.A.; Lechner, A.; Sastry, A.; Bordbar, A.; et al. Multi-omic data integration enables discovery of hidden biological regularities. Nat. Commun. 2016, 7, 13091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, G.J.; Burkhardt, D.H.; Kelly, J.W.; Powers, E.T. Translation efficiency is maintained at elevated temperature in Escherichia coli. J. Biol. Chem. 2018, 293, 777–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niess, A.; Siemann-Herzberg, M.; Takors, R. Protein production in Escherichia coli is guided by the trade-off between intracellular substrate availability and energy cost. Microb. Cell Fact. 2019, 18, 8. [Google Scholar] [CrossRef] [PubMed]
- Kustatscher, G.; Grabowski, P.; Rappsilber, J. Pervasive coexpression of spatially proximal genes is buffered at the protein level. Mol. Syst. Biol. 2017, 13, 937. [Google Scholar] [CrossRef] [PubMed]



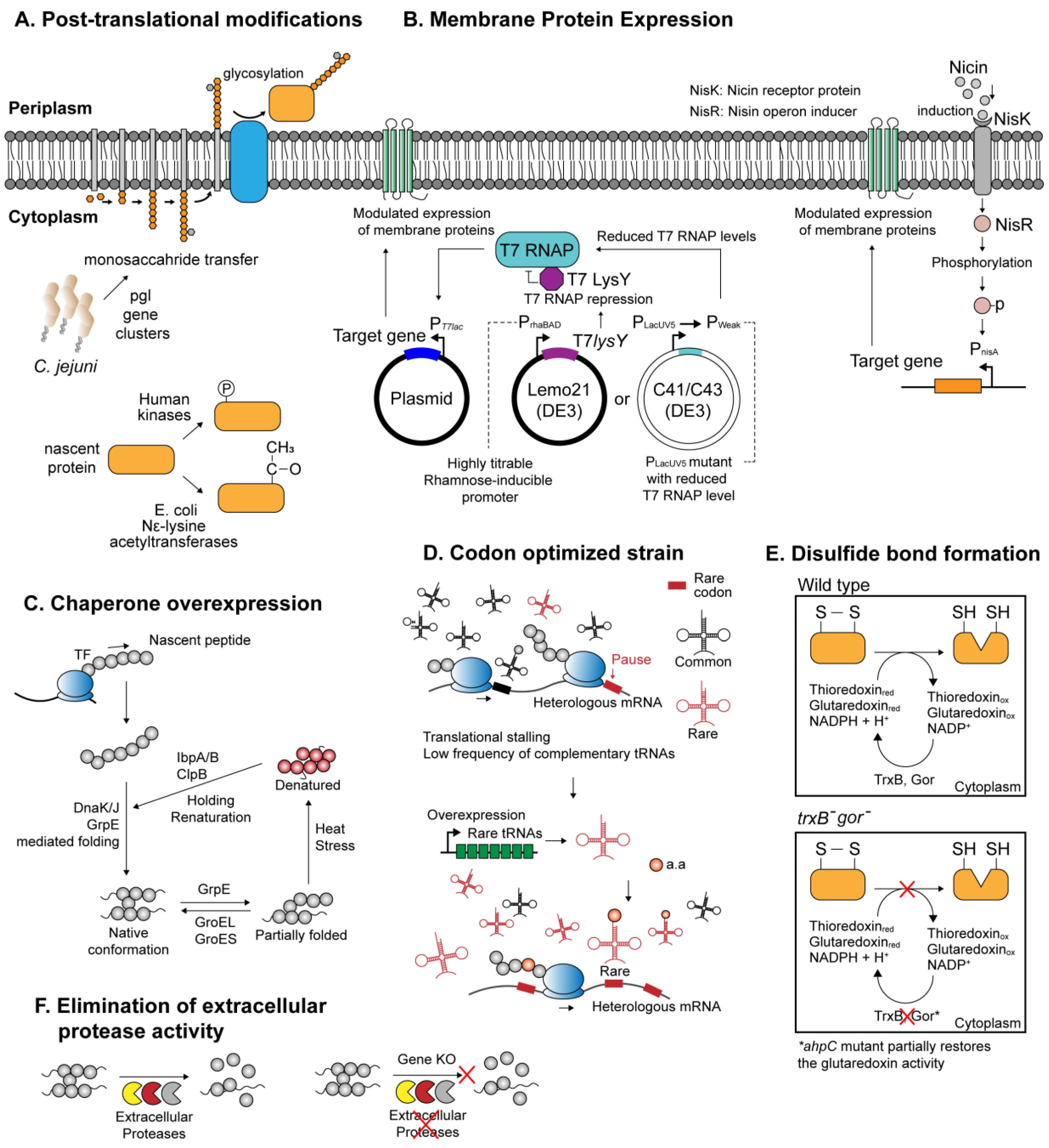{kind=link}
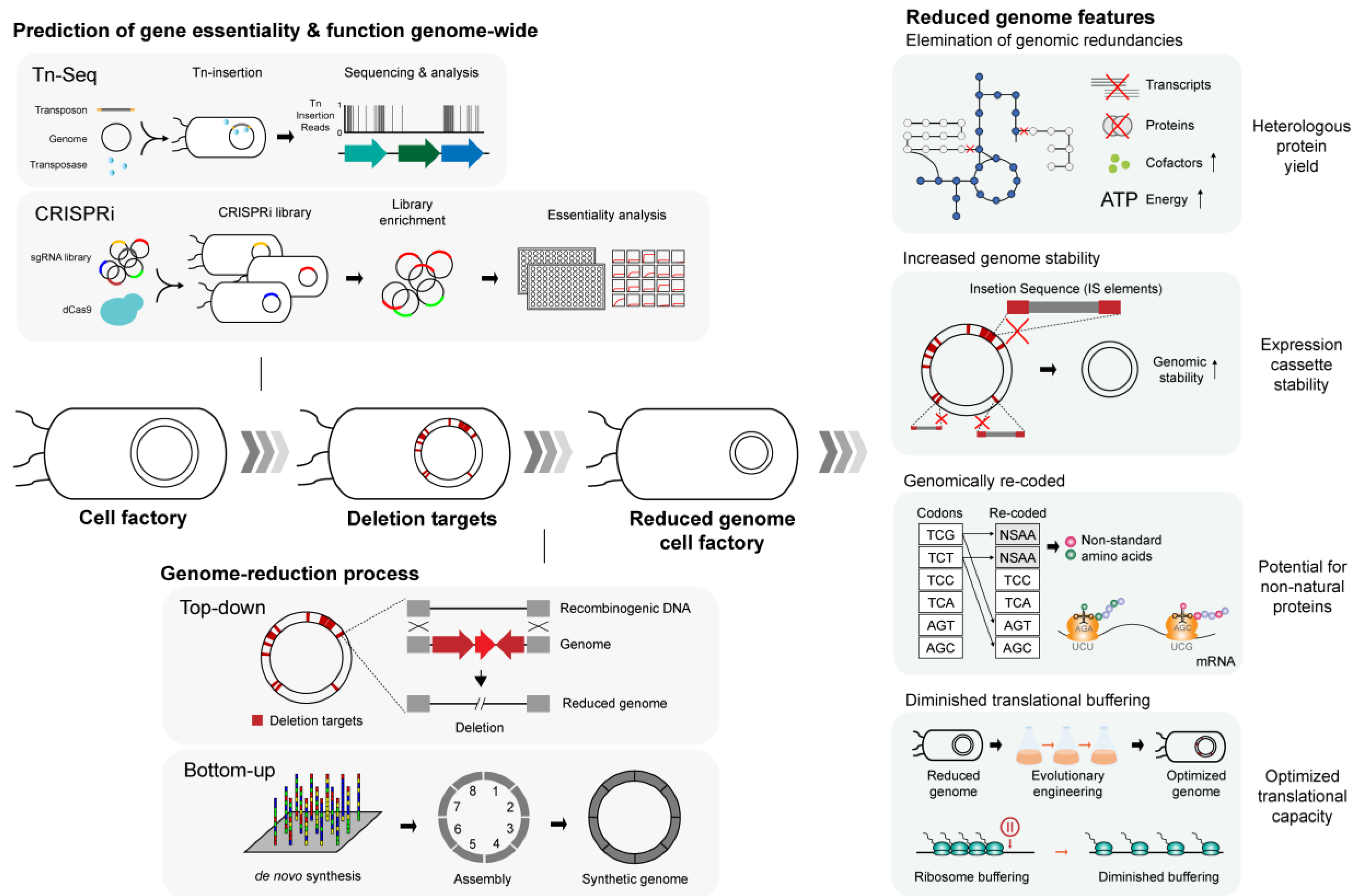{kind=link}
| Strain | Defining Features | Reference |
|---|---|---|
| E. coli (pgl cluster) | Expression of N-glycosylation pathway of C. jejuni origin, catalyzing glycosylation of recombinant proteins where appropriate. | [45] |
| E. coli (human JNK1) | Coexpression of human Jun N-terminal kinase 1 (JNK1) effectively catalyzes recombinant protein phosphorylation | [46] |
| E. coli (rimJ) | Use of the native acetylation machinery in E. coli to acetylate human proteins. | [47] |
| E. coli (yeast NatA NatB) | Coexpression of yeast-derived NatA NatB acetylation enzymes for amino-terminal acetylation | [48] |
| E. coli (Nε-acetyllysine) | Site-directed incorporation of Nε-acetyllysine using a three plasmid system expressing recoded target genes, suppressor tRNA, and evolutionarily engineered aminoacyl-tRNA synthetase. | [49] |
| E. coli (Chaperone overexpression) | Coordinated co-overexpression of E. coli native molecular chaperones—GroEL/ES, DnaK/J/GrpE, IbaA/B, and ClpB—significantly improves the solubility of the recombinant proteins. | [50] |
| E. coli Origami | Facilitates formation of disulfide bonds within the cytoplasmic compartment, through inactivation of thioredoxin and glutathione reductase pathways (ΔtrxB, Δgor, aphC) | [51] |
