RhoA/ROCK Regulates Prion Pathogenesis by Controlling Connexin 43 Activity

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
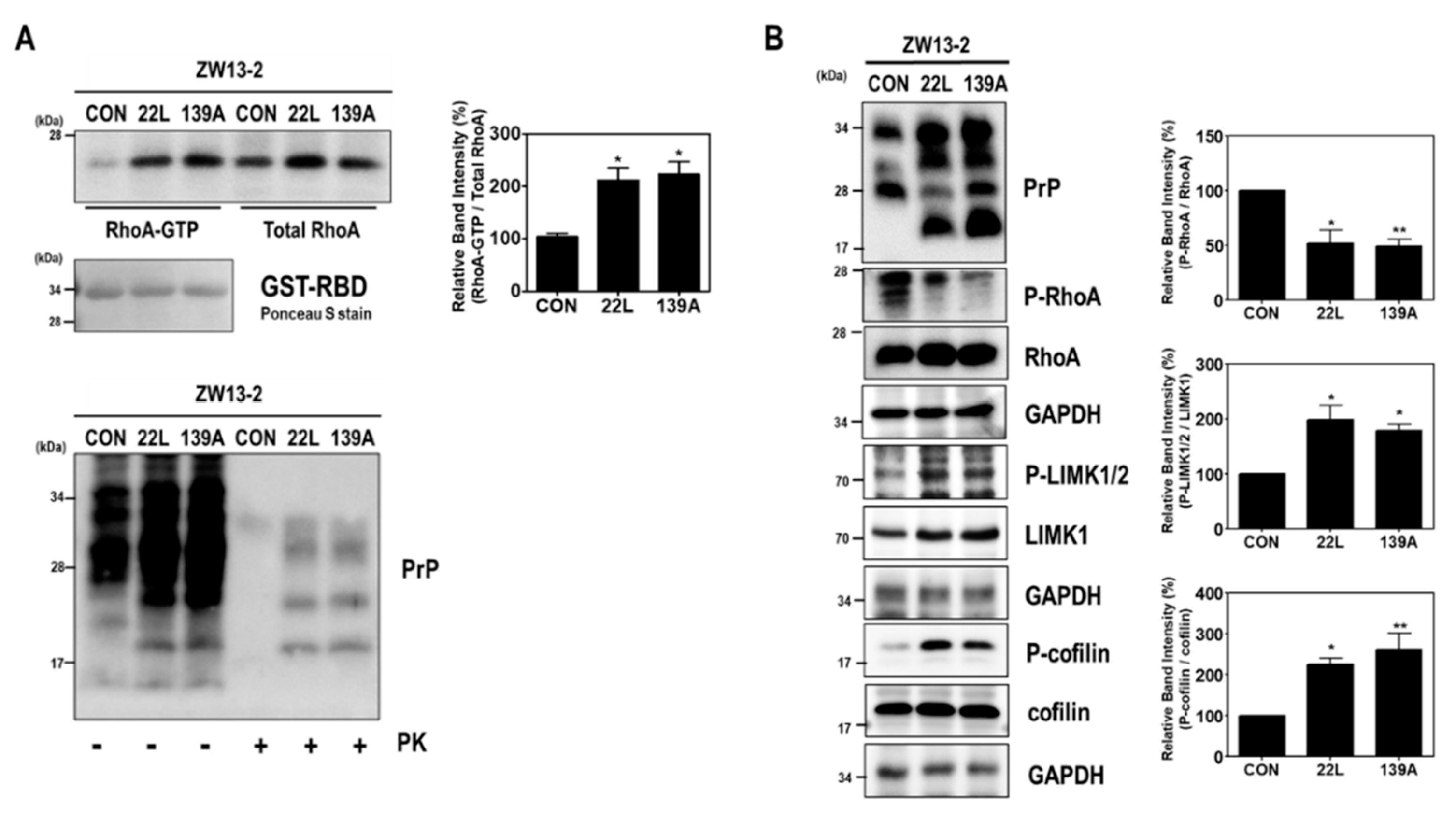
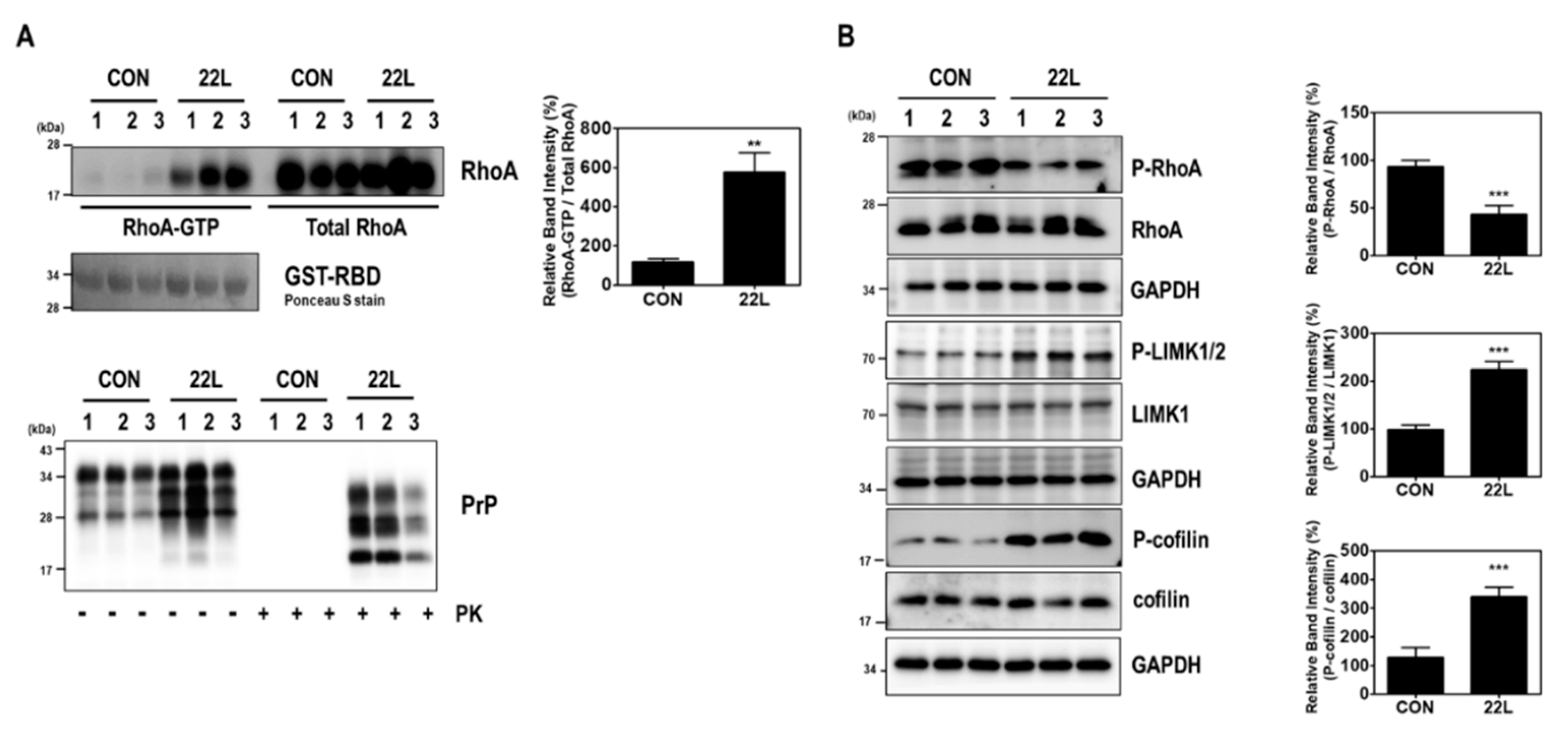
2.1. Scrapie Infection Alters RhoA Activity and the RhoA-ROCK-LIMK-Cofilin Pathway
2.2. Scrapie Infection Controls F-Actin Formation through RhoA Activity
2.3. Scrapie Infection Induces the Activation of RhoA and the RhoA-ROCK-LIMK-Cofilin Pathway by Reducing the Interaction between RhoA and p190RhoGAP
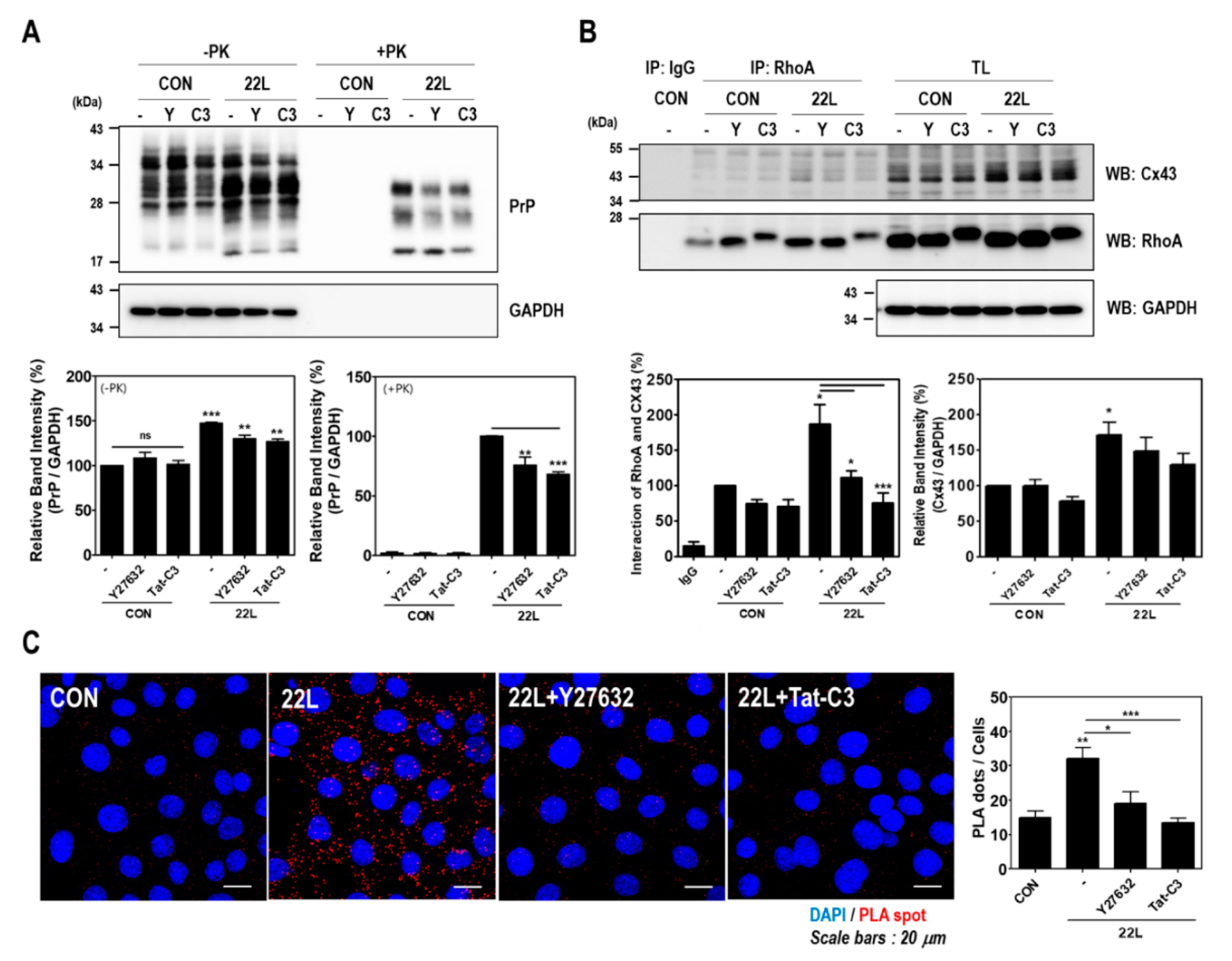
2.4. Scrapie Infection Enhances the Interaction between RhoA and Cx43
2.5. Inhibition of RhoA and ROCK Reduces PrPSc Accumulation and the RhoA-Cx43 Interaction in Scrapie-Infected Hippocampal Neuronal Cells
2.6. Inhibition of RhoA and ROCK Reduces EtBr Uptake and Dye Transfer in Scrapie-Infected Hippocampal Neuronal Cells
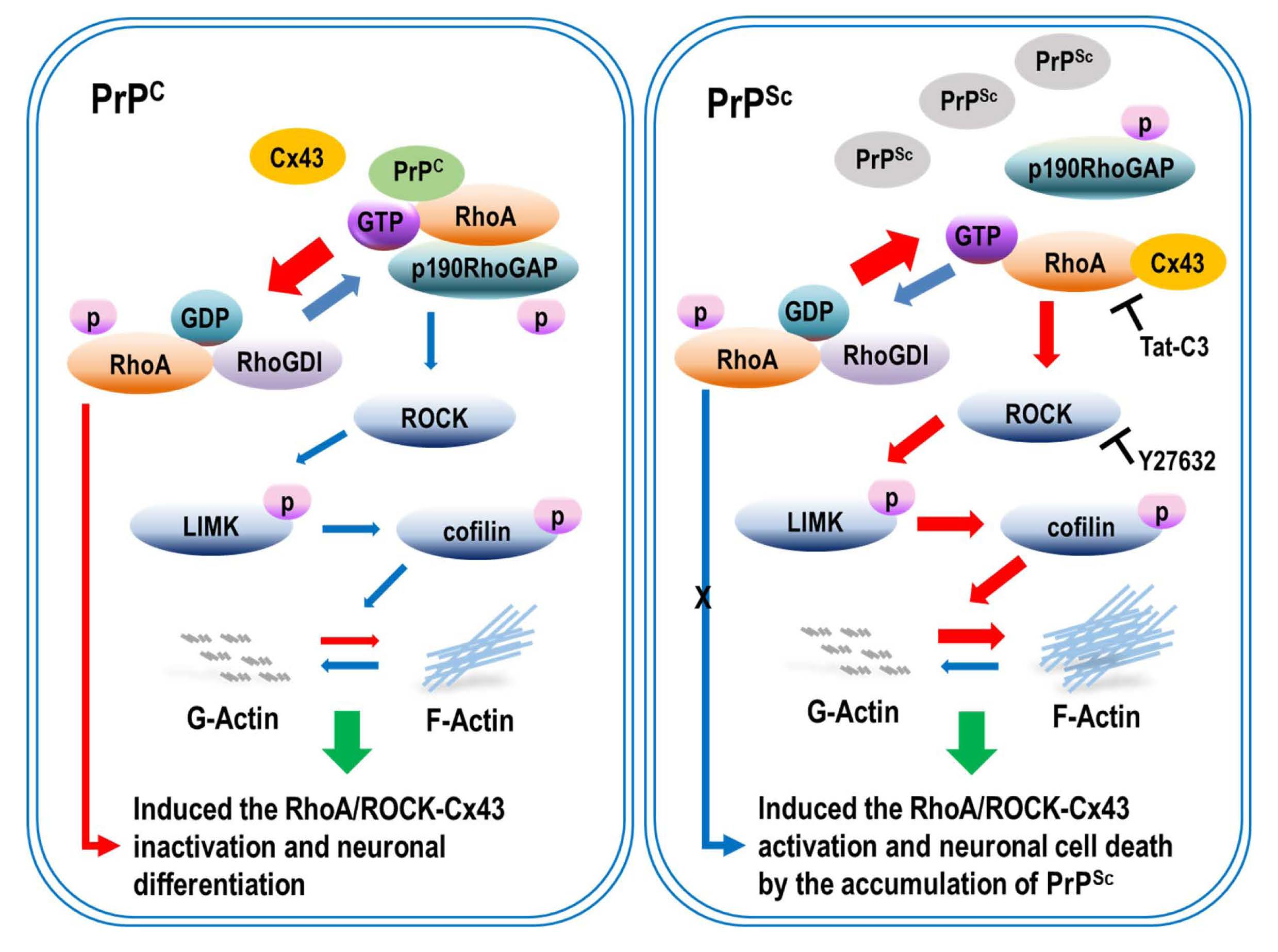
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Maintenance of Scrapie-Infected Cultured Cell Lines
4.3. Animals
4.4. Western Blot Analysis
4.5. Coimmunoprecipitation
4.6. Glutathione-s-Transferase (GST) Pull-Down Assay for Detecting RhoA, Rac1, and Cdc42 Activity
4.7. Immunofluorescence Staining
4.8. In Situ Proximity Ligation Assay
4.9. Dye-Uptake Assay
4.10. Scrape-Loading Dye Transfer Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| GAP | GTPase-activating protein |
| ROCK | Rho-associated kinase |
| RhoGDI | Rho GDP-dissociation inhibitor |
| GST-Rhotekin-RBD | Glutathione S-transferase-Rhotekin-Rho-binding domain |
| GST-Rhotekin-PBD | Glutathione S-transferase-p21 binding domain |
| GJA1 | Gap junction alpha-1 protein |
| PDK1 | ROCK-3-phosphoinositide-dependent kinase 1 |
References
- Eghiaian, F.; Grosclaude, J.; Lesceu, S.; Debey, P.; Doublet, B.; Treguer, E.; Rezaei, H.; Knossow, M. Insight into the PrPC-->PrPSc conversion from the structures of antibody-bound ovine prion scrapie-susceptibility variants. Proc. Natl. Acad. Sci. USA 2004, 101, 10254–10259. [Google Scholar] [CrossRef] [Green Version]
- Riesner, D. Biochemistry and structure of PrPC and PrPSc. Br. Med. Bull. 2003, 66, 21–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geschwind, M.D. Prion Diseases. Continuum (Minneapolis, Minn.) 2015, 21, 1612–1638. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Satani, N. The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol. Med. 2011, 17, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [Green Version]
- Alleaume-Butaux, A.; Nicot, S.; Pietri, M.; Baudry, A.; Dakowski, C.; Tixador, P.; Ardila-Osorio, H.; Haeberle, A.M.; Bailly, Y.; Peyrin, J.M.; et al. Double-Edge Sword of Sustained ROCK Activation in Prion Diseases through Neuritogenesis Defects and Prion Accumulation. PLoS Pathog. 2015, 11, e1005073. [Google Scholar] [CrossRef] [PubMed]
- Loubet, D.; Dakowski, C.; Pietri, M.; Pradines, E.; Bernard, S.; Callebert, J.; Ardila-Osorio, H.; Mouillet-Richard, S.; Launay, J.M.; Kellermann, O.; et al. Neuritogenesis: the prion protein controls beta1 integrin signaling activity. FASEB J. 2012, 26, 678–690. [Google Scholar] [CrossRef]
- Schmitt-Ulms, G.; Legname, G.; Baldwin, M.A.; Ball, H.L.; Bradon, N.; Bosque, P.J.; Crossin, K.L.; Edelman, G.M.; DeArmond, S.J.; Cohen, F.E.; et al. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 2001, 314, 1209–1225. [Google Scholar] [CrossRef]
- Santuccione, A.; Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 2005, 169, 341–354. [Google Scholar] [CrossRef] [Green Version]
- Hundt, C.; Peyrin, J.M.; Haik, S.; Gauczynski, S.; Leucht, C.; Rieger, R.; Riley, M.L.; Deslys, J.P.; Dormont, D.; Lasmezas, C.I.; et al. Identification of interaction domains of the prion protein with its 37-kDa/67-kDa laminin receptor. EMBO J. 2001, 20, 5876–5886. [Google Scholar] [CrossRef]
- West, D.C.; Rees, C.G.; Duchesne, L.; Patey, S.J.; Terry, C.J.; Turnbull, J.E.; Delehedde, M.; Heegaard, C.W.; Allain, F.; Vanpouille, C.; et al. Interactions of multiple heparin binding growth factors with neuropilin-1 and potentiation of the activity of fibroblast growth factor-2. J. Biol. Chem. 2005, 280, 13457–13464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanata, S.M.; Lopes, M.H.; Mercadante, A.F.; Hajj, G.N.; Chiarini, L.B.; Nomizo, R.; Freitas, A.R.; Cabral, A.L.; Lee, K.S.; Juliano, M.A.; et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002, 21, 3307–3316. [Google Scholar] [CrossRef] [PubMed]
- Spielhaupter, C.; Schatzl, H.M. PrPC directly interacts with proteins involved in signaling pathways. J. Biol. Chem. 2001, 276, 44604–44612. [Google Scholar] [CrossRef] [Green Version]
- Mouillet-Richard, S.; Ermonval, M.; Chebassier, C.; Laplanche, J.L.; Lehmann, S.; Launay, J.M.; Kellermann, O. Signal transduction through prion protein. Science 2000, 289, 1925–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieger, R.; Edenhofer, F.; Lasmezas, C.I.; Weiss, S. The human 37-kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat. Med. 1997, 3, 1383–1388. [Google Scholar] [CrossRef]
- Beraldo, F.H.; Arantes, C.P.; Santos, T.G.; Machado, C.F.; Roffe, M.; Hajj, G.N.; Lee, K.S.; Magalhaes, A.C.; Caetano, F.A.; Mancini, G.L.; et al. Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma1 chain. FASEB J. 2011, 25, 265–279. [Google Scholar] [CrossRef]
- Kim, H.J.; Choi, H.S.; Park, J.H.; Kim, M.J.; Lee, H.G.; Petersen, R.B.; Kim, Y.S.; Park, J.B.; Choi, E.K. Regulation of RhoA activity by the cellular prion protein. Cell Death Dis. 2017, 8, e2668. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.K.; Kim, J.G.; Kim, H.J.; Cho, J.Y.; Jeong, H.; Park, Y.; Islam, R.; Cap, C.K.; Park, J.B. Regulation of RhoA GTPase and novel target proteins for ROCK. Small GTPases 2017, 1–8. [Google Scholar] [CrossRef]
- Coleman, M.L.; Marshall, C.J.; Olson, M.F. RAS and RHO GTPases in G1-phase cell-cycle regulation. Nat. Rev. Mol. Cell Biol. 2004, 5, 355–366. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef]
- Spiering, D.; Hodgson, L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh. Migr. 2011, 5, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Govek, E.E.; Newey, S.E.; Van Aelst, L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005, 19, 1–49. [Google Scholar] [CrossRef] [Green Version]
- Stout, C.E.; Costantin, J.L.; Naus, C.C.; Charles, A.C. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 2002, 277, 10482–10488. [Google Scholar] [CrossRef] [Green Version]
- Lurtz, M.M.; Louis, C.F. Intracellular calcium regulation of connexin43. Am. J. Physiol. Cell Physiol. 2007, 293, C1806–C1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.; Kang, N.; Lovatt, D.; Torres, A.; Zhao, Z.; Lin, J.; Nedergaard, M. Connexin 43 hemichannels are permeable to ATP. J. Neurosci. 2008, 28, 4702–4711. [Google Scholar] [CrossRef] [PubMed]
- Dermietzel, R.; Hertberg, E.L.; Kessler, J.A.; Spray, D.C. Gap junctions between cultured astrocytes: Immunocytochemical, molecular, and electrophysiological analysis. J. Neurosci. 1991, 11, 1421–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, J.I.; Dudek, F.E.; Rash, J.E. Update on connexins and gap junctions in neurons and glia in the mammalian nervous system. Brain Res. Rev. 2004, 47, 191–215. [Google Scholar] [CrossRef]
- Nualart-Marti, A.; Solsona, C.; Fields, R.D. Gap junction communication in myelinating glia. Biochim. Biophys. Acta 2013, 1828, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Mei, X.; Ezan, P.; Giaume, C.; Koulakoff, A. Astroglial connexin immunoreactivity is specifically altered at beta-amyloid plaques in beta-amyloid precursor protein/presenilin1 mice. Neuroscience 2010, 171, 92–105. [Google Scholar] [CrossRef]
- Rufer, M.; Wirth, S.B.; Hofer, A.; Dermietzel, R.; Pastor, A.; Kettenmann, H.; Unsicker, K. Regulation of connexin-43, GFAP, and FGF-2 is not accompanied by changes in astroglial coupling in MPTP-lesioned, FGF-2-treated parkinsonian mice. J. Neurosci. Res. 1996, 46, 606–617. [Google Scholar] [CrossRef]
- Masaki, K.; Suzuki, S.O.; Matsushita, T.; Matsuoka, T.; Imamura, S.; Yamasaki, R.; Suzuki, M.; Suenaga, T.; Iwaki, T.; Kira, J. Connexin 43 astrocytopathy linked to rapidly progressive multiple sclerosis and neuromyelitis optica. PLoS ONE 2013, 8, e72919. [Google Scholar] [CrossRef]
- Fonseca, C.G.; Green, C.R.; Nicholson, L.F. Upregulation in astrocytic connexin 43 gap junction levels may exacerbate generalized seizures in mesial temporal lobe epilepsy. Brain Res. 2002, 929, 105–116. [Google Scholar] [CrossRef]
- Lee, G.H.; Jang, B.; Choi, H.S.; Kim, H.J.; Park, J.H.; Jeon, Y.C.; Carp, R.I.; Kim, Y.S.; Choi, E.K. Upregulation of Connexin 43 Expression Via C-Jun N-Terminal Kinase Signaling in Prion Disease. J. Alzheimers Dis. 2016, 49, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Derangeon, M.; Bourmeyster, N.; Plaisance, I.; Pinet-Charvet, C.; Chen, Q.; Duthe, F.; Popoff, M.R.; Sarrouilhe, D.; Herve, J.C. RhoA GTPase and F-actin dynamically regulate the permeability of Cx43-made channels in rat cardiac myocytes. J. Biol. Chem. 2008, 283, 30754–30765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gago-Fuentes, R.; Fernandez-Puente, P.; Megias, D.; Carpintero-Fernandez, P.; Mateos, J.; Acea, B.; Fonseca, E.; Blanco, F.J.; Mayan, M.D. Proteomic Analysis of Connexin 43 Reveals Novel Interactors Related to Osteoarthritis. Mol. Cell. Proteomics 2015, 14, 1831–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.H.; Kim, J.I.; Choi, E.K.; Carp, R.I.; Kim, Y.S. A neuronal cell line that does not express either prion or doppel proteins. Neuroreport 2005, 16, 425–429. [Google Scholar] [CrossRef]
- Sit, S.T.; Manser, E. Rho GTPases and their role in organizing the actin cytoskeleton. J. Cell Sci. 2011, 124, 679–683. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Chen, C.; Huang, K.; Wang, S.; Hao, J.; Huang, J.; Huang, H. RhoA/rho kinase signaling reduces connexin43 expression in high glucose-treated glomerular mesangial cells with zonula occludens-1 involvement. Exp. Cell Res. 2014, 327, 276–286. [Google Scholar] [CrossRef]
- Ponsaerts, R.; D’Hondt, C.; Hertens, F.; Parys, J.B.; Leybaert, L.; Vereecke, J.; Himpens, B.; Bultynck, G. RhoA GTPase switch controls Cx43-hemichannel activity through the contractile system. PLoS ONE 2012, 7, e42074. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.C.; Stone, C.; Tkach, L.; SundarRaj, N. Rho and Rho-kinase (ROCK) signaling in adherens and gap junction assembly in corneal epithelium. Investig. Ophthalmol. Vis. Sci. 2002, 43, 978–986. [Google Scholar]
- Vicente-Steijn, R.; Kelder, T.P.; Tertoolen, L.G.; Wisse, L.J.; Pijnappels, D.A.; Poelmann, R.E.; Schalij, M.J.; deRuiter, M.C.; Gittenberger-de Groot, A.C.; Jongbloed, M.R.M. RHOA-ROCK signalling is necessary for lateralization and differentiation of the developing sinoatrial node. Cardiovasc. Res. 2017, 113, 1186–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmerling, D.; Hegyi, I.; Fischer, M.; Blattler, T.; Brandner, S.; Gotz, J.; Rulicke, T.; Flechsig, E.; Cozzio, A.; von Mering, C.; et al. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 1998, 93, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Baumann, F.; Tolnay, M.; Brabeck, C.; Pahnke, J.; Kloz, U.; Niemann, H.H.; Heikenwalder, M.; Rulicke, T.; Burkle, A.; Aguzzi, A. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007, 26, 538–547. [Google Scholar] [CrossRef]
- Brouns, M.R.; Matheson, S.F.; Hu, K.Q.; Delalle, I.; Caviness, V.S.; Silver, J.; Bronson, R.T.; Settleman, J. The adhesion signaling molecule p190 RhoGAP is required for morphogenetic processes in neural development. Development 2000, 127, 4891–4903. [Google Scholar]
- Brouns, M.R.; Matheson, S.F.; Settleman, J. p190 RhoGAP is the principal Src substrate in brain and regulates axon outgrowth, guidance and fasciculation. Nat. Cell. Biol. 2001, 3, 361–367. [Google Scholar] [CrossRef]
- Jeon, C.Y.; Kim, H.J.; Lee, J.Y.; Kim, J.B.; Kim, S.C.; Park, J.B. p190RhoGAP and Rap-dependent RhoGAP (ARAP3) inactivate RhoA in response to nerve growth factor leading to neurite outgrowth from PC12 cells. Exp. Mol. Med. 2010, 42, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Lee, W.H.; Kim, M.J.; Shin, S.; Jang, B.; Park, J.B.; Wasco, W.; Buxbaum, J.D.; Kim, Y.S.; Choi, E.K. Calsenilin, a Presenilin Interactor, Regulates RhoA Signaling and Neurite Outgrowth. Int. J. Mol. Sci. 2018, 19, 1196. [Google Scholar] [CrossRef] [Green Version]
- Thurnherr, T.; Benninger, Y.; Wu, X.; Chrostek, A.; Krause, S.M.; Nave, K.A.; Franklin, R.J.; Brakebusch, C.; Suter, U.; Relvas, J.B. Cdc42 and Rac1 signaling are both required for and act synergistically in the correct formation of myelin sheaths in the CNS. J. Neurosci. 2006, 26, 10110–10119. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.; Gomez, T.M. Rac1 and RhoA promote neurite outgrowth through formation and stabilization of growth cone point contacts. J. Neurosci. 2006, 26, 1418–1428. [Google Scholar] [CrossRef] [Green Version]
- Gallo, G. RhoA-kinase coordinates F-actin organization and myosin II activity during semaphorin-3A-induced axon retraction. J. Cell Sci. 2006, 119, 3413–3423. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.C.; Wyeth, M.S.; Baltan-Tekkok, S.; Ransom, B.R. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J. Neurosci. 2003, 23, 3588–3596. [Google Scholar] [CrossRef] [Green Version]
- Orellana, J.A.; Shoji, K.F.; Abudara, V.; Ezan, P.; Amigou, E.; Saez, P.J.; Jiang, J.X.; Naus, C.C.; Saez, J.C.; Giaume, C. Amyloid beta-induced death in neurons involves glial and neuronal hemichannels. J. Neurosci. 2011, 31, 4962–4977. [Google Scholar] [CrossRef]
- Le, H.T.; Sin, W.C.; Lozinsky, S.; Bechberger, J.; Vega, J.L.; Guo, X.Q.; Saez, J.C.; Naus, C.C. Gap junction intercellular communication mediated by connexin43 in astrocytes is essential for their resistance to oxidative stress. J. Biol. Chem. 2014, 289, 1345–1354. [Google Scholar] [CrossRef] [Green Version]
- Santiago, M.F.; Alcami, P.; Striedinger, K.M.; Spray, D.C.; Scemes, E. The carboxyl-terminal domain of connexin43 is a negative modulator of neuronal differentiation. J. Biol. Chem. 2010, 285, 11836–11845. [Google Scholar] [CrossRef] [Green Version]
- Liebmann, M.; Stahr, A.; Guenther, M.; Witte, O.W.; Frahm, C. Astrocytic Cx43 and Cx30 differentially modulate adult neurogenesis in mice. Neurosci. Lett. 2013, 545, 40–45. [Google Scholar] [CrossRef]
- Liu, X.; Sun, L.; Torii, M.; Rakic, P. Connexin 43 controls the multipolar phase of neuronal migration to the cerebral cortex. Proc. Natl. Acad. Sci. USA 2012, 109, 8280–8285. [Google Scholar] [CrossRef] [Green Version]
- Nagy, J.I.; Li, W.; Hertzberg, E.L.; Marotta, C.A. Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer’s disease. Brain Res. 1996, 717, 173–178. [Google Scholar] [CrossRef]
- Kyle, J.W.; Minogue, P.J.; Thomas, B.C.; Domowicz, D.A.; Berthoud, V.M.; Hanck, D.A.; Beyer, E.C. An intact connexin N-terminus is required for function but not gap junction formation. J. Cell Sci. 2008, 121, 2744–2750. [Google Scholar] [CrossRef] [Green Version]
- Gemel, J.; Lin, X.; Veenstra, R.D.; Beyer, E.C. N-terminal residues in Cx43 and Cx40 determine physiological properties of gap junction channels, but do not influence heteromeric assembly with each other or with Cx26. J. Cell Sci. 2006, 119, 2258–2268. [Google Scholar] [CrossRef] [Green Version]
- Marquez-Rosado, L.; Solan, J.L.; Dunn, C.A.; Norris, R.P.; Lampe, P.D. Connexin43 phosphorylation in brain, cardiac, endothelial and epithelial tissues. Biochim. Biophys. Acta 2012, 1818, 1985–1992. [Google Scholar] [CrossRef] [Green Version]
- Lampe, P.D.; Lau, A.F. The effects of connexin phosphorylation on gap junctional communication. Int J. Biochem. Cell. Biol. 2004, 36, 1171–1186. [Google Scholar] [CrossRef] [Green Version]
- Solan, J.L.; Lampe, P.D. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 2014, 588, 1423–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattii, L.; Pardini, C.; Ippolito, C.; Bianchi, F.; Sabbatini, A.R.M.; Vaglini, F. Rho-inhibition and neuroprotective effect on rotenone-treated dopaminergic neurons in vitro. Neurotoxicology 2019, 72, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, A.; Hayashi, T.; Nakachi, K.; Trosko, J.E.; Sugihara, K.; Kotake, Y.; Ohta, S. Modulation of connexin 43 in rotenone-induced model of Parkinson’s disease. Neuroscience 2009, 160, 61–68. [Google Scholar] [CrossRef]
- Choi, J.K.; Park, S.J.; Jun, Y.C.; Oh, J.M.; Jeong, B.H.; Lee, H.P.; Park, S.N.; Carp, R.I.; Kim, Y.S. Generation of monoclonal antibody recognized by the GXXXG motif (glycine zipper) of prion protein. Hybridoma (2005) 2006, 25, 271–277. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.-J.; Kim, M.-J.; Mostafa, M.N.; Park, J.-H.; Choi, H.-S.; Kim, Y.-S.; Choi, E.-K. RhoA/ROCK Regulates Prion Pathogenesis by Controlling Connexin 43 Activity. Int. J. Mol. Sci. 2020, 21, 1255. https://doi.org/10.3390/ijms21041255
Kim H-J, Kim M-J, Mostafa MN, Park J-H, Choi H-S, Kim Y-S, Choi E-K. RhoA/ROCK Regulates Prion Pathogenesis by Controlling Connexin 43 Activity. International Journal of Molecular Sciences. 2020; 21(4):1255. https://doi.org/10.3390/ijms21041255
Chicago/Turabian StyleKim, Hee-Jun, Mo-Jong Kim, Mohd Najib Mostafa, Jeong-Ho Park, Hong-Seok Choi, Yong-Sun Kim, and Eun-Kyoung Choi. 2020. "RhoA/ROCK Regulates Prion Pathogenesis by Controlling Connexin 43 Activity" International Journal of Molecular Sciences 21, no. 4: 1255. https://doi.org/10.3390/ijms21041255
APA StyleKim, H. -J., Kim, M. -J., Mostafa, M. N., Park, J. -H., Choi, H. -S., Kim, Y. -S., & Choi, E. -K. (2020). RhoA/ROCK Regulates Prion Pathogenesis by Controlling Connexin 43 Activity. International Journal of Molecular Sciences, 21(4), 1255. https://doi.org/10.3390/ijms21041255