Chemical and Light Inducible Epigenome Editing


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
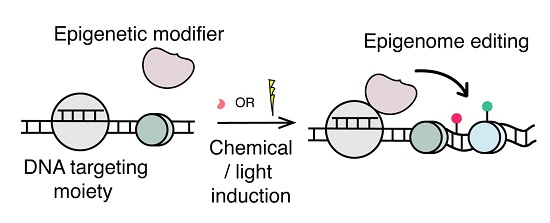
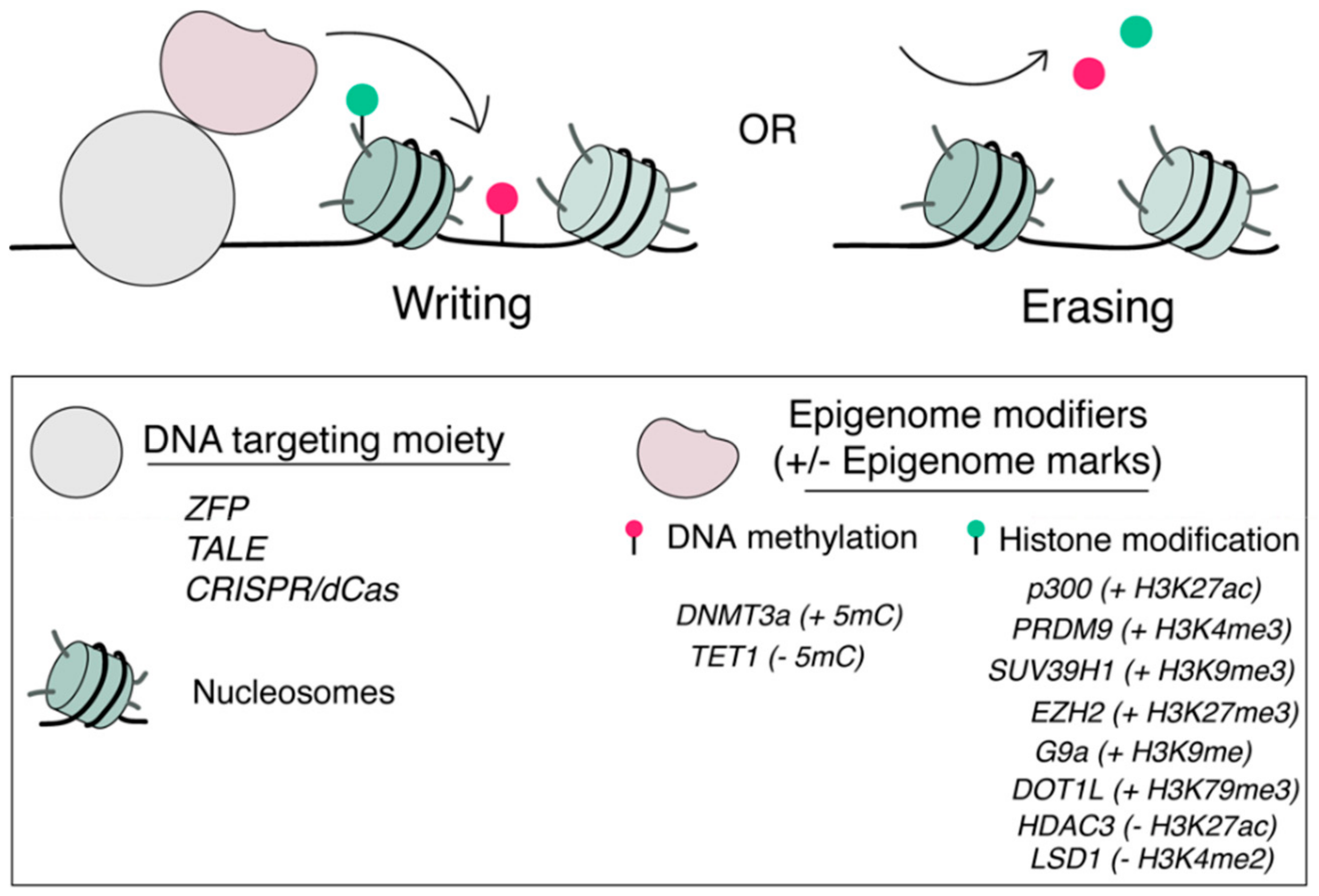
2. Overview of Epigenome Editing
3. Conditional Epigenome Editing
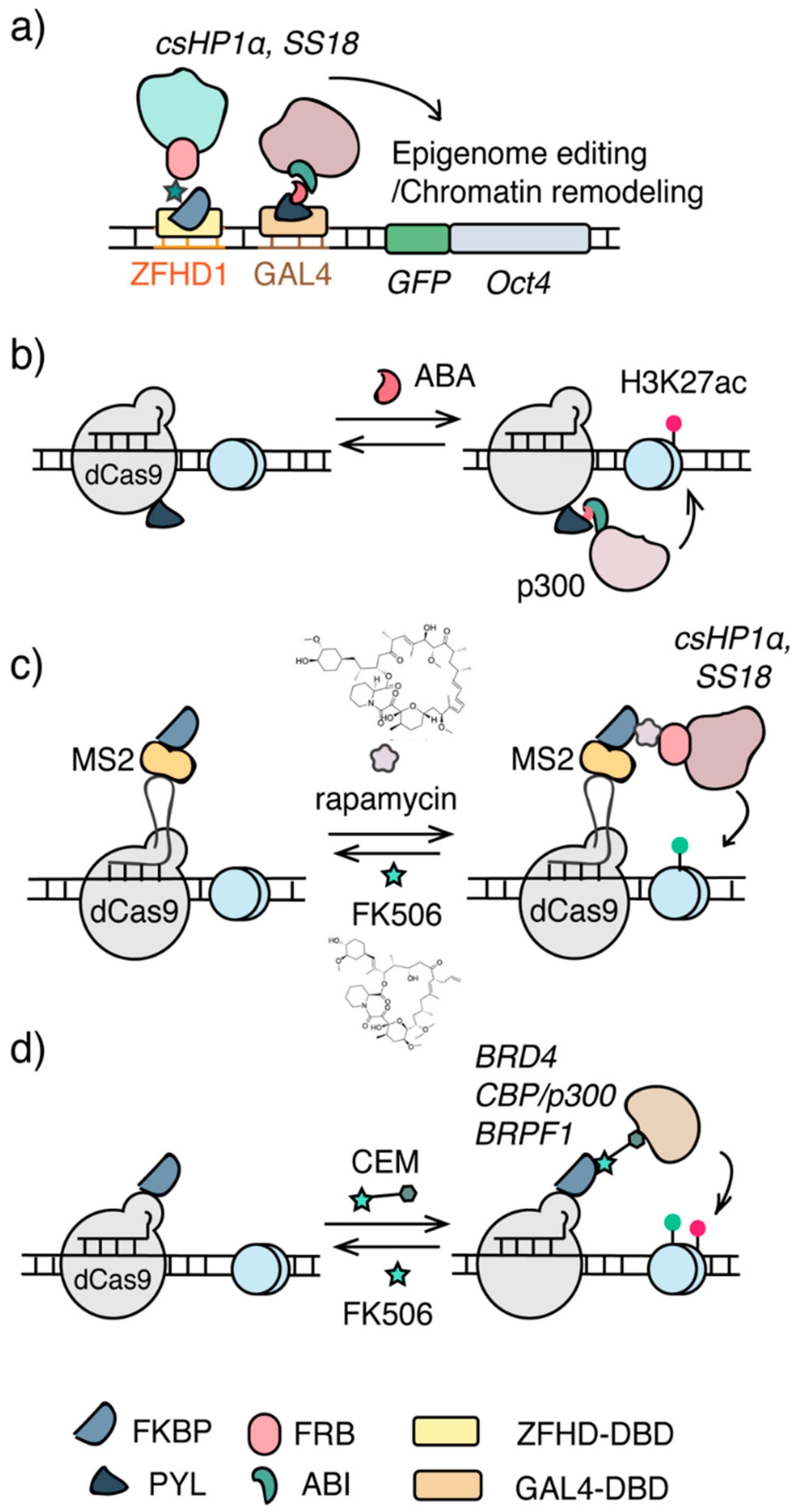
3.1. Chemically Induced Proximity (CIP)-Based Editing
3.2. Chemical Epigenetic Modifier (CEM)-Based Editing
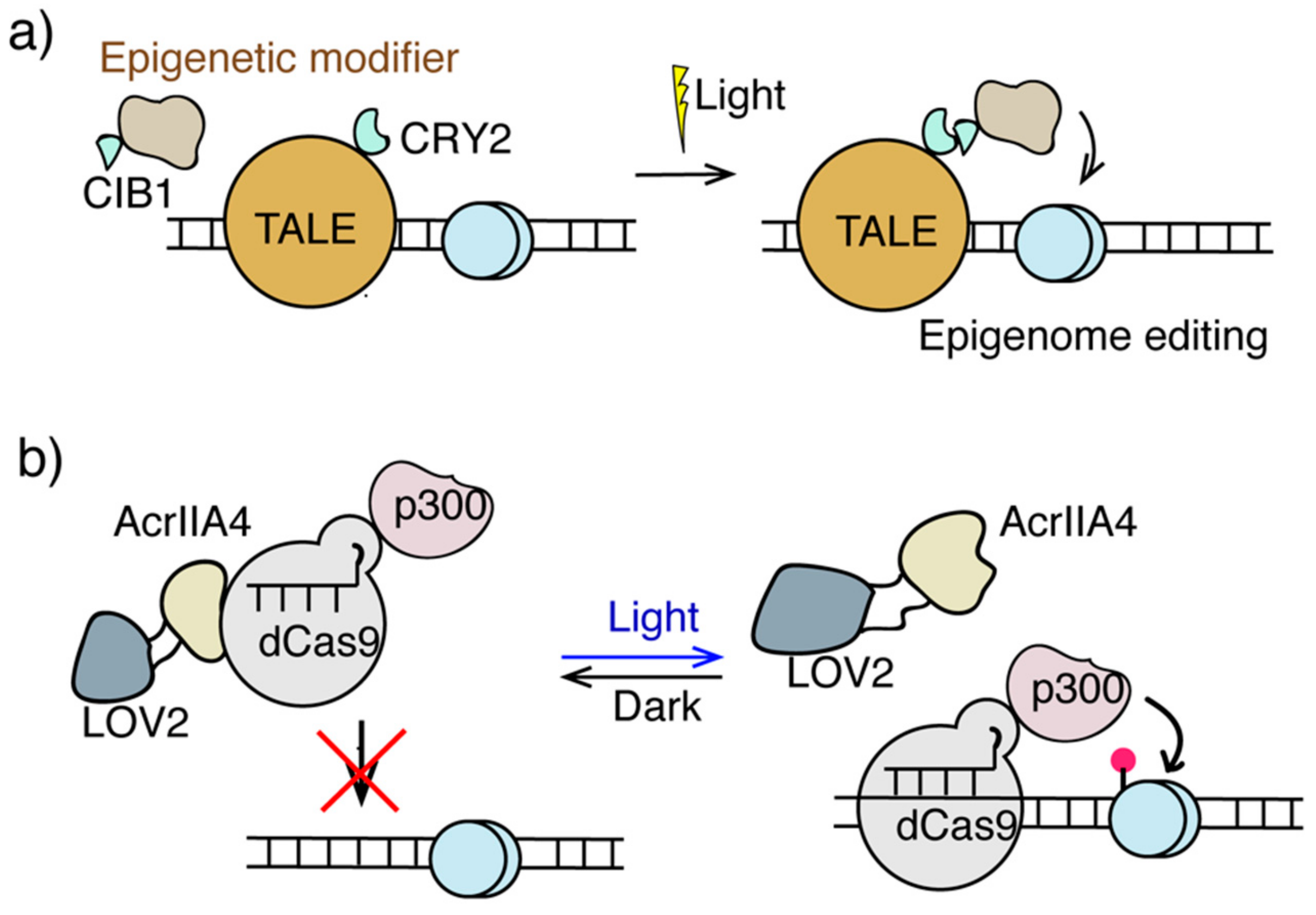
3.3. Light Inducible Epigenome Editing
3.4. Inducible Regulation of Higher-Order Epigenome Organization
4. Other Inducible Approaches Applicable to Epigenome Editing
5. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Cantone, I.; Fisher, A.G. Epigenetic programming and reprogramming during development. Nat. Struct. Mol. Biol. 2013, 20, 282–289. [Google Scholar] [CrossRef]
- Rinaldi, L.; Benitah, S.A. Epigenetic regulation of adult stem cell function. FEBS J. 2015, 282, 1589–1604. [Google Scholar] [CrossRef] [Green Version]
- Atlasi, Y.; Stunnenberg, H.G. The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet. 2017, 18, 643–658. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-h.; Bressler, J.; Beaudet, A.L. Epigenetics and human disease. Ann. Rev. Genom. Hum. Genet. 2004, 5, 479–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.G.; Allis, C.D.; Chi, P. Chromatin remodeling and cancer, part II: ATP-dependent chromatin remodeling. Trends Mol. Med. 2007, 13, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.G.; Allis, C.D.; Chi, P. Chromatin remodeling and cancer, part I: Covalent histone modifications. Trends Mol. Med. 2007, 13, 363–372. [Google Scholar] [CrossRef]
- Tsankova, N.; Renthal, W.; Kumar, A.; Nestler, E.J. Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 2007, 8, 355–367. [Google Scholar] [CrossRef]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications—Miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef] [Green Version]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Hakre, S.; Chavez, L.; Shirakawa, K.; Verdin, E. Epigenetic regulation of HIV latency. Curr. Opin. HIV AIDS 2011, 6, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer Epigenetics: From Mechanism to Therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, M.A.; Kouzarides, T.; Huntly, B.J.P. Targeting Epigenetic Readers in Cancer. N. Engl. J. Med. 2012, 367, 647–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K. The Hope and Hype of CRISPR-Cas9 Genome Editing: A Review. JAMA Cardiol. 2017, 2, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, E.N.; Scaffidi, P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 2017, 3, 372–386. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P. The Key Role of Epigenetics in Human Disease Prevention and Mitigation. N. Engl. J. Med. 2018, 378, 1323–1334. [Google Scholar] [CrossRef]
- Hnisz, D.; Schuijers, J.; Li, C.H.; Young, R.A. Regulation and Dysregulation of Chromosome Structure in Cancer. Ann. Rev. Cancer Biol. 2018, 2, 21–40. [Google Scholar] [CrossRef] [Green Version]
