Dose-Dependent Effects of Long-Term Administration of Hydrogen Sulfide on Myocardial Ischemia–Reperfusion Injury in Male Wistar Rats: Modulation of RKIP, NF-κB, and Oxidative Stress




Abstract
:1. Introduction
2. Results
2.1. Effect of NaSH Administration on Body and Heart Weights
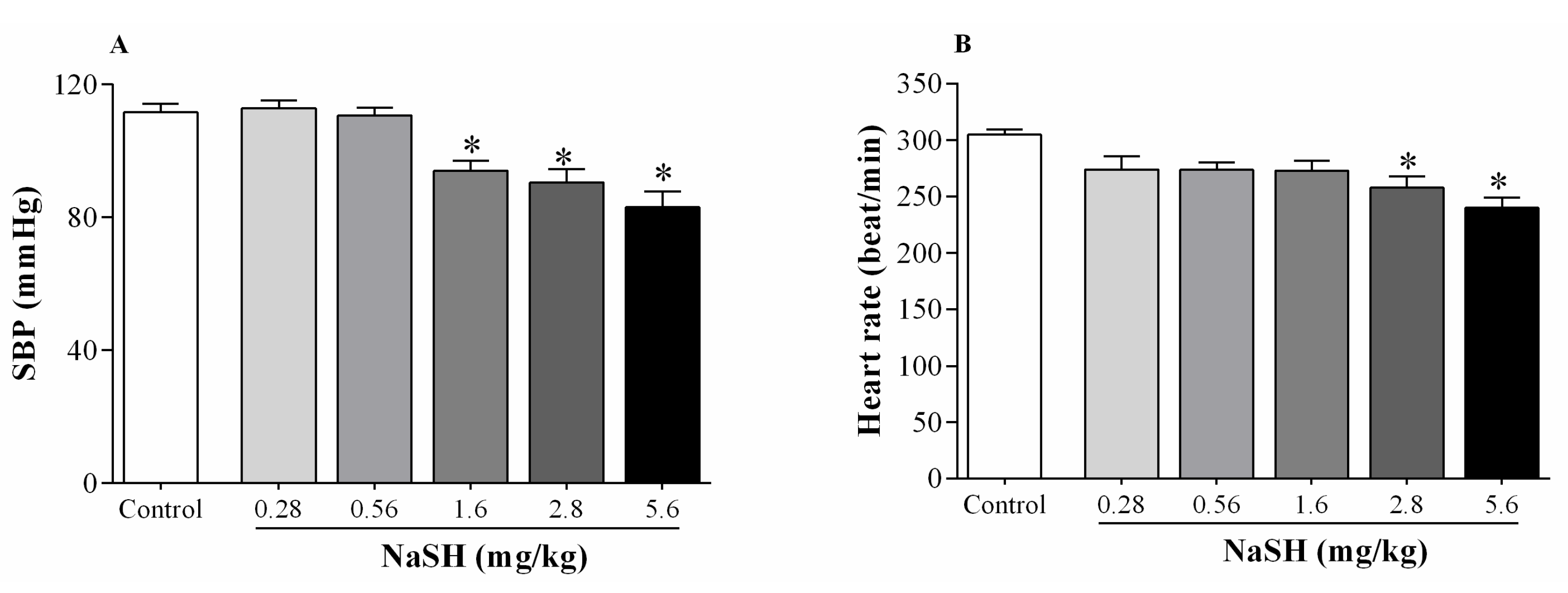
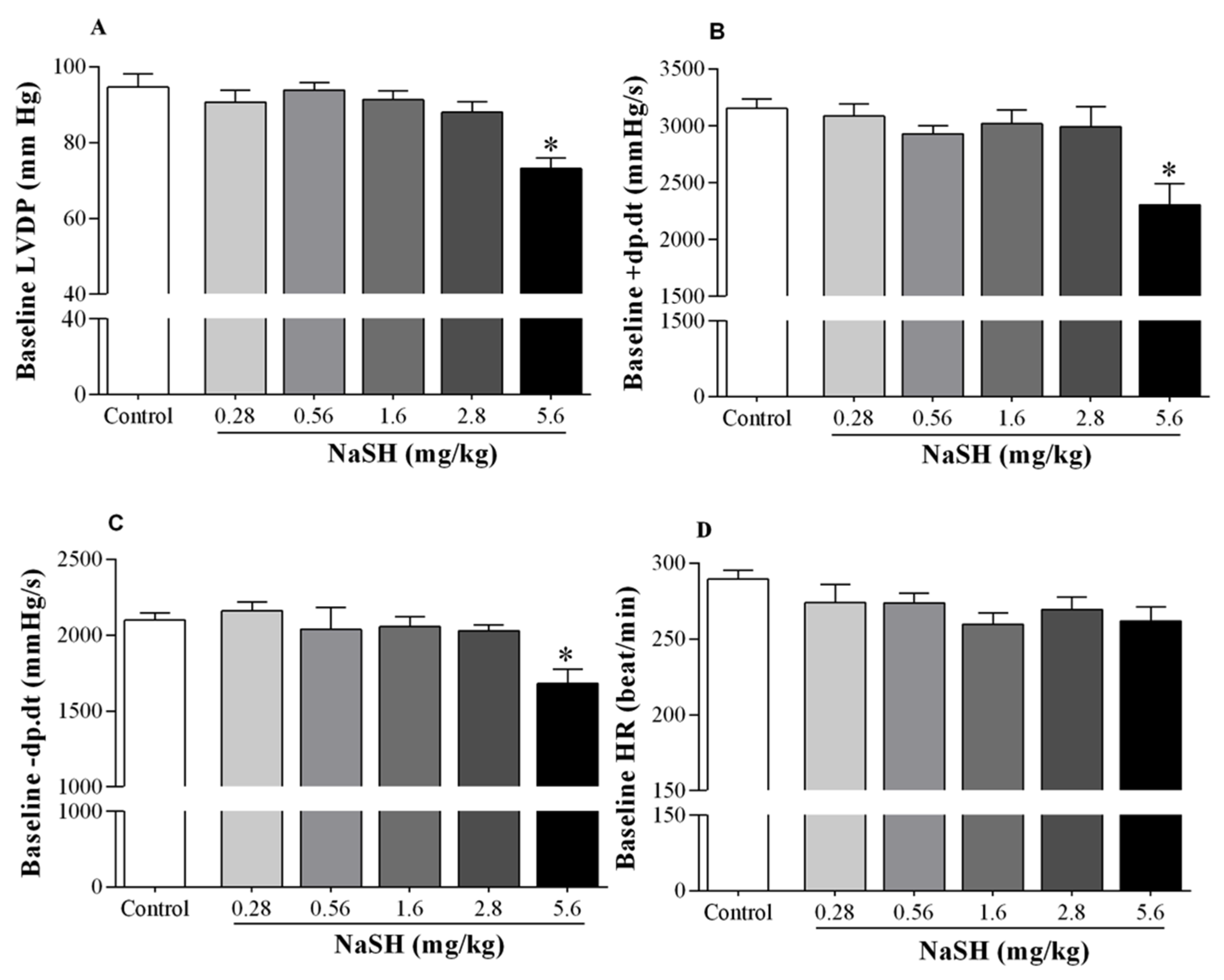
2.2. Effect of NaSH on Systolic Blood Pressure, Heart Rate, and Hemodynamic Parameters
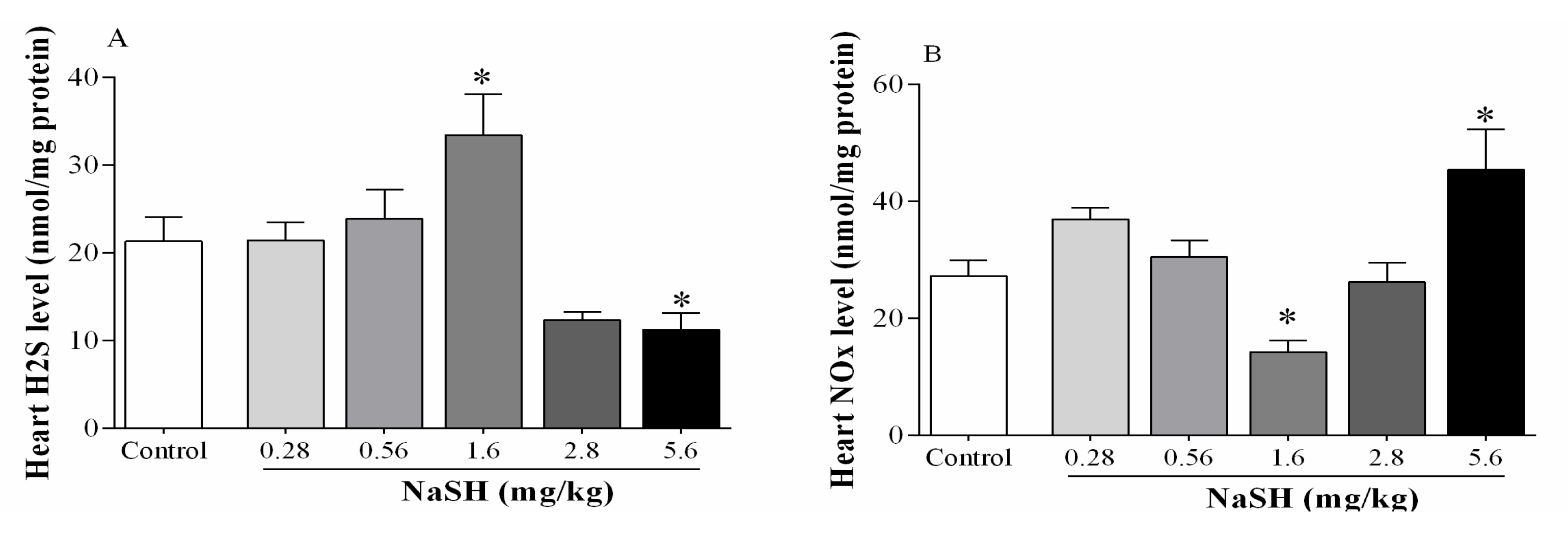
2.3. Effect of NaSH on H2S and Nitrite + Nitrate (NOx) Levels in Heart Tissue
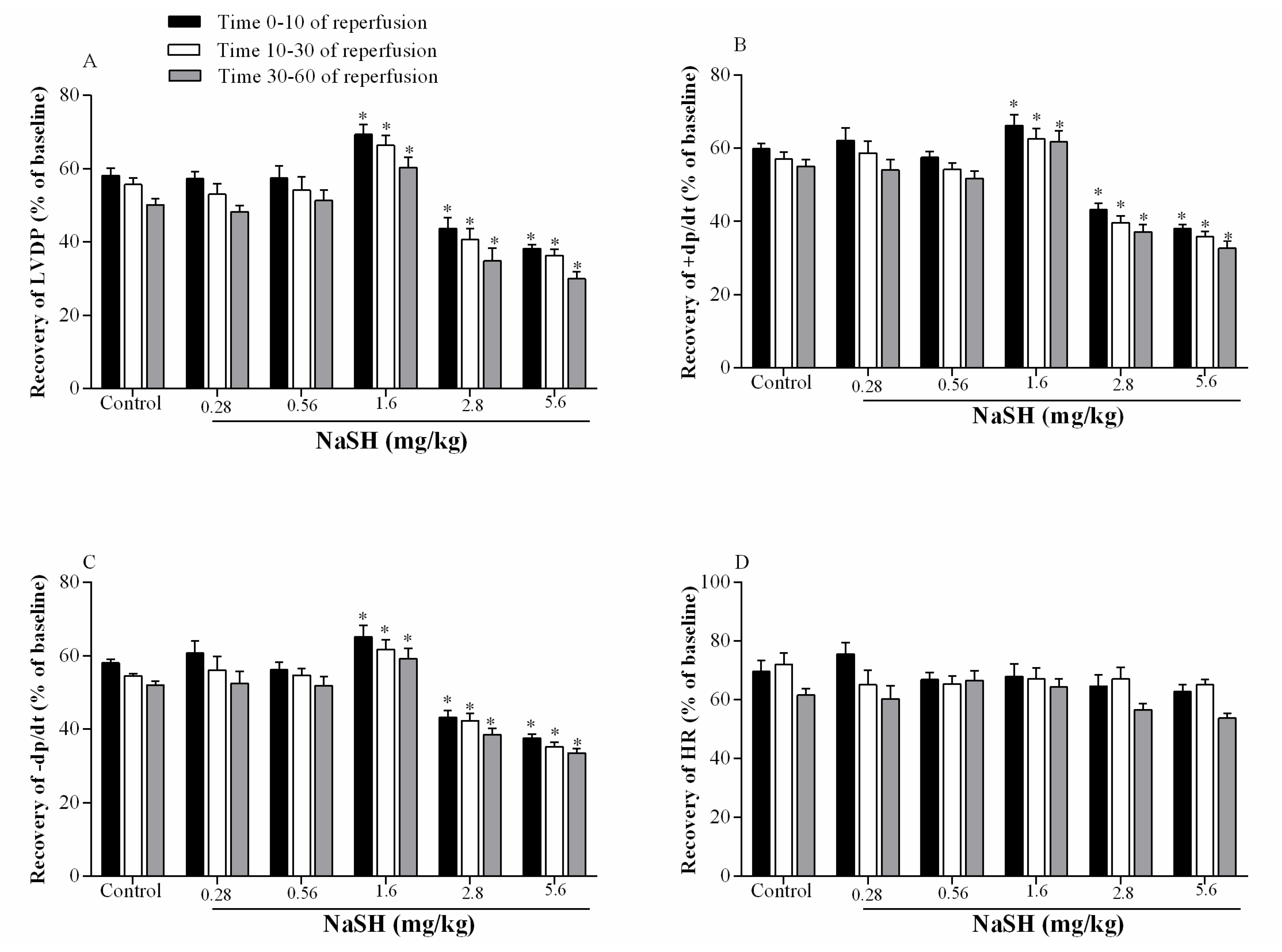
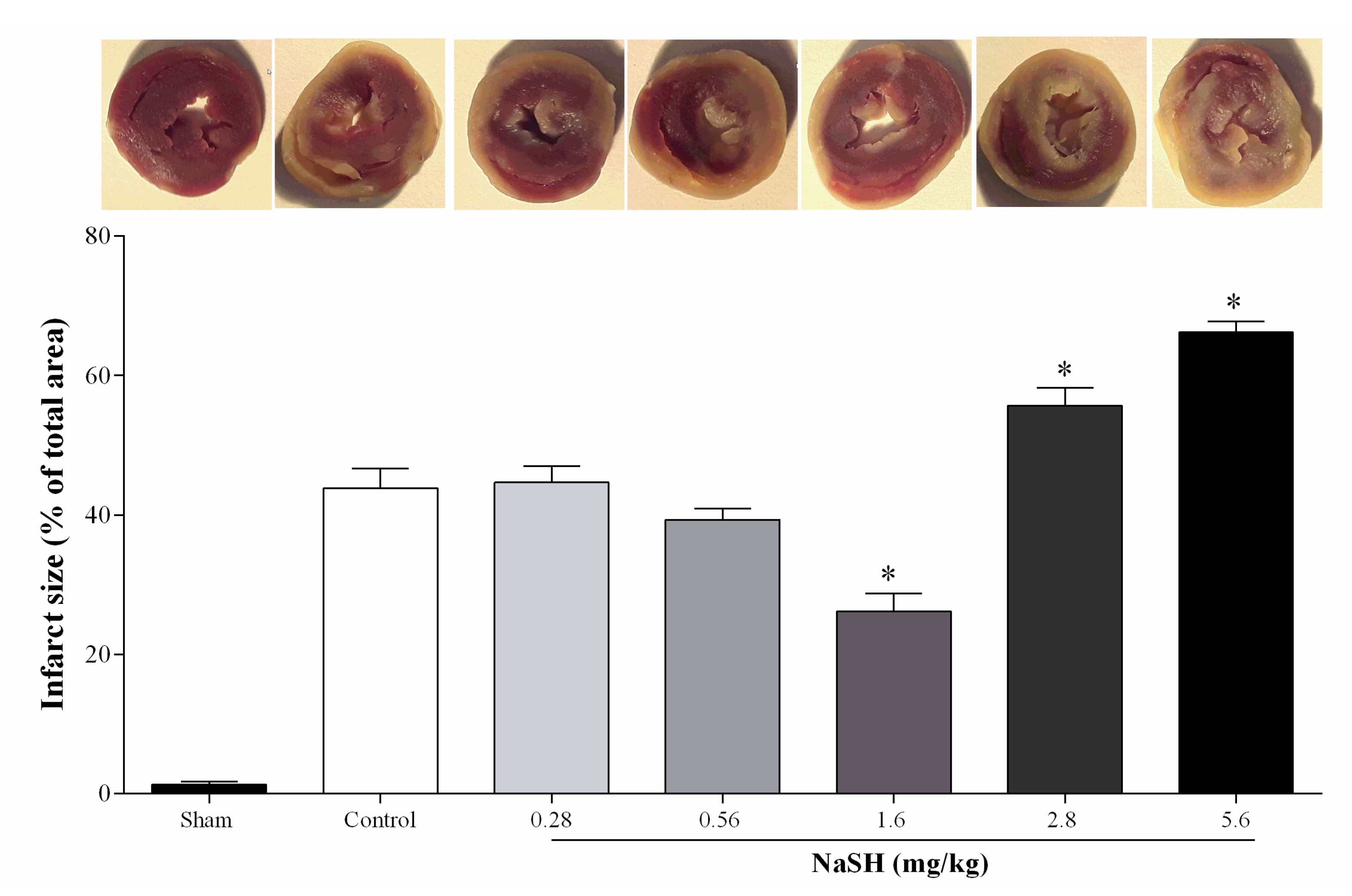
2.4. Effect of NaSH on Infarct Size
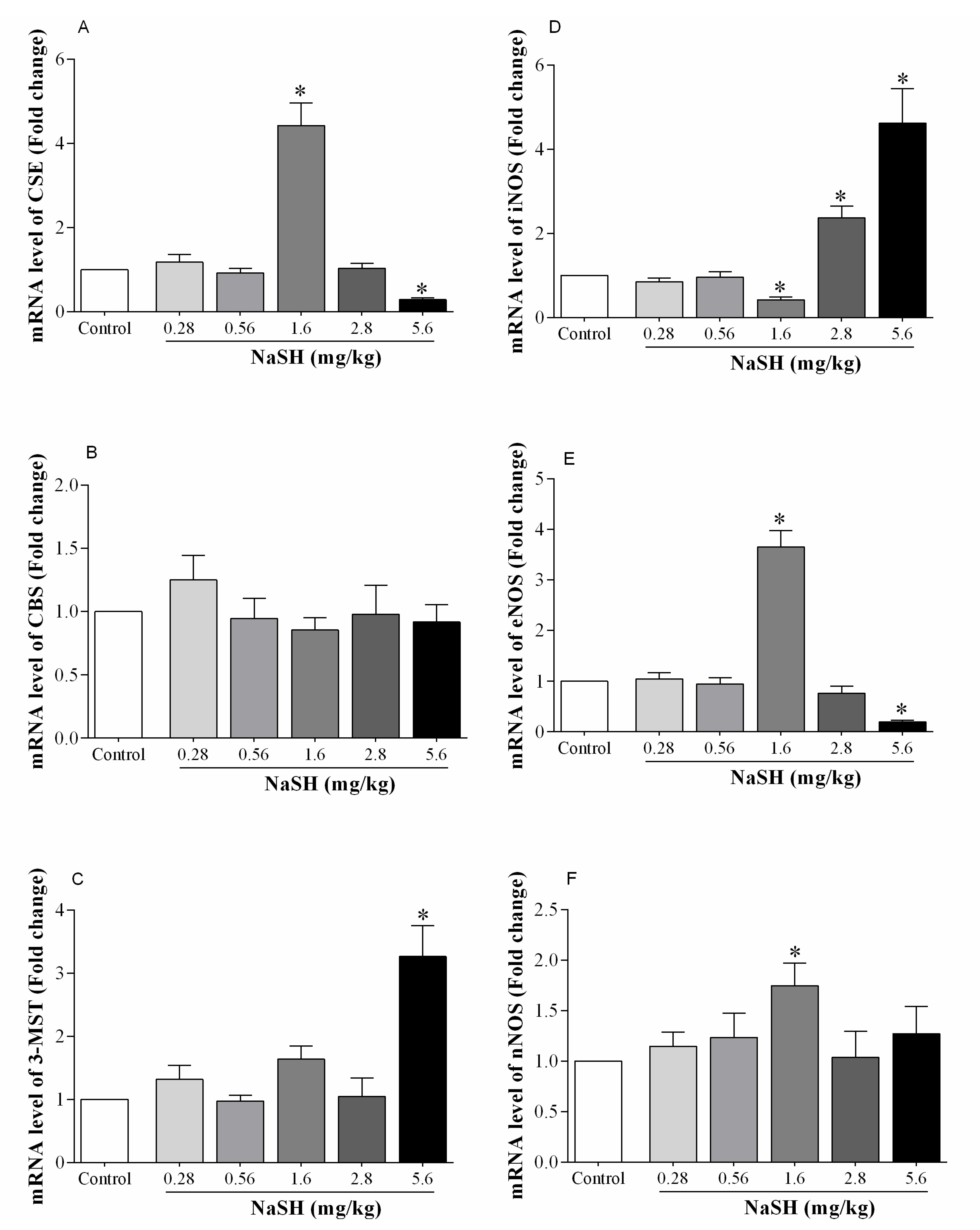
2.5. Effect of NaSH on mRNA Expression of H2S- and NO-Producing Enzymes in the Heart
2.6. Effect of NaSH on mRNA Expression of Inflammation-Related Markers in the Heart
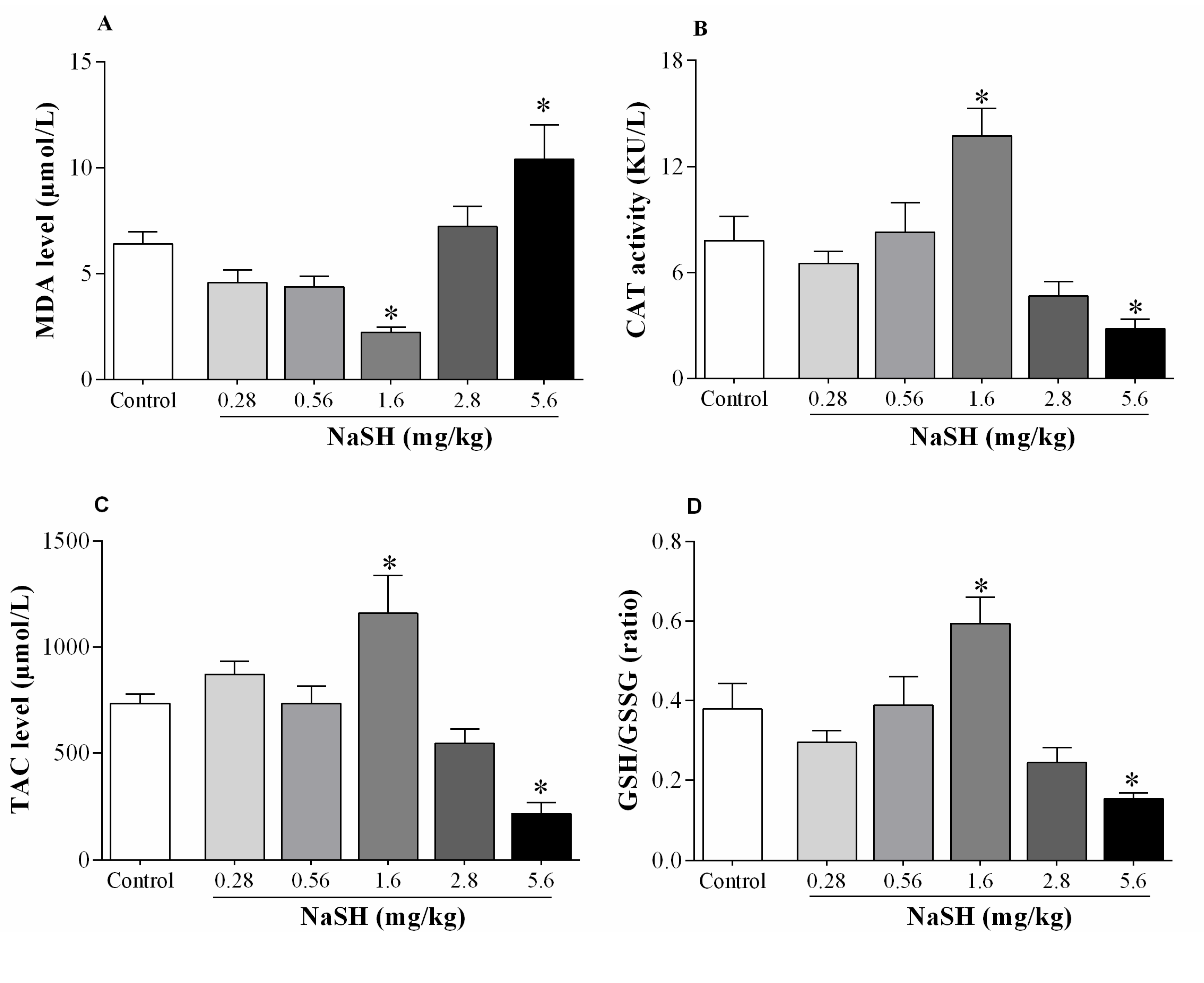
2.7. Effect of NaSH on Oxidative Stress Indices in Heart Tissue
3. Discussion
4. Materials and Methods
4.1. Animals
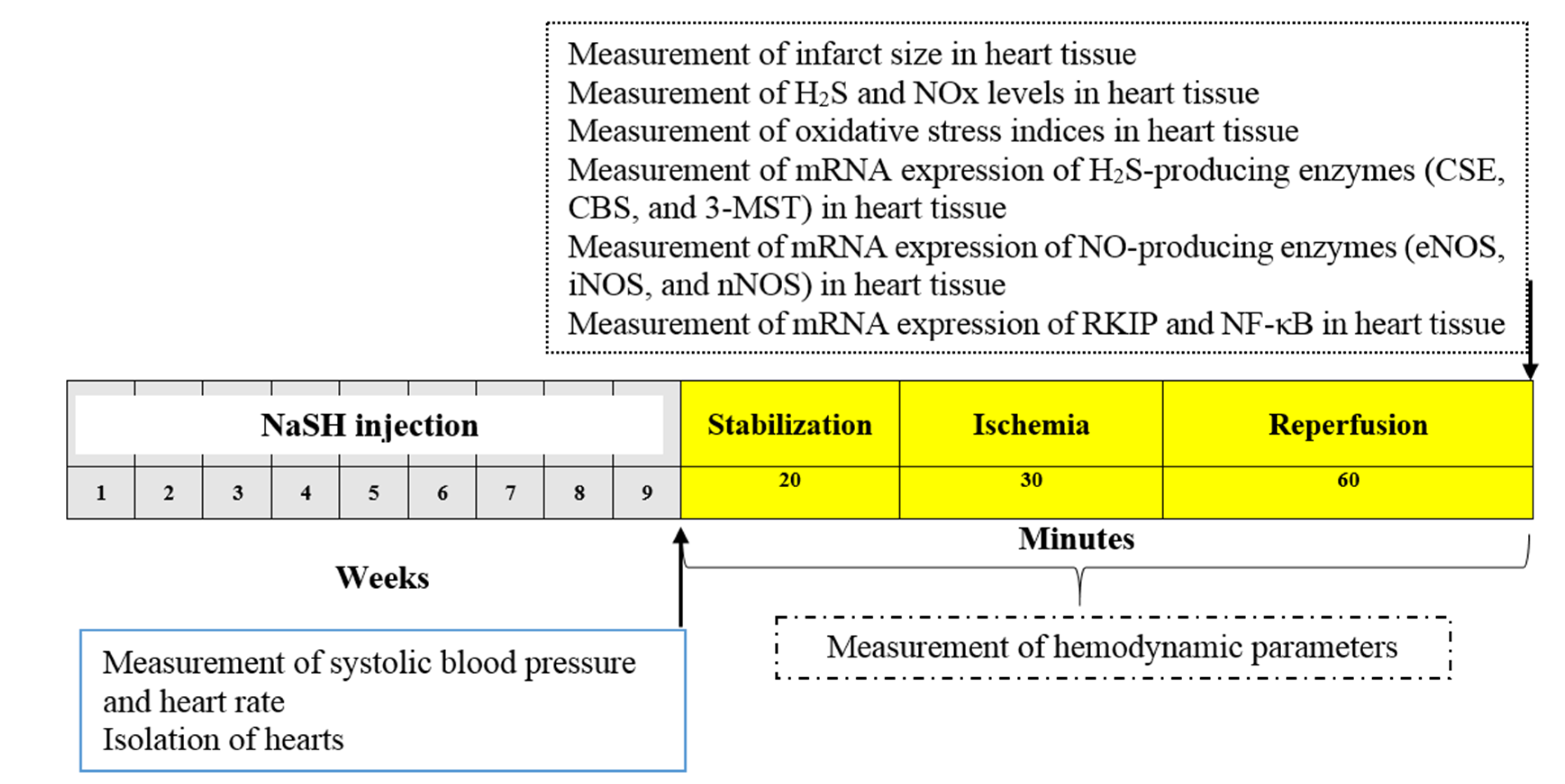
4.2. Experimental Design
4.3. Measurement of Hemodynamic Parameters
4.4. Measurements of H2S and NOx Levels in Heart Tissues
4.5. Measurement of Infarct Size
4.6. Measurement of mRNA Expression
4.7. Measurement of Oxidative Stress Indices in the Heart Tissue
4.8. Statistical Analyses
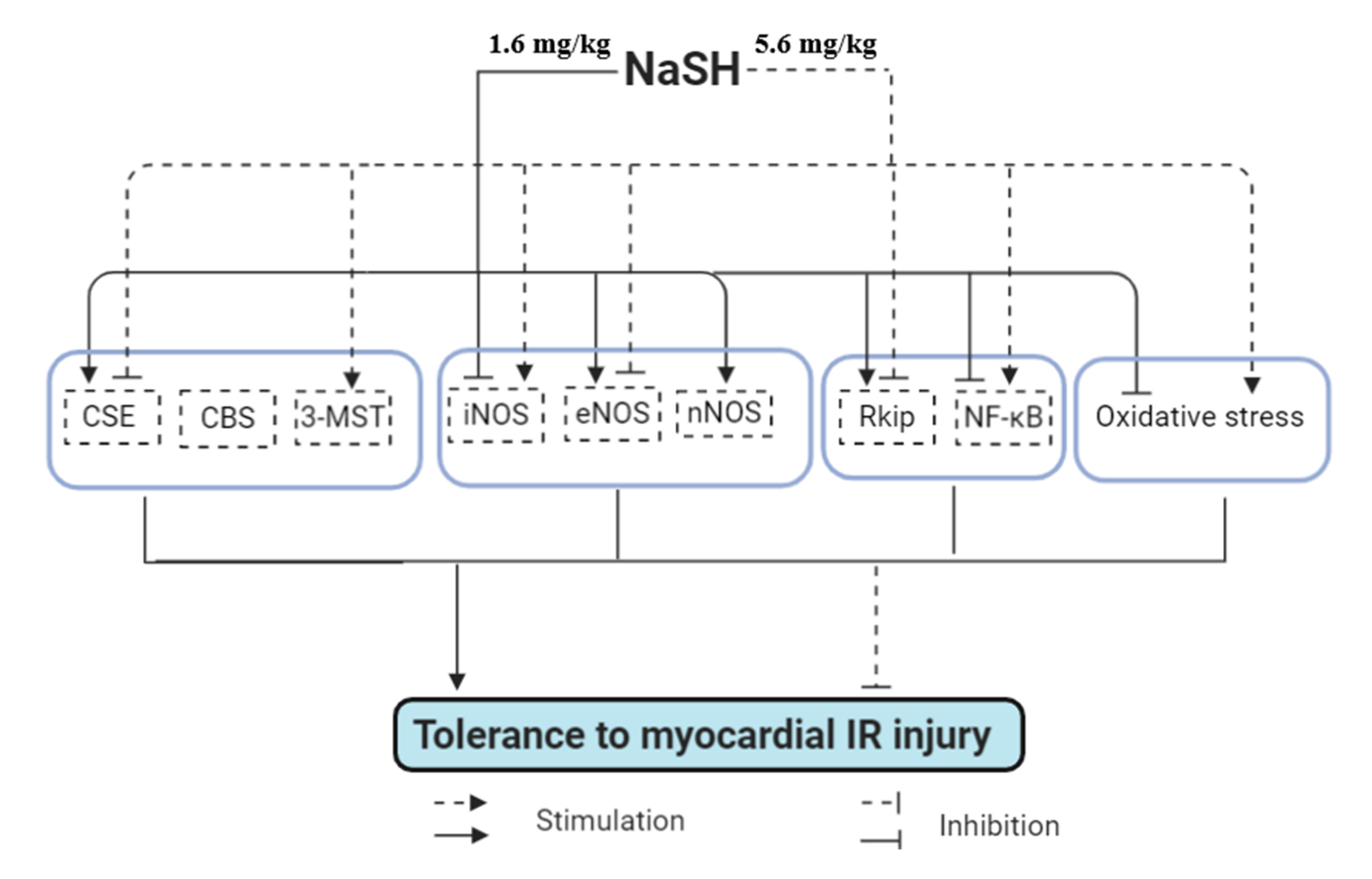
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dalen, J.E.; Alpert, J.S.; Goldberg, R.J.; Weinstein, R.S. The epidemic of the 20th century: Coronary heart disease. Am. J. Med. 2014, 127, 807–812. [Google Scholar] [CrossRef]
- Silvestri, P.; di Russo, C.; Rigattieri, S.; Fedele, S.; Todaro, D.; Ferraiuolo, G.; Altamura, G.; Loschiavo, P. MicroRNAs and ischemic heart disease: Towards a better comprehension of pathogenesis, new diagnostic tools and new therapeutic targets. Recent Pat. Cardiovasc. Drug Discov. 2009, 4, 109–118. [Google Scholar] [CrossRef]
- Ovize, M.; Baxter, G.F.; di Lisa, F.; Ferdinandy, P.; Garcia-Dorado, D.; Hausenloy, D.J.; Heusch, G.; Vinten-Johansen, J.; Yellon, D.M.; Schulz, R. Postconditioning and protection from reperfusion injury: Where do we stand? Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc. Res. 2010, 87, 406–423. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G. Cardioprotection: Chances and challenges of its translation to the clinic. Lancet 2013, 381, 166–175. [Google Scholar] [CrossRef]
- Sluijter, J.P.; Condorelli, G.; Davidson, S.M.; Engel, F.B.; Ferdinandy, P.; Hausenloy, D.J.; Lecour, S.; Madonna, R.; Ovize, M.; Ruiz-Meana, M.; et al. Novel therapeutic strategies for cardioprotection. Pharmacol. Ther. 2014, 144, 60–70. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Schulz, R.; Baxter, G.F. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol. Rev. 2007, 59, 418–458. [Google Scholar] [CrossRef]
