In Steatotic Cells, ATP-Citrate Lyase mRNA Is Efficiently Translated through a Cap-Independent Mechanism, Contributing to the Stimulation of De Novo Lipogenesis

 , ,
, , 
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture
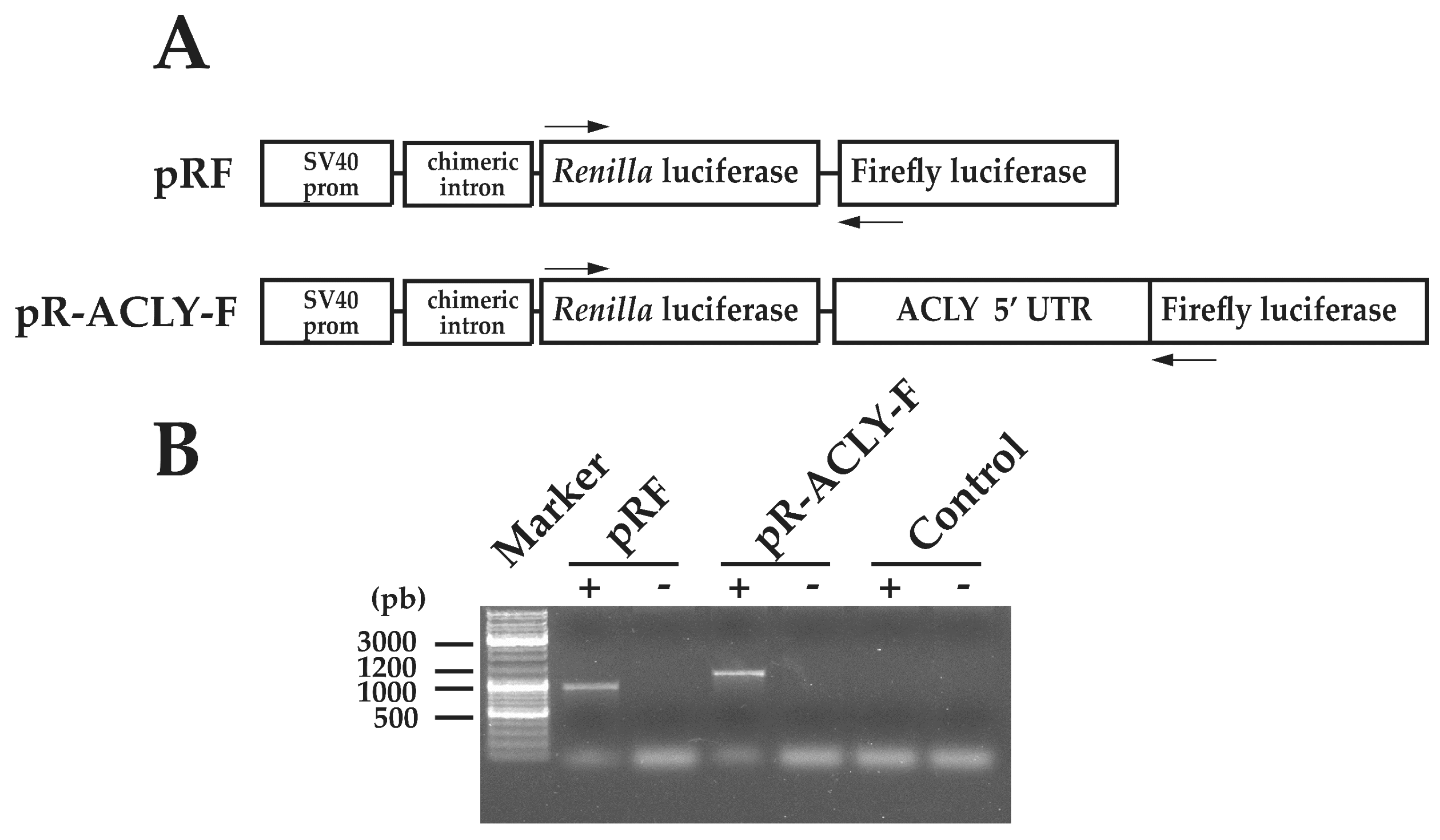
2.2. Synthesis of Mono- and Di-Cistronic Constructs
2.3. Transient Transfection Assay
2.4. Isolation of RNA and qRT-PCR Analysis
2.5. Immunoelectrophoretic Analysis
2.6. ACLY Half-Life Analysis
2.7. Statistical Analysis
3. Results
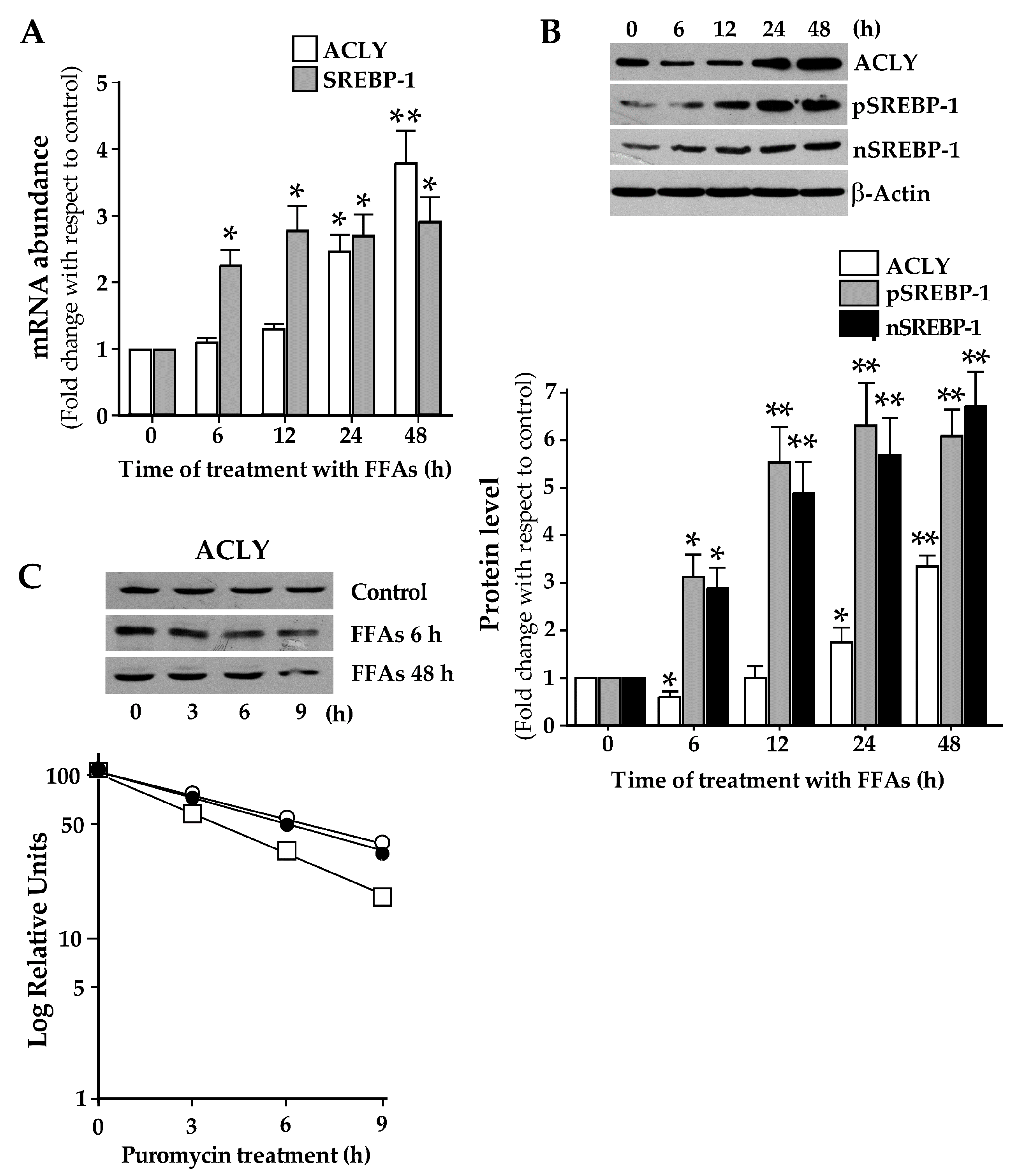
3.1. ACLY Expression Changes in HepG2 Cells upon Lipid Accumulation.
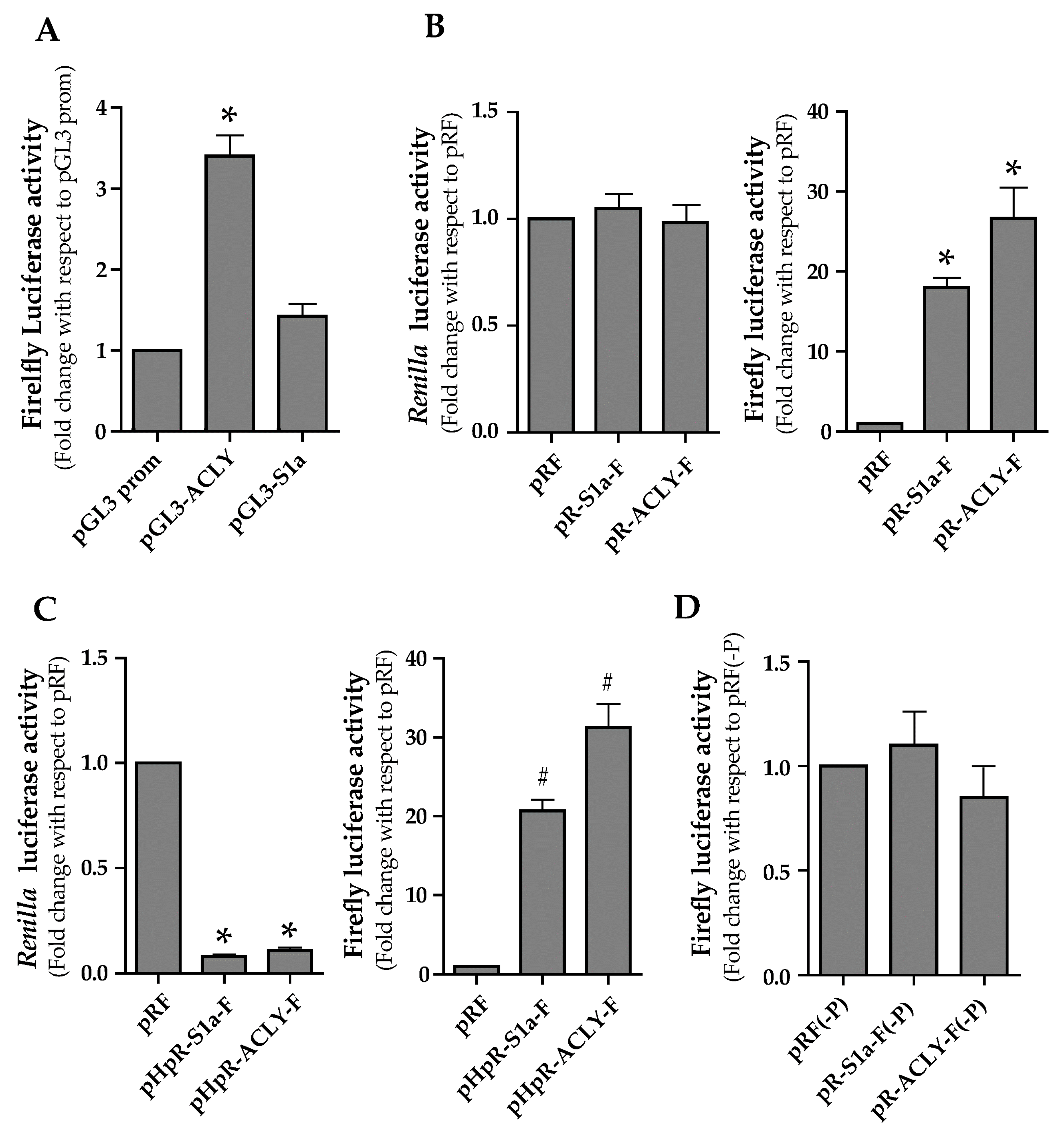
3.2. Functional Characterization of 5′ Untraslated Region of ACLY mRNA
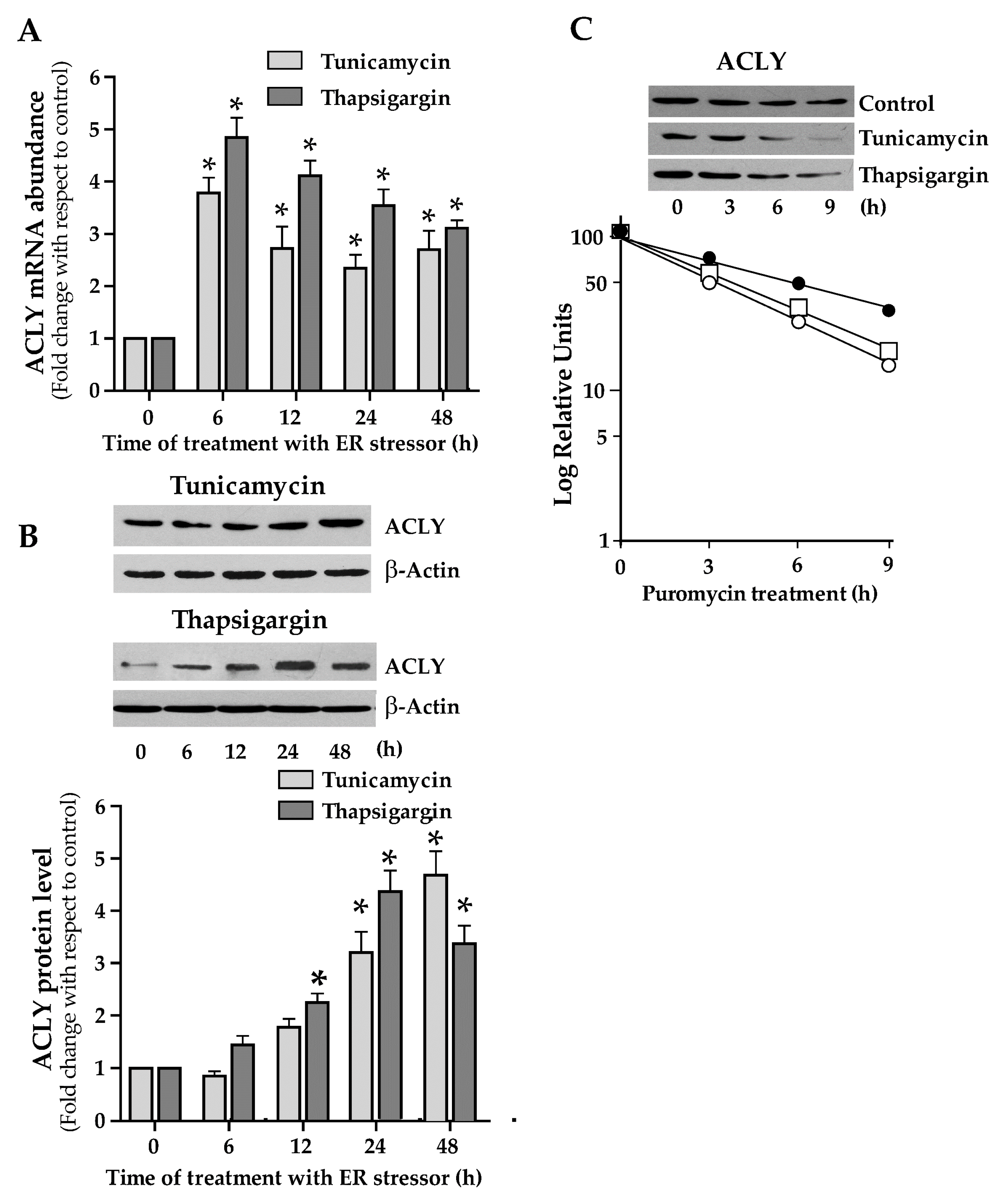
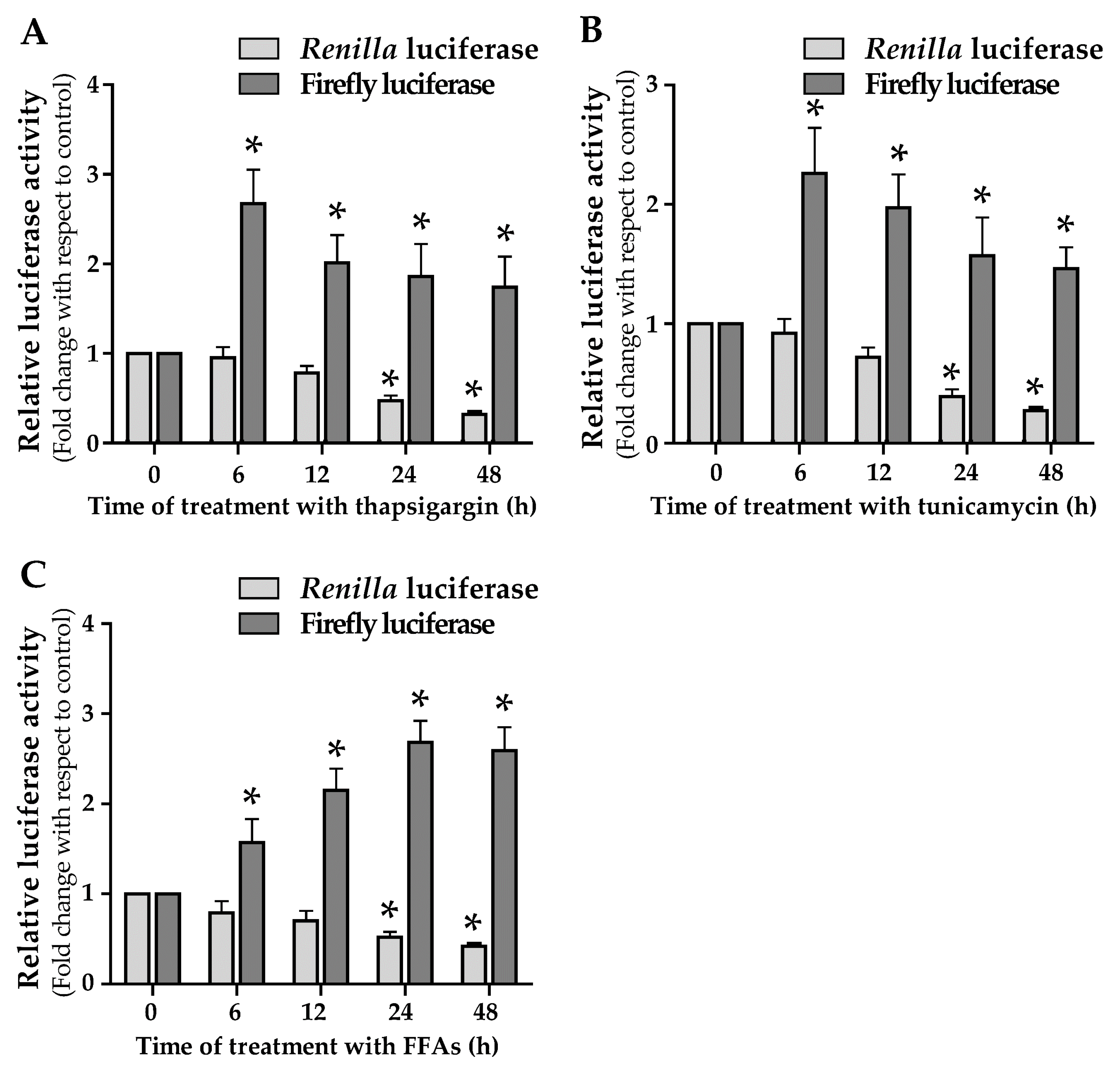
3.3. Endoplasmic Reticulum-Stress Promotes the ACLY Expression
3.4. ER Stress and Lipid Droplets Accumulation Drive ACLY IRES Activity
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Association for the study of liver diseases, American college of gastroenterology, and the American gastroenterological association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Linfoot, P.; Dare, D.; Aghajanian, K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am. J. Clin. Nutr. 2003, 77, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Otero, Y.F.; Stafford, J.M.; McGuinness, O.P. Pathway-selective insulin resistance and metabolic disease: The importance of nutrient flux. J. Biol. Chem. 2014, 289, 20462–20469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, R. ER stress and hepatic lipid metabolism. Front. Genet. 2014, 5, 112. [Google Scholar] [CrossRef]
- Siculella, L.; Tocci, R.; Rochira, A.; Testini, M.; Gnoni, A.; Damiano, F. Lipid accumulation stimulates the cap-independent translation of SREBP-1a mRNA by promoting hnRNP A1 binding to its 5’-UTR in a cellular model of hepatic steatosis. Biochim. Biophys. Acta 2016, 1861, 471–481. [Google Scholar] [CrossRef]
- Koteish, A.; Diehl, A.M. Animal models of steatosis. Semin. Liver Dis. 2001, 21, 89–104. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Donato, M.T.; Martínez-Romero, A.; Jiménez, N.; Castell, J.V.; O’Connor, J.E. A human hepatocellular in vitro model to investigate steatosis. Chem. Biol. Interact. 2007, 165, 106–116. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Pinkosky, S.L.; Groot, P.H.E.; Lalwani, N.D.; Steinberg, G.R. Targeting ATP-Citrate Lyase in Hyperlipidemia and Metabolic Disorders. Trends Mol. Med. 2017, 23, 1047–1063. [Google Scholar] [CrossRef] [PubMed]
- Giudetti, A.M.; Stanca, E.; Siculella, L.; Gnoni, G.V.; Damiano, F. Nutritional and Hormonal Regulation of Citrate and Carnitine/Acylcarnitine Transporters: Two Mitochondrial Carriers Involved in Fatty Acid Metabolism. Int. J. Mol. Sci. 2016, 17, 817. [Google Scholar] [CrossRef] [PubMed]
- DeBose-Boyd, R.A. Feedback regulation of cholesterol synthesis: Sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 2008, 18, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-Citrate Lyase: A Key Player in Cancer Metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Tao, R.; Gao, X.; Li, T.; Zhou, X.; Guan, K.L.; Xiong, Y.; Lei, Q.Y. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell 2013, 51, 506–518. [Google Scholar] [CrossRef] [Green Version]
- Sato, R.; Okamoto, A.; Inoue, J.; Miyamoto, W.; Sakai, Y.; Emoto, N.; Shimano, H.; Maeda, M. Transcriptional regulation of the ATP-citrate lyase gene by sterol regulatory element-binding proteins. J. Biol. Chem. 2000, 275, 12497–12502. [Google Scholar] [CrossRef] [Green Version]
- Damiano, F.; Testini, M.; Tocci, R.; Gnoni, G.V.; Siculella, L. Translational control of human acetyl-CoA carboxylase 1 mRNA is mediated by an internal ribosome entry site in response to ER stress, serum deprivation or hypoxia mimetic CoCl2. Biochim. Biophys. Acta 2018, 1863, 388–398. [Google Scholar] [CrossRef]
- Damiano, F.; Alemanno, S.; Gnoni, G.V.; Siculella, L. Translational control of the sterol-regulatory transcription factor SREBP-1 mRNA in response to serum starvation or ER stress is mediated by an internal ribosome entry site. Biochem. J. 2010, 429, 603–612. [Google Scholar] [CrossRef] [Green Version]
- Damiano, F.; Gnoni, G.V.; Siculella, L. Functional analysis of rat liver citrate carrier promoter: Differential responsiveness to polyunsaturated fatty acids. Biochem. J. 2009, 417, 561–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochira, A.; Damiano, F.; Marsigliante, S.; Gnoni, G.V.; Siculella, L. 3,5-Diiodo-l-thyronine induces SREBP-1 proteolytic cleavage block and apoptosis in human hepatoma (Hepg2) cells. Biochim. Biophys. Acta 2013, 1831, 1679–1689. [Google Scholar] [CrossRef]
- Grasselli, E.; Voci, A.; Canesi, L.; Salis, A.; Damonte, G.; Compalati, A.D.; Goglia, F.; Gallo, G.; Vergani, L. 3,5-Diiodo-L-thyronine modifies the lipid droplet composition in a model of hepatosteatosis. Cell. Physiol. Biochem. 2014, 33, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Stoneley, M.; Paulin, F.E.M.; Le Quesne, J.P.C.; Chappell, S.A.; Willis, A.E. c-Myc 5 untranslated region contains an internal ribosome entry segment. Oncogene 1998, 16, 423–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacerda, R.; Menezes, J.; Candeias, M.M. Alternative Mechanisms of mRNA Translation Initiation in Cellular Stress Response and Cancer. Adv. Exp. Med. Biol. 2019, 1157, 117–132. [Google Scholar]
- Lewis, G.F.; Carpentier, A.; Adeli, K.; Giacca, A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr. Rev. 2002, 23, 201–229. [Google Scholar] [CrossRef]
- Savage, D.B.; Choi, C.S.; Samuel, V.T.; Liu, Z.X.; Zhang, D.; Wang, A.; Zhang, X.M.; Cline, G.W.; Yu, X.X.; Geisler, J.G.; et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J. Clin. Investig. 2006, 116, 817–824. [Google Scholar] [CrossRef] [Green Version]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Li, S.; Brown, M.S.; Goldstein, J.L. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3441–3446. [Google Scholar] [CrossRef] [Green Version]
- Sachithanandan, N.; Fam, B.C.; Fynch, S.; Dzamko, N.; Watt, M.J.; Wormald, S.; Honeyman, J.; Galic, S.; Proietto, J.; Andrikopoulos, S.; et al. Liver-specific suppressor of cytokine signaling-3 deletion in mice enhances hepatic insulin sensitivity and lipogenesis resulting in fatty liver and obesity. Hepatology 2010, 52, 1632–1642. [Google Scholar] [CrossRef]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [Green Version]
- Gruben, N.; Shiri-Sverdlov, R.; Koonen, D.P.; Hofker, M.H. Nonalcoholic fatty liver disease: A main driver of insulin resistance or a dangerous liaison? Biochim. Biophys. Acta 2014, 1842, 2329–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Jiang, L.; Wang, J.; Li, S.; Yu, Y.; You, J.; Zeng, R.; Gao, X.; Rui, L.; Li, W.; et al. Abrogation of hepatic ATP-citrate lyase protects against fatty liver and ameliorates hyperglycemia in leptin receptor deficient mice. Hepatology 2009, 49, 1166–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahrens, M.; Ammerpohl, O.; von Schönfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrannini, E.; Barrett, E.J.; Bevilacqua, S.; Fronzo, R.A. Effect of fatty acids on glucose production and utilization in man. J. Clin. Investig. 1983, 72, 1737–1747. [Google Scholar] [CrossRef] [Green Version]
- Godet, A.C.; David, F.; Hantelys, F.; Tatin, F.; Lacazette, E.; Garmy-Susini, B.; Prats, A.C. IRES Trans-Acting Factors, Key Actors of the Stress Response. Int. J. Mol. Sci. 2019, 20, 924. [Google Scholar] [CrossRef] [Green Version]
- Diallo, L.H.; Tatin, F.; David, F.; Godet, A.C.; Zamora, A.; Prats, A.C.; Garmy-Susini, B.; Lacazette, E. How are circRNAs translated by non-canonical initiation mechanisms? Biochimie 2019, 164, 45–52. [Google Scholar] [CrossRef]
- Dara, L.; Ji, C.; Kaplowitz, N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 2011, 53, 1752–1763. [Google Scholar] [CrossRef] [Green Version]
- Basseri, S.; Austin, R.C. Endoplasmic reticulum stress and lipid metabolism: Mechanisms and therapeutic potential. Biochem. Res. Int. 2012, 2012, 841362. [Google Scholar] [CrossRef]
- Damiano, F.; Rochira, A.; Tocci, R.; Alemanno, S.; Gnoni, A.; Siculella, L. HnRNP A1 mediates the activation of the IRES-dependent SREBP-1a mRNA translation in response to endoplasmic reticulum stress. Biochem. J. 2013, 449, 543–553. [Google Scholar] [CrossRef]
- Fiorino, E.; Giudici, M.; Ferrari, A.; Mitro, N.; Caruso, D.; De Fabiani, E.; Crestani, M. The sirtuin class of histone deacetylases: Regulation and roles in lipid metabolism. IUBMB Life 2014, 66, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnocchi, D.; Pedrelli, M.; Hurt-Camejo, E.; Parini, P. Lipids around the Clock: Focus on Circadian Rhythms and Lipid Metabolism. Biology 2015, 4, 104–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pehar, M.; Lehnus, M.; Karst, A.; Puglielli, L. Proteomic assessment shows that many endoplasmic reticulum (ER)-resident proteins are targeted by Nε-lysine acetylation in the lumen of the organelle and predicts broad biological impact. J. Biol. Chem. 2012, 287, 22436–22440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiano, F.; Tocci, R.; Gnoni, G.V.; Siculella, L. Expression of citrate carrier gene is activated by ER stress effectors XBP1 and ATF6α, binding to an UPRE in its promoter. Biochim. Biophys. Acta 2015, 1849, 23–31. [Google Scholar] [CrossRef]
- Shyu, P., Jr.; Wong, X.F.A.; Crasta, K.; Thibault, G. Dropping in on lipid droplets: Insights into cellular stress and cancer. Biosci. Rep. 2018, 38, pii:BSR20180764. [Google Scholar] [CrossRef] [Green Version]
- Chypre, M.; Zaidi, N.; Smans, K. ATP-citrate lyase: A mini-review. Biochem. Biophys. Res. Commun. 2012, 422, 1–4. [Google Scholar] [CrossRef]
- Pinkosky, S.L.; Newton, R.S.; Day, E.A.; Ford, R.J.; Lhotak, S.; Austin, R.C.; Birch, C.M.; Smith, B.K.; Filippov, S.; Groot, P.H.E.; et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat. Commun. 2016, 7, 13457. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Oligonucleotide Sequence |
|---|---|
| ACLY-RTFor | 5′-TACATCTGCAAAGTGAAGTGG-3′ |
| ACLY-RTRev | 5′-TTCAGCAAGGTCAGCTTCAG-3′ |
| SREBP-1For | 5′-ACACCATGGGGAAGCACAC-3′ |
| SREBP-1Rev | 5′-CTTCACTCTCAATGCGCC-3′ |
| rRNA18SFor | 5′-GTTGGTTTTCGGAACTGAGGC-3′ |
| rRNA18SRev | 5′-CGGTCGGCATCGTTTATGGT-3′ |
| ACLY For1-pRF/pHpRF | 5′-AAGCTTGAATTCAGCCGATGGGGGCGGGGAAA-3′ |
| ACLY Rev1-pRF/pHpRF | 5′-GAATTCCATGGCTGCAGAGAGACCTGCTC-3′ |
| ACLY For1-pGL3prom | 5′-GAATTCAAGCTTAGCCGATGGGGGCGGGGAAA-3′ |
| ACLY Rev1-pGL3prom | 5′-GAATTCCATGGCTGCAGAGAGACCTGCTC-3′ |
| CSFor | 5′-GGCTTCCAAGGTGTACGACCCCGAG-3′ |
| CSRev | 5′-GGGCCCTTCTTAATGTTCTTAGCAT-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siculella, L.; Giannotti, L.; Testini, M.; Gnoni, G.V.; Damiano, F. In Steatotic Cells, ATP-Citrate Lyase mRNA Is Efficiently Translated through a Cap-Independent Mechanism, Contributing to the Stimulation of De Novo Lipogenesis. Int. J. Mol. Sci. 2020, 21, 1206. https://doi.org/10.3390/ijms21041206
Siculella L, Giannotti L, Testini M, Gnoni GV, Damiano F. In Steatotic Cells, ATP-Citrate Lyase mRNA Is Efficiently Translated through a Cap-Independent Mechanism, Contributing to the Stimulation of De Novo Lipogenesis. International Journal of Molecular Sciences. 2020; 21(4):1206. https://doi.org/10.3390/ijms21041206
Chicago/Turabian StyleSiculella, Luisa, Laura Giannotti, Mariangela Testini, Gabriele V. Gnoni, and Fabrizio Damiano. 2020. "In Steatotic Cells, ATP-Citrate Lyase mRNA Is Efficiently Translated through a Cap-Independent Mechanism, Contributing to the Stimulation of De Novo Lipogenesis" International Journal of Molecular Sciences 21, no. 4: 1206. https://doi.org/10.3390/ijms21041206
APA StyleSiculella, L., Giannotti, L., Testini, M., Gnoni, G. V., & Damiano, F. (2020). In Steatotic Cells, ATP-Citrate Lyase mRNA Is Efficiently Translated through a Cap-Independent Mechanism, Contributing to the Stimulation of De Novo Lipogenesis. International Journal of Molecular Sciences, 21(4), 1206. https://doi.org/10.3390/ijms21041206