- Woodcock, C.L.; Ghosh, R.P. Chromatin higher-order structure and dynamics. Cold Spring Harb. Perspect. Biol. 2010, 2, a000596. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Erdel, F.; Rippe, K. Chromatin remodelling in mammalian cells by ISWI-type complexes—Where, when and why? FEBS J. 2011, 278, 3608–3618. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Tsuchiya, S.; Meltzer, S.J.; Shimizu, K. MicroRNAs and epigenetics. FEBS J. 2011, 278, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Rothbart, S.B.; Strahl, B.D. Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta 2014, 1839, 627–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Rowley, M.J.; Corces, V.G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018, 19, 789–800. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Kelsey, G.; Stegle, O.; Reik, W. Single-cell epigenomics: Recording the past and predicting the future. Science 2017, 358, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Cermakova, K.; Hodges, H.C. Next-Generation Drugs and Probes for Chromatin Biology: From Targeted Protein Degradation to Phase Separation. Molecules 2018, 23, 1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuvier, O.; Fierz, B. Dynamic chromatin technologies: From individual molecules to epigenomic regulation in cells. Nat. Rev. Genet. 2017, 18, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Coussens, N.P.; Sittampalam, G.S.; Guha, R.; Brimacombe, K.; Grossman, A.; Chung, T.D.Y.; Weidner, J.R.; Riss, T.; Trask, O.J.; Auld, D.; et al. Assay Guidance Manual: Quantitative Biology and Pharmacology in Preclinical Drug Discovery. Clin. Transl. Sci. 2018, 11, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, S.H.; Doudna, J.A. Expanding the Biologist’s Toolkit with CRISPR-Cas9. Mol. Cell 2015, 58, 568–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rots, M.G.; Jeltsch, A. Editing the Epigenome: Overview, Open Questions, and Directions of Future Development. In Epigenome Editing: Methods and Protocols; Jeltsch, A., Rots, M.G., Eds.; Springer: New York, NY, USA, 2018; pp. 3–18. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef] [PubMed]
- Kungulovski, G.; Jeltsch, A. Epigenome Editing: State of the Art, Concepts, and Perspectives. Trends Genet. 2016, 32, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Pulecio, J.; Verma, N.; Mejía-Ramírez, E.; Huangfu, D.; Raya, A. CRISPR/Cas9-Based Engineering of the Epigenome. Cell Stem Cell 2017, 21, 431–447. [Google Scholar] [CrossRef] [Green Version]
- Holtzman, L.; Gersbach, C.A. Editing the Epigenome: Reshaping the Genomic Landscape. Ann. Rev. Genom. Hum. Genet. 2018, 19, 43–71. [Google Scholar] [CrossRef]
- Knauert, M.P.; Glazer, P.M. Triplex forming oligonucleotides: Sequence-specific tools for gene targeting. Hum. Mol. Genet. 2001, 10, 2243–2251. [Google Scholar] [CrossRef] [Green Version]
- Waryah, C.B.; Moses, C.; Arooj, M.; Blancafort, P. Zinc Fingers, TALEs, and CRISPR Systems: A Comparison of Tools for Epigenome Editing. Methods Mol. Biol. 2018, 1767, 19–63. [Google Scholar] [CrossRef]