- Granfeldt, A.; Lefer, D.J.; Vinten-Johansen, J. Protective ischaemia in patients: Preconditioning and postconditioning. Cardiovasc. Res. 2009, 83, 234–246. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.L.; Liu, X.H.; Gong, Q.H.; Yang, H.B.; Zhu, Y.Z. Role of cystathionine γ-lyase/hydrogen sulfide pathway in cardiovascular disease: A novel therapeutic strategy? Antioxid. Redox Signal. 2012, 17, 106–118. [Google Scholar] [CrossRef] [Green Version]
- King, A.L.; Polhemus, D.J.; Bhushan, S.; Otsuka, H.; Kondo, K.; Nicholson, C.K.; Bradley, J.M.; Islam, K.N.; Calvert, J.W.; Tao, Y.-X.; et al. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc. Natl. Acad. Sci. USA 2014, 111, 3182–3187. [Google Scholar] [CrossRef] [Green Version]
- Elrod, J.W.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef] [Green Version]
- Kolluru, G.K.; Shen, X.; Kevil, C.G. A tale of two gases: NO and H2S, foes or friends for life? Redox Biol. 2013, 1, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Hu, Q.; Zhu, D. An Update on Hydrogen Sulfide and Nitric Oxide Interactions in the Cardiovascular System. Oxidative Med. Cell. Longev. 2018, 2018, 4579140. [Google Scholar] [CrossRef] [Green Version]
- Burger, D.E.; Lu, X.; Lei, M.; Xiang, F.L.; Hammoud, L.; Jiang, M.; Wang, H.; Jones, D.L.; Sims, S.M.; Feng, Q. Neuronal nitric oxide synthase protects against myocardial infarction-induced ventricular arrhythmia and mortality in mice. Circulation 2009, 120, 1345–1354. [Google Scholar] [CrossRef] [Green Version]
- Xia, R.; Zhao, B.; Wu, Y.; Hou, J.B.; Zhang, L.; Xu, J.J.; Xia, Z.Y. Ginsenoside Rb1 preconditioning enhances eNOS expression and attenuates myocardial ischemia/reperfusion injury in diabetic rats. J. BioMed. Biotechnol. 2011, 2011, 767930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parlakpinar, H.; Ozer, M.K.; Acet, A. Effect of aminoguanidine on ischemia-reperfusion induced myocardial injury in rats. Mol. Cell. Biochem. 2005, 277, 137–142. [Google Scholar] [CrossRef]
- Fan, Q.; Yang, X.C.; Liu, Y.; Wang, L.F.; Liu, S.H.; Ge, Y.G.; Chen, M.L.; Wang, W.; Zhang, L.K.; Irwin, M.G.; et al. Postconditioning attenuates myocardial injury by reducing nitro-oxidative stress in vivo in rats and in humans. Clin. Sci. 2011, 120, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yan, Y.; Wang, Y.; Tong, F. The interaction between CSE/H2S and the iNOS/NO-mediated resveratrol/poly(ethylene glycol)-poly(phenylalanine) complex alleviates intestinal ischemia/reperfusion injuries in diabetic rats. BioMed. Pharmacother. 2019, 112, 108736. [Google Scholar] [CrossRef] [PubMed]
- Issa, K.; Kimmoun, A.; Collin, S.; Ganster, F.; Fremont-Orlowski, S.; Asfar, P.; Mertes, P.M.; Levy, B. Compared effects of inhibition and exogenous administration of hydrogen sulphide in ischaemia-reperfusion injury. Crit. Care 2013, 17, R129. [Google Scholar] [CrossRef] [Green Version]