| E. coli CyDisCo | Introduction of eukaryotic thiol oxidase and disulfide isomerase encourages formation of disulfide bonds within the E. coli cytoplasm. | [52] |
| E. coli C41(DE3), C43(DE3) | BL21(DE3) derivative with mutations that confer increased tolerance to toxic membrane proteins. | [53] |
| E. coli Lemo21(DE3) | Harbors a gene expression system that allows fine-tuning of overexpression intensity. Suitable for membrane protein production. | [54] |
| E. coli Rosetta | Alleviates codon-bias by overexpression of tRNA species orthogonal to rare codons in E. coli—AUA, AGG, AGA, CGG, CUA, CCC, and GGA. | [55] |
| B. subtilis WB600 | Strain that lacks six out of seven extracellular proteases to circumvent host-mediated proteolysis | [56] |
| B. subtilis (dlt-) | Inactivation of D-anlanylation in dlt- B. subtilis increases the availability of folding factors Mg2+, Ca2+ and Fe3+ around the cell membrane microenvironment, alleviating protease-mediated recombinant protein degradation. | [57] |
| L. lactis NZ9000 | Microbial expression system that effectively supports the production of various prokaryotic and eukaryotic membrane proteins. | [58] |
| Haloferax volcanii | Halophilic archeon that stably overexpresses seven transmembrane helix proteins such as bacteroiopsins. The transmembrane protein expression machinery can be exploited to express eukaryotic proteins with similar protein topology. | [59,60] |
| Geobacillus kaustophilus | Thermophilic bacteria with an array of heat-stable, sugar-inducible promoters demonstrated soluble expression of heterologous enzymes otherwise insoluble in mesophilic host. With maximal protein yield of 59 mg/L | [61] |
| Strain | Designation | Genome Reduction | Notable Characteristics | Reference |
|---|---|---|---|---|
| E. coli MG1655 | MDS42 | 663 kbp (14.3%) | IS-free strain with increased stability of exogenous genetic construct. Increased yield of chimeric fusion protein. Improved efficiency of electroporation comparable to commercial DH10B strain. Additional engineering yielded improved recombinant protein productivity | [124] |
| E. coli MG1655 | MS56 | 1068 kbp (23.0%) | IS-free strain with increased stability of exogenous genetic construct. Higher electroporation efficiency. | [127] |
| E. coli MG1655 | eMS57 | 1089 kbp (23.5%) | Evolutionary engineering of MS56 restored the growth on minimal medium. Diminished translational buffering predicted to increase production of recombinant proteins. | [148] |
| E. coli W3110 | MGF-01 | 1030 kbp (22.2%) | Increased growth density. | [125] |
| E. coli W3110 | DGF-298 | 1670 kbp (35.9%) | Higher genome stability, increased growth rate. | [149] |
| B. subtilis 168 | MGIM | 991 kbp (23.5%) | Small reduction in growth rate, comparable enzyme production. | [135] |
| B. subtilis 168 | MBG874 | 874 kbp (20.7%) | Protein productivity increased up to 2.5-fold. Enhanced nutrient utilization. | [113] |
| B. subtilis 168 | PG10 | 1460 kbp (~36%) | Improved secretory protein production, including that of some of the difficult-to-produce proteins. | [137] |
| L. lactis NZ9000 | 9k-4 | 72 kbp (2.8%) | 2.2- to 2.5-fold increase in recombinant protein activities. Higher final cell density and growth rates. | [139] |
| P. putida KT2440 | EM383 | 266 kbp (4.3%) | Higher growth rate and final cell density. Showed as much as 41% increase in recombinant protein yield depending on the carbon source used. | [141] |
| C. glutamicum ATCC 13032 | MB001 | 205 kbp (6.2%) | Increased recombinant protein activity and transformation efficiency. | [142] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.; Choe, D.; Lee, D.-H.; Cho, B.-K. Engineering Biology to Construct Microbial Chassis for the Production of Difficult-to-Express Proteins. Int. J. Mol. Sci. 2020, 21, 990. https://doi.org/10.3390/ijms21030990
Kim K, Choe D, Lee D-H, Cho B-K. Engineering Biology to Construct Microbial Chassis for the Production of Difficult-to-Express Proteins. International Journal of Molecular Sciences. 2020; 21(3):990. https://doi.org/10.3390/ijms21030990
Chicago/Turabian StyleKim, Kangsan, Donghui Choe, Dae-Hee Lee, and Byung-Kwan Cho. 2020. "Engineering Biology to Construct Microbial Chassis for the Production of Difficult-to-Express Proteins" International Journal of Molecular Sciences 21, no. 3: 990. https://doi.org/10.3390/ijms21030990
APA StyleKim, K., Choe, D., Lee, D. -H., & Cho, B. -K. (2020). Engineering Biology to Construct Microbial Chassis for the Production of Difficult-to-Express Proteins. International Journal of Molecular Sciences, 21(3), 990. https://doi.org/10.3390/ijms21030990