- Nomura, W. Development of Toolboxes for Precision Genome/Epigenome Editing and Imaging of Epigenetics. Chem. Rec. 2018, 18, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qi, L.S. A CRISPR–dCas Toolbox for Genetic Engineering and Synthetic Biology. J. Mol. Biol. 2019, 431, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Porteus, M.H.; Carroll, D. Gene targeting using zinc finger nucleases. Nat. Biotechnol. 2005, 23, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Bogdanove, A.J.; Voytas, D.F. TAL Effectors: Customizable Proteins for DNA Targeting. Science 2011, 333, 1843–1846. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Dancy, B.M.; Cole, P.A. Protein Lysine Acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Shrimp, J.H.; Grose, C.; Widmeyer, S.R.T.; Thorpe, A.L.; Jadhav, A.; Meier, J.L. Chemical Control of a CRISPR-Cas9 Acetyltransferase. ACS Chem. Biol. 2018, 13, 455–460. [Google Scholar] [CrossRef]
- O’Geen, H.; Ren, C.; Nicolet, C.M.; Perez, A.A.; Halmai, J.; Le, V.M.; Mackay, J.P.; Farnham, P.J.; Segal, D.J. dCas9-based epigenome editing suggests acquisition of histone methylation is not sufficient for target gene repression. Nucleic Acids Res. 2017, 45, 9901–9916. [Google Scholar] [CrossRef] [Green Version]
- O’Geen, H.; Bates, S.L.; Carter, S.S.; Nisson, K.A.; Halmai, J.; Fink, K.D.; Rhie, S.K.; Farnham, P.J.; Segal, D.J. Ezh2-dCas9 and KRAB-dCas9 enable engineering of epigenetic memory in a context-dependent manner. Epigenet. Chromatin 2019, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Cano-Rodriguez, D.; Gjaltema, R.A.; Jilderda, L.J.; Jellema, P.; Dokter-Fokkens, J.; Ruiters, M.H.; Rots, M.G. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat. Commun. 2016, 7, 12284. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.Y.; Zhao, Y.T.; Lamonica, J.M.; Zhou, Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun. 2017, 8, 15315. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Angstman, J.F.; Richardson, M.E.; Linder, S.J.; Cascio, V.M.; Tsai, S.Q.; Ho, Q.H.; Sander, J.D.; Reyon, D.; Bernstein, B.E.; et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat. Biotechnol. 2013, 31, 1137–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, D.L.; Le Lay, J.E.; Ruano, E.G.; Kaestner, K.H. TALE-mediated epigenetic suppression of CDKN2A increases replication in human fibroblasts. J. Clin. Investig. 2015, 125, 1998–2006. [Google Scholar] [CrossRef]
- Amabile, A.; Migliara, A.; Capasso, P.; Biffi, M.; Cittaro, D.; Naldini, L.; Lombardo, A. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016, 167, 219–232. [Google Scholar] [CrossRef] [Green Version]
- McDonald, J.I.; Celik, H.; Rois, L.E.; Fishberger, G.; Fowler, T.; Rees, R.; Kramer, A.; Martens, A.; Edwards, J.R.; Challen, G.A. Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biol. Open 2016, 5, 866. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Stepper, P.; Kungulovski, G.; Jurkowska, R.Z.; Chandra, T.; Krueger, F.; Reinhardt, R.; Reik, W.; Jeltsch, A.; Jurkowski, T.P. Efficient targeted DNA methylation with chimeric dCas9–Dnmt3a–Dnmt3L methyltransferase. Nucleic Acids Res. 2016, 45, 1703–1713. [Google Scholar] [CrossRef]
- Vojta, A.; Dobrinic, P.; Tadic, V.; Bockor, L.; Korac, P.; Julg, B.; Klasic, M.; Zoldos, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, S.R.; Cui, Y.; Lubecka, K.; Stefanska, B.; Irudayaraj, J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 2016, 7, 46545–46556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, T.; Meister, G.E.; Workman, R.E.; Kato, N.C.; Spellberg, M.J.; Turker, F.; Timp, W.; Ostermeier, M.; Novina, C.D. Targeted DNA methylation in human cells using engineered dCas9-methyltransferases. Sci. Rep. 2017, 7, 6732. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, E.M.; Williamson, K.E.; Reyon, D.; Zou, J.Y.; Ram, O.; Joung, J.K.; Bernstein, B.E. Locus-specific editing of histone modifications at endogenous enhancers. Nat. Biotechnol. 2013, 31, 1133–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearns, N.A.; Pham, H.; Tabak, B.; Genga, R.M.; Silverstein, N.J.; Garber, M.; Maehr, R. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 2015, 12, 401–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.K.; Hatanaka, F.; Araoka, T.; Reddy, P.; Wu, M.Z.; Sui, Y.; Yamauchi, T.; Sakurai, M.; O’Keefe, D.D.; Nunez-Delicado, E.; et al. In Vivo Target Gene Activation via CRISPR/Cas9-Mediated Trans-epigenetic Modulation. Cell 2017, 171, 1495–1507.e15. [Google Scholar] [CrossRef] [Green Version]
- Gestwicki, J.E.; Marinec, P.S. Chemical control over protein-protein interactions: Beyond inhibitors. Comb. Chem. High Throughput Screen. 2007, 10, 667–675. [Google Scholar] [CrossRef]
- Fegan, A.; White, B.; Carlson, J.C.; Wagner, C.R. Chemically controlled protein assembly: Techniques and applications. Chem. Rev. 2010, 110, 3315–3336. [Google Scholar] [CrossRef]
- Shekhawat, S.S.; Ghosh, I. Split-protein systems: Beyond binary protein–protein interactions. Curr. Opin. Chem. Biol. 2011, 15, 789–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putyrski, M.; Schultz, C. Protein translocation as a tool: The current rapamycin story. FEBS Lett. 2012, 586, 2097–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeRose, R.; Miyamoto, T.; Inoue, T. Manipulating signaling at will: Chemically-inducible dimerization (CID) techniques resolve problems in cell biology. Pflug. Arch. Eur. J. Physiol. 2013, 465, 409–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voß, S.; Klewer, L.; Wu, Y.-W. Chemically induced dimerization: Reversible and spatiotemporal control of protein function in cells. Curr. Opin. Chem. Biol. 2015, 28, 194–201. [Google Scholar] [CrossRef]
- Stanton, B.Z.; Chory, E.J.; Crabtree, G.R. Chemically induced proximity in biology and medicine. Science 2018, 359, eaao5902. [Google Scholar] [CrossRef] [Green Version]
- Klewer, L.; Wu, Y.W. Light-Induced Dimerization Approaches to Control Cellular Processes. Chem. Eur. J. 2019, 25, 12452–12463. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Calderon, A.; Konstantinidis, G.; Hou, J.; Voss, S.; Chen, X.; Li, F.; Banerjee, S.; Hoffmann, J.E.; Theiss, C.; et al. A bioorthogonal small-molecule-switch system for controlling protein function in live cells. Angew. Chem. Int. Ed. 2014, 53, 10049–10055. [Google Scholar] [CrossRef]
- Zhao, W.; Nguyen, H.; Zeng, G.; Gao, D.; Yan, H.; Liang, F.-S. A chemically induced proximity system engineered from the plant auxin signaling pathway. Chem. Sci. 2018, 9, 5822–5827. [Google Scholar] [CrossRef] [Green Version]
- Zeng, G.; Wang, Y.; Bruchez, M.P.; Liang, F.-S. Self-Reporting Chemically Induced Protein Proximity System Based on a Malachite Green Derivative and the L5** Fluorogen Activating Protein. Bioconjugate Chem. 2018, 29, 3010–3015. [Google Scholar] [CrossRef]
- Hathaway, N.A.; Bell, O.; Hodges, C.; Miller, E.L.; Neel, D.S.; Crabtree, G.R. Dynamics and memory of heterochromatin in living cells. Cell 2012, 149, 1447–1460. [Google Scholar] [CrossRef] [Green Version]
- Hodges, C.; Kirkland, J.G.; Crabtree, G.R. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadoch, C.; Williams, R.T.; Calarco, J.P.; Miller, E.L.; Weber, C.M.; Braun, S.M.; Pulice, J.L.; Chory, E.J.; Crabtree, G.R. Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat. Genet. 2017, 49, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.L.; Hargreaves, D.C.; Kadoch, C.; Chang, C.Y.; Calarco, J.P.; Hodges, C.; Buenrostro, J.D.; Cui, K.; Greenleaf, W.J.; Zhao, K.; et al. TOP2 synergizes with BAF chromatin remodeling for both resolution and formation of facultative heterochromatin. Nat. Struct. Mol. Biol. 2017, 24, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Stanton, B.Z.; Hodges, C.; Calarco, J.P.; Braun, S.M.; Ku, W.L.; Kadoch, C.; Zhao, K.; Crabtree, G.R. Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat. Genet. 2017, 49, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Gao, D.; Zhang, R.; Zeng, G.; Yan, H.; Lim, E.; Liang, F.-S. Chemically Controlled Epigenome Editing through an Inducible dCas9 System. J. Am. Chem. Soc. 2017, 139, 11337–11340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, E.I.; Stafford, J.M.; Reinberg, D. Epigenetic inheritance: Histone bookmarks across generations. Trends Cell Biol. 2014, 24, 664–674. [Google Scholar] [CrossRef] [Green Version]
- D’Urso, A.; Brickner, J.H. Mechanisms of epigenetic memory. Trends Genet. 2014, 30, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Braun, S.M.G.; Kirkland, J.G.; Chory, E.J.; Husmann, D.; Calarco, J.P.; Crabtree, G.R. Rapid and reversible epigenome editing by endogenous chromatin regulators. Nat. Commun. 2017, 8, 560. [Google Scholar] [CrossRef]
- Gao, Y.; Xiong, X.; Wong, S.; Charles, E.J.; Lim, W.A.; Qi, L.S. Complex transcriptional modulation with orthogonal and inducible dCas9 regulators. Nat. Methods 2016, 13, 1043–1049. [Google Scholar] [CrossRef]
- Bao, Z.; Jain, S.; Jaroenpuntaruk, V.; Zhao, H. Orthogonal Genetic Regulation in Human Cells Using Chemically Induced CRISPR/Cas9 Activators. ACS Synth. Biol. 2017, 6, 686–693. [Google Scholar] [CrossRef]
- Butler, K.V.; Chiarella, A.M.; Jin, J.; Hathaway, N.A. Targeted Gene Repression Using Novel Bifunctional Molecules to Harness Endogenous Histone Deacetylation Activity. ACS Synth. Biol. 2018, 7, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Chiarella, A.M.; Butler, K.V.; Gryder, B.E.; Lu, D.; Wang, T.A.; Yu, X.; Pomella, S.; Khan, J.; Jin, J.; Hathaway, N.A. Dose-dependent activation of gene expression is achieved using CRISPR and small molecules that recruit endogenous chromatin machinery. Nat. Biotechnol. 2019, 38, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.; Hsu, P.D.; Heidenreich, M.; Cong, L.; Platt, R.J.; Scott, D.A.; Church, G.M.; Zhang, F. Optical control of mammalian endogenous transcription and epigenetic states. Nature 2013, 500, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Polstein, L.R.; Gersbach, C.A. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat. Chem. Biol. 2015, 11, 198–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubeck, F.; Hoffmann, M.D.; Harteveld, Z.; Aschenbrenner, S.; Bietz, A.; Waldhauer, M.C.; Borner, K.; Fakhiri, J.; Schmelas, C.; Dietz, L.; et al. Engineered anti-CRISPR proteins for optogenetic control of CRISPR-Cas9. Nat. Methods 2018, 15, 924–927. [Google Scholar] [CrossRef]
- Ellis-Davies, G.C.R. Caged compounds: Photorelease technology for control of cellular chemistry and physiology. Nat. Methods 2007, 4, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Sadovski, O.; Jaikaran, A.S.; Samanta, S.; Fabian, M.R.; Dowling, R.J.; Sonenberg, N.; Woolley, G.A. A collection of caged compounds for probing roles of local translation in neurobiology. Bioorg. Med. Chem. 2010, 18, 7746–7752. [Google Scholar] [CrossRef] [Green Version]
- Umeda, N.; Ueno, T.; Pohlmeyer, C.; Nagano, T.; Inoue, T. A Photocleavable Rapamycin Conjugate for Spatiotemporal Control of Small GTPase Activity. J. Am. Chem. Soc. 2011, 133, 12–14. [Google Scholar] [CrossRef] [Green Version]
- Karginov, A.V.; Zou, Y.; Shirvanyants, D.; Kota, P.; Dokholyan, N.V.; Young, D.D.; Hahn, K.M.; Deiters, A. Light Regulation of Protein Dimerization and Kinase Activity in Living Cells Using Photocaged Rapamycin and Engineered FKBP. J. Am. Chem. Soc. 2011, 133, 420–423. [Google Scholar] [CrossRef] [Green Version]
- Ballister, E.R.; Aonbangkhen, C.; Mayo, A.M.; Lampson, M.A.; Chenoweth, D.M. Localized light-induced protein dimerization in living cells using a photocaged dimerizer. Nat. Commun. 2014, 5, 5475. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.W.; Guo, Z.-F.; Liang, F.-S. Light Control of Cellular Processes by Using Photocaged Abscisic Acid. ChemBioChem 2015, 16, 254–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schelkle, K.M.; Griesbaum, T.; Ollech, D.; Becht, S.; Buckup, T.; Hamburger, M.; Wombacher, R. Light-Induced Protein Dimerization by One- and Two-Photon Activation of Gibberellic Acid Derivatives in Living Cells. Angew. Chem. Int. Ed. 2015, 54, 2825–2829. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; Zou, Y.; Shirvanyants, D.; Zhang, J.; Samanta, S.; Mantravadi, P.K.; Dokholyan, N.V.; Deiters, A. Light-cleavable rapamycin dimer as an optical trigger for protein dimerization. Chem. Commun. 2015, 51, 5702–5705. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Zhang, R.; Xuan, W.; Wang, W.; Liang, F.-S. Constructing de Novo H2O2 Signaling via Induced Protein Proximity. ACS Chem. Biol. 2015, 10, 1404–1410. [Google Scholar] [CrossRef] [Green Version]
- Zeng, G.; Li, H.; Wei, Y.; Xuan, W.; Zhang, R.; Breden, L.E.; Wang, W.; Liang, F.-S. Engineering Iron Responses in Mammalian Cells by Signal-Induced Protein Proximity. ACS Synth. Biol. 2017, 6, 921–927. [Google Scholar] [CrossRef]
- Chen, X.; Venkatachalapathy, M.; Kamps, D.; Weigel, S.; Kumar, R.; Orlich, M.; Garrecht, R.; Hirtz, M.; Niemeyer, C.M.; Wu, Y.-W.; et al. “Molecular Activity Painting”: Switch-like, Light-Controlled Perturbations inside Living Cells. Angew. Chem. Int. Ed. 2017, 56, 5916–5920. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Aonbangkhen, C.; Tarasovetc, E.V.; Ballister, E.R.; Chenoweth, D.M.; Lampson, M.A. Optogenetic control of kinetochore function. Nat. Chem. Biol. 2017, 13, 1096–1101. [Google Scholar] [CrossRef]
- Kowalik, L.; Chen, J.K. Illuminating developmental biology through photochemistry. Nat. Chem. Biol. 2017, 13, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Risca, V.I.; Greenleaf, W.J. Unraveling the 3D genome: Genomics tools for multiscale exploration. Trends Genet. 2015, 31, 357–372. [Google Scholar] [CrossRef] [Green Version]
- Dekker, J.; Heard, E. Structural and functional diversity of Topologically Associating Domains. FEBS Lett. 2015, 589, 2877–2884. [Google Scholar] [CrossRef] [Green Version]
- Schoenfelder, S.; Fraser, P. Long-range enhancer–promoter contacts in gene expression control. Nat. Rev. Genet. 2019, 20, 437–455. [Google Scholar] [CrossRef]
- Lopes Novo, C.; Rugg-Gunn, P. Crosstalk between pluripotency factors and higher-order chromatin organization. Nucleus 2016, 7, 447–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, S.L.; Mariano, N.C.; Bermudez, A.; Arruda, N.L.; Wu, F.; Luo, Y.; Shankar, G.; Jia, L.; Chen, H.; Hu, J.-F.; et al. Manipulation of nuclear architecture through CRISPR-mediated chromosomal looping. Nat. Commun. 2017, 8, 15993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Xu, X.; Nguyen, C.M.; Liu, Y.; Gao, Y.; Lin, X.; Daley, T.; Kipniss, N.H.; La Russa, M.; Qi, L.S. CRISPR-Mediated Programmable 3D Genome Positioning and Nuclear Organization. Cell 2018, 175, 1405–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Cheong, S.A.; Lee, J.G.; Lee, S.W.; Lee, M.S.; Baek, I.J.; Sung, Y.H. Generation of knockout mice by Cpf1-mediated gene targeting. Nat. Biotechnol. 2016, 34, 808–810. [Google Scholar] [CrossRef]
- Zetsche, B.; Heidenreich, M.; Mohanraju, P.; Fedorova, I.; Kneppers, J.; DeGennaro, E.M.; Winblad, N.; Choudhury, S.R.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol. 2017, 35, 31–34. [Google Scholar] [CrossRef]
- Nihongaki, Y.; Otabe, T.; Ueda, Y.; Sato, M. A split CRISPR-Cpf1 platform for inducible genome editing and gene activation. Nat. Chem. Biol. 2019, 15, 882–888. [Google Scholar] [CrossRef]
- Kawano, F.; Suzuki, H.; Furuya, A.; Sato, M. Engineered pairs of distinct photoswitches for optogenetic control of cellular proteins. Nat. Commun. 2015, 6, 6256. [Google Scholar] [CrossRef]
- Kundert, K.; Lucas, J.E.; Watters, K.E.; Fellmann, C.; Ng, A.H.; Heineike, B.M.; Fitzsimmons, C.M.; Oakes, B.L.; Qu, J.; Prasad, N.; et al. Controlling CRISPR-Cas9 with ligand-activated and ligand-deactivated sgRNAs. Nat. Commun. 2019, 10, 2127. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, M.; Bjorklund, T.; Lundberg, C.; Kirik, D.; Wandless, T.J. A general chemical method to regulate protein stability in the mammalian central nervous system. Chem. Biol. 2010, 17, 981–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, Y.; Imoto, H.; Chen, L.-C.; Wandless, T.J. Destabilizing domains derived from the human estrogen receptor. J. Am. Chem. Soc. 2012, 134, 3942–3945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maji, B.; Moore, C.L.; Zetsche, B.; Volz, S.E.; Zhang, F.; Shoulders, M.D.; Choudhary, A. Multidimensional chemical control of CRISPR-Cas9. Nat. Chem. Biol. 2017, 13, 9–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balboa, D.; Weltner, J.; Eurola, S.; Trokovic, R.; Wartiovaara, K.; Otonkoski, T. Conditionally Stabilized dCas9 Activator for Controlling Gene Expression in Human Cell Reprogramming and Differentiation. Stem Cell Rep. 2015, 5, 448–459. [Google Scholar] [CrossRef] [Green Version]
- Tague, E.P.; Dotson, H.L.; Tunney, S.N.; Sloas, D.C.; Ngo, J.T. Chemogenetic control of gene expression and cell signaling with antiviral drugs. Nat. Methods 2018, 15, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Pei, W.-D.; Zhang, Y.; Yin, T.-L.; Yu, Y. Epigenome editing by CRISPR/Cas9 in clinical settings: Possibilities and challenges. Brief. Funct. Genom. 2019. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, W.; Wang, Y.; Liang, F.-S. Chemical and Light Inducible Epigenome Editing. Int. J. Mol. Sci. 2020, 21, 998. https://doi.org/10.3390/ijms21030998
Zhao W, Wang Y, Liang F-S. Chemical and Light Inducible Epigenome Editing. International Journal of Molecular Sciences. 2020; 21(3):998. https://doi.org/10.3390/ijms21030998
Chicago/Turabian StyleZhao, Weiye, Yufan Wang, and Fu-Sen Liang. 2020. "Chemical and Light Inducible Epigenome Editing" International Journal of Molecular Sciences 21, no. 3: 998. https://doi.org/10.3390/ijms21030998
APA StyleZhao, W., Wang, Y., & Liang, F. -S. (2020). Chemical and Light Inducible Epigenome Editing. International Journal of Molecular Sciences, 21(3), 998. https://doi.org/10.3390/ijms21030998