- Predmore, B.L.; Kondo, K.; Bhushan, S.; Zlatopolsky, M.A.; King, A.L.; Aragon, J.P.; Grinsfelder, D.B.; Condit, M.E.; Lefer, D.J. The polysulfide diallyl trisulfide protects the ischemic myocardium by preservation of endogenous hydrogen sulfide and increasing nitric oxide bioavailability, American journal of physiology. Heart Circ. Physiol. 2012, 302, H2410–H2418. [Google Scholar] [CrossRef]
- Al-Salam, S.; Hashmi, S. Myocardial Ischemia Reperfusion Injury: Apoptotic, Inflammatory and Oxidative Stress Role of Galectin-3. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 50, 1123–1139. [Google Scholar] [CrossRef]
- Lorenz, K.; Rosner, M.R.; Brand, T.; Schmitt, J.P. Raf kinase inhibitor protein: Lessons of a better way for beta-adrenergic receptor activation in the heart. J. Physiol. 2017, 595, 4073–4087. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.C.; Rose, D.W.; Dhillon, A.S.; Yaros, D.; Gustafsson, M.; Chatterjee, D.; McFerran, B.; Wyche, J.; Kolch, W.; Sedivy, J.M. Raf kinase inhibitor protein interacts with NF-kappaB-inducing kinase and TAK1 and inhibits NF-kappaB activation. Mol. Cell. Biol. 2001, 21, 7207–7217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valen, G.; Yan, Z.-Q.; Hansson, G.K. Nuclear factor kappa-B and the heart. J. Am. Coll. Cardiol. 2001, 38, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Sivarajah, A.; Collino, M.; Yasin, M.; Benetti, E.; Gallicchio, M.; Mazzon, E.; Cuzzocrea, S.; Fantozzi, R.; Thiemermann, C. Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock 2009, 31, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yao, X.; Zhang, Y.; Li, W.; Kang, K.; Sun, L.; Sun, X. The protective role of hydrogen sulfide in myocardial ischemia-reperfusion-induced injury in diabetic rats. Int. J. Cardiol. 2011, 152, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Karwi, Q.G.; Bice, J.S.; Baxter, G.F. Pre- and postconditioning the heart with hydrogen sulfide (H2S) against ischemia/reperfusion injury in vivo: A systematic review and meta-analysis. Basic Res. Cardiol. 2018, 113, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.; Li, W.; Xi, W.; Yi, Y.; Ciren, Y.; Shen, H.; Zhang, Y.; Jiang, H.; Xiao, J.; Wang, Z. Hydrogen Sulfide Protects Cardiomyocytes against Apoptosis in Ischemia/Reperfusion through MiR-1-Regulated Histone Deacetylase 4 Pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 41, 10–21. [Google Scholar] [CrossRef]
- Yao, X.; Tan, G.; He, C.; Gao, Y.; Pan, S.; Jiang, H.; Zhang, Y.; Sun, X. Hydrogen sulfide protects cardiomyocytes from myocardial ischemia-reperfusion injury by enhancing phosphorylation of apoptosis repressor with caspase recruitment domain. Tohoku J. Exp. Med. 2012, 226, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.Z.; Wang, Z.J.; Ho, P.; Loke, Y.Y.; Zhu, Y.C.; Huang, S.H.; Tan, C.S.; Whiteman, M.; Lu, J.; Moore, P.K. Hydrogen sulfide and its possible roles in myocardial ischemia in experimental rats. J. Appl. Physiol. 2007, 102, 261–268. [Google Scholar] [CrossRef]
- Sivarajah, A.; McDonald, M.C.; Thiemermann, C. The production of hydrogen sulfide limits myocardial ischemia and reperfusion injury and contributes to the cardioprotective effects of preconditioning with endotoxin, but not ischemia in the rat. Shock 2006, 26, 154–161. [Google Scholar] [CrossRef]
- Zhuo, Y.; Chen, P.F.; Zhang, A.Z.; Zhong, H.; Chen, C.Q.; Zhu, Y.Z. Cardioprotective effect of hydrogen sulfide in ischemic reperfusion experimental rats and its influence on expression of survivin gene. Biol. Pharm. Bull. 2009, 32, 1406–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.; Tu, C.; Zhao, J.; Ou, D.; Chen, G.; Liu, Y.; Xiao, X. Exogenous hydrogen sulfide protects against global cerebral ischemia/reperfusion injury via its anti-oxidative, anti-inflammatory and anti-apoptotic effects in rats. Brain Res. 2013, 1491, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.B.; de la Cruz, S.H.; Medina-Terol, G.J.; Beltran-Ornelas, J.H.; Sánchez-López, A.; Silva-Velasco, D.L.; Centurión, D. Chronic administration of NaHS and L-Cysteine restores cardiovascular changes induced by high-fat diet in rats. Eur. J. Pharmacol. 2019, 863, 172707. [Google Scholar] [CrossRef] [PubMed]
- Swan, K.W.; Song, B.M.; Chen, A.L.; Chen, T.J.; Chan, R.A.; Guidry, B.T.; Katakam, P.V.G.; Kerut, E.K.; Giles, T.D.; Kadowitz, P.J. Analysis of decreases in systemic arterial pressure and heart rate in response to the hydrogen sulfide donor sodium sulfide, American journal of physiology. Heart Circ. Physiol. 2017, 313, H732–H743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, D.; Jupiter, R.C.; Pankey, E.A.; Reddy, V.G.; Edward, J.A.; Swan, K.W.; Peak, T.C.; Mostany, R.; Kadowitz, P.J. Analysis of cardiovascular responses to the H2S donors Na2S and NaHS in the rat, American journal of physiology. Heart Circ. Physiol. 2015, 309, H605–H614. [Google Scholar] [CrossRef] [Green Version]
- Volpato, G.P.; Searles, R.; Yu, B.; Scherrer-Crosbie, M.; Bloch, K.D.; Ichinose, F.; Zapol, W.M. Inhaled hydrogen sulfide: A rapidly reversible inhibitor of cardiac and metabolic function in the mouse. Anesthesiology 2008, 108, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Gheibi, S.; Jeddi, S.; Kashfi, K.; Ghasemi, A. Effects of Hydrogen Sulfide on Carbohydrate Metabolism in Obese Type 2 Diabetic Rats. Molecules 2019, 24, 190. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Saito, Y.; Kuwahara, K.; Rong, X.; Kishimoto, I.; Harada, M.; Horiuchi, M.; Murray, M.; Nakao, K. Vasodilator therapy with hydralazine induces angiotensin AT receptor-mediated cardiomyocyte growth in mice lacking guanylyl cyclase-A. Br. J. Pharmacol. 2010, 159, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.L.; Huang, X.W.; Wang, Y.G.; Cao, Y.X.; Zhang, C.C.; Zhu, Y.C. Hydrogen sulfide protects cardiomyocytes from hypoxia/reoxygenation-induced apoptosis by preventing GSK-3beta-dependent opening of Mptp. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1310–H1319. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Wang, W.; Dai, J.; Jin, S.; Huang, J.; Guo, C.; Wang, C.; Pang, L.; Wang, Y. A Long-Term and Slow-Releasing Hydrogen Sulfide Donor Protects against Myocardial Ischemia/Reperfusion Injury. Sci. Rep. 2017, 7, 3541. [Google Scholar] [CrossRef] [PubMed]
- Geng, B.; Chang, L.; Pan, C.; Qi, Y.; Zhao, J.; Pang, Y.; Du, J.; Tang, C. Endogenous hydrogen sulfide regulation of myocardial injury induced by isoproterenol. Biochem. Biophys. Res. Commun. 2004, 318, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Ransy, C.; Modis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 2014, 171, 2099–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.Y.; Yan, X.H.; Chen, S.J. H(2)S protects myocardium against ischemia/reperfusion injury and its effect on c-Fos protein expression in rats. Sheng Li Xue Bao (Acta Physiol. Sin.) 2008, 60, 221–227. [Google Scholar]
- Pan, T.T.; Chen, Y.Q.; Bian, J.S. All in the timing: A comparison between the cardioprotection induced by H2S preconditioning and post-infarction treatment. Eur. J. Pharmacol. 2009, 616, 160–165. [Google Scholar] [CrossRef]
- Li, C.; Hu, M.; Wang, Y.; Lu, H.; Deng, J.; Yan, X. Hydrogen sulfide preconditioning protects against myocardial ischemia/reperfusion injury in rats through inhibition of endo/sarcoplasmic reticulum stress. Int. J. Clin. Exp. Pathol. 2015, 8, 7740–7751. [Google Scholar]
- Johansen, D.; Ytrehus, K.; Baxter, G.F. Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia–reperfusion injury. Basic Res. Cardiol. 2006, 101, 53–60. [Google Scholar] [CrossRef]
- Zheng, Y.; Yu, B.; de la Cruz, L.K.; Choudhury, M.R.; Anifowose, A.; Wang, B. Toward Hydrogen Sulfide Based Therapeutics: Critical Drug Delivery and Developability Issues. Med. Res. Rev. 2018, 38, 57–100. [Google Scholar] [CrossRef]
- Kabil, O.; Vitvitsky, V.; Xie, P.; Banerjee, R. The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid. Redox Signal. 2011, 15, 36–372. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Nandi, S.S.; Mishra, P.K. H(2)S and homocysteine control a novel feedback regulation of cystathionine beta synthase and cystathionine gamma lyase in cardiomyocytes. Sci. Rep. 2017, 7, 3639. [Google Scholar] [CrossRef]
- Jain, A.K.; Singh, D.; Dubey, K.; Maurya, R.; Mittal, S.; Pandey, A.K. Chapter 3—Models and Methods for In Vitro Toxicity. In In Vitro Toxicology; Dhawan, A., Kwon, S., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 45–65. [Google Scholar]
- Cox, L.A.; Popken, D.A.; Kaplan, A.M.; Plunkett, L.M.; Becker, R.A. How well can in vitro data predict in vivo effects of chemicals? Rodent carcinogenicity as a case study. Regul. Toxicol. Pharmacol. 2016, 77, 54–64. [Google Scholar]
- Chen, T.; Vunjak-Novakovic, G. In vitro Models of Ischemia-Reperfusion Injury. Regen. Eng. Transl. Med. 2018, 4, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-E.; Tang, Z.-H.; Xie, W.; Shen, X.-T.; Liu, M.-H.; Peng, X.-P.; Zhao, Z.-Z.; Nie, D.E.B.; Liu, L.-S.; Jiang, Z.-S. Endogenous hydrogen sulfide mediates the cardioprotection induced by ischemic postconditioning in the early reperfusion phase. Exp. Ther. Med. 2012, 4, 1117–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliksøen, M.; Kaljusto, M.-L.; Vaage, J.; Stensløkken, K.-O. Effects of hydrogen sulphide on ischaemia–reperfusion injury and ischaemic preconditioning in the isolated, perfused rat heart. Eur. J. Cardio-Thorac. Surg. 2008, 34, 344–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Wang, M.J.; Jin, S.; Bai, Y.D.; Hou, C.L.; Ma, F.F.; Li, X.H.; Zhu, Y.C. The H2S Donor NaHS Changes the Expression Pattern of H2S-Producing Enzymes after Myocardial Infarction. Oxid. Med. Cell. Longev. 2016, 2016, 6492469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venditti, P.; de Rosa, R.; Cigliano, L.; Agnisola, C.; di Meo, S. Role of nitric oxide in the functional response to ischemia-reperfusion of heart mitochondria from hyperthyroid rats. Cell. Mol. Life Sci. 2004, 61, 2244–2252. [Google Scholar] [CrossRef]
- Masullo, P.; Venditti, P.; Agnisola, C.; di Meo, S. Role of nitric oxide in the reperfusion induced injury in hyperthyroid rat hearts. Free Radic. Res. 2000, 32, 411–421. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Schulz, R. Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia-reperfusion injury and preconditioning. Br. J. Pharmacol. 2003, 138, 532–543. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Wang, J.; Zhu, H.; Wu, X.; Zhou, L.; Song, Y.; Zhu, S.; Hao, M.; Liu, C.; Fan, Y.; et al. Ischemic postconditioning protects the heart against ischemia-reperfusion injury via neuronal nitric oxide synthase in the sarcoplasmic reticulum and mitochondria. Cell Death Dis. 2016, 7, e2222. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Qu, Y.; Tao, L.; Liu, H.; Hu, A.; Gao, F.; Sharifi-Azad, S.; Grunwald, Z.; Ma, X.L.; Sun, J.Z. Inhibition of iNOS protects the aging heart against beta-adrenergic receptor stimulation-induced cardiac dysfunction and myocardial ischemic injury. J. Surg. Res. 2006, 131, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Mungrue, I.N.; Gros, R.; You, X.; Pirani, A.; Azad, A.; Csont, T.; Schulz, R.; Butany, J.; Stewart, D.J.; Husain, M. Cardiomyocyte overexpression of iNOS in mice results in peroxynitrite generation, heart block, and sudden death. J. Clin. Investig. 2002, 109, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Carretero, O.A.; Xu, J.; Rhaleb, N.-E.; Wang, F.; Lin, C.; Yang, J.J.; Pagano, P.J.; Yang, X.-P. Lack of inducible NO synthase reduces oxidative stress and enhances cardiac response to isoproterenol in mice with deoxycorticosterone acetate-salt hypertension. Hypertension 2005, 46, 1355–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpure, B.V.; Bian, J.S. Interaction of Hydrogen Sulfide with Nitric Oxide in the Cardiovascular System. Oxid. Med. Cell. Longev. 2016, 2016, 6904327. [Google Scholar] [CrossRef] [Green Version]
- Kazakov, A.; Hall, R.A.; Werner, C.; Meier, T.; Trouvain, A.; Rodionycheva, S.; Nickel, A.; Lammert, F.; Maack, C.; Bohm, M.; et al. Raf kinase inhibitor protein mediates myocardial fibrosis under conditions of enhanced myocardial oxidative stress. Basic Res. Cardiol. 2018, 113, 42. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.-H.; Liu, F.; Chen, Y.; Zhu, Y.-C. Hydrogen sulfide decreases the levels of ROS by inhibiting mitochondrial complex IV and increasing SOD activities in cardiomyocytes under ischemia/reperfusion. Biochem. Biophys. Res. Commun. 2012, 421, 164–169. [Google Scholar] [CrossRef]
- Bai, Y.-D.; Yang, Y.-R.; Mu, X.-P.; Lin, G.; Wang, Y.-P.; Jin, S.; Chen, Y.; Wang, M.-J.; Zhu, Y.-C. Hydrogen Sulfide Alleviates Acute Myocardial Ischemia Injury by Modulating Autophagy and Inflammation Response under Oxidative Stress. Oxidative Med. and Cell. Longev. 2018, 2018, 3402809. [Google Scholar] [CrossRef] [Green Version]
- Kondo, K.; Bhushan, S.; King, A.L.; Prabhu, S.D.; Hamid, T.; Koenig, S.; Murohara, T.; Predmore, B.L.; Gojon, G., Sr.; Gojon, G., Jr.; et al. H₂S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 2013, 127, 1116–1127. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H. Signaling molecules: Hydrogen sulfide and polysulfide. Antioxid. Redox Signal. 2015, 22, 362–376. [Google Scholar] [CrossRef] [Green Version]
- Szabo, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007, 6, 917–935. [Google Scholar] [CrossRef]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T.T.; Neo, K.L.; Hu, L.F.; Yong, Q.C.; Bian, J.S. H2S preconditioning-induced PKC activation regulates intracellular calcium handling in rat cardiomyocytes. Am. J. Physiol.-Cell Physiol. 2008, 294, C169–C177. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Li, L.; Rose, P.; Tan, C.-H.; Parkinson, D.B.; Moore, P.K. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid. Redox Signal. 2010, 12, 1147–1154. [Google Scholar] [CrossRef]
- Hackam, D.G.; Redelmeier, D.A. Translation of research evidence from animals to humans. JAMA 2006, 296, 1731–1732. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R. Comparing rat’s to human’s age: How old is my rat in people years? Nutrition 2005, 21, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari, M.; Jeddi, S.; Bagheripuor, F.; Ghasemi, A. The effect of maternal hypothyroidism on cardiac function and tolerance to ischemia-reperfusion injury in offspring male and female rats. J. Endocrinol. Investig. 2015, 38, 915–922. [Google Scholar] [CrossRef]
- Shen, X.; Pattillo, C.B.; Pardue, S.; Bir, S.C.; Wang, R.; Kevil, C.G. Measurement of plasma hydrogen sulfide in vivo and in vitro. Free Radic. Biol. Med. 2011, 50, 1021–1031. [Google Scholar] [CrossRef] [Green Version]
- Lee, Z.W.; Zhou, J.; Chen, C.S.; Zhao, Y.; Tan, C.H.; Li, L.; Moore, P.K.; Deng, L.W. The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS ONE 2011, 6, e21077. [Google Scholar] [CrossRef] [Green Version]
- Kodela, R.; Chattopadhyay, M.; Kashfi, K. Synthesis and biological activity of NOSH-naproxen (AVT-219) and NOSH-sulindac (AVT-18A) as potent anti-inflammatory agents with chemotherapeutic potential. MedChemComm 2013, 4, 1472–1481. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, A.; Zahediasl, S. Preanalytical and analytical considerations for measuring nitric oxide metabolites in serum or plasma using the Griess method. Clin. Lab. 2012, 58, 615–624. [Google Scholar]
- Kruger, N.J. The Bradford method for protein quantitation. Methods Mol. Biol. 1994, 32, 9–15. [Google Scholar] [PubMed]
- Zaman, J.; Jeddi, S.; Daneshpour, M.S.; Zarkesh, M.; Daneshian, Z.; Ghasemi, A. Ischemic postconditioning provides cardioprotective and antiapoptotic effects against ischemia-reperfusion injury through iNOS inhibition in hyperthyroid rats. Gene 2015, 570, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Gholami, H.; Jeddi, S.; Zadeh-Vakili, A.; Farrokhfall, K.; Rouhollah, F.; Zarkesh, M.; Ghanbari, M.; Ghasemi, A. Transient Congenital Hypothyroidism Alters Gene Expression of Glucose Transporters and Impairs Glucose Sensing Apparatus in Young and Aged Offspring Rats. Cell. Physiol. Biochem. 2017, 43, 2338–2352. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K. Serum lipid peroxide in cerebrovascular disorders determined by a new colorimetric method. Clin. Chim. Acta 1978, 90, 37–43. [Google Scholar] [PubMed]
- Hadwan, M.H. New Method for Assessment of Serum Catalase Activity. Indian J. Sci. Technol. 2016, 9, 1–5. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. Ferric reducing/antioxidant power assay: Direct measure of total antioxidant activity of biological fluids and modified version for simultaneous measurement of total antioxidant power and ascorbic acid concentration. Methods Enzymol. 1999, 299, 15–27. [Google Scholar]
- Sedlak, J.; Lindsay, R.H. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal. Biochem. 1968, 25, 192–205. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Groups | |||||
|---|---|---|---|---|---|---|
| Control | NaSH (mg/kg/day) | |||||
| 0.28 | 0.56 | 1.6 | 2.8 | 5.6 | ||
| Initial body weight (g) | 239.7 ± 2.8 | 238.0 ± 2.2 | 245.7 ± 2.5 | 241.7 ± 1.9 | 245.2 ± 1.6 | 242.8 ± 2.2 |
| Final body weight (g) | 291.3 ± 4.6 | 290.5 ± 3.3 | 290.2 ± 2.6 | 295.7 ± 2.2 | 299.8 ± 2.5 | 291.0 ± 2.7 |
| Body weight gain (g) | 51.67 ± 3.8 | 52.5 ± 5.2 | 44.5 ± 3.9 | 54.0 ± 3.6 | 54.7 ± 2.8 | 48.2 ± 2.3 |
| Heart weight (g) | 1.01 ± 0.05 | 1.01 ± 0.05 | 0.99 ± 0.40 | 1.10 ± 0.05 | 1.10 ± 0.04 | 1.37 ± 0.06 * |
| Heart weight/Body weight (%) | 0.35 ± 0.02 | 0.35 ± 0.02 | 0.34 ± 0.01 | 0.37 ± 0.02 | 0.37 ± 0.01 | 0.47 ± 0.02 * |
| Study | Year | Rat strain | Dose (μmol/kg) # | Duration ** | Mechanism | Administration Route | Ref. |
|---|---|---|---|---|---|---|---|
| Geng et al. | 2004 | Wistar | 2.8 and 14 | 5 days | Inhibition of oxidative stress | Daily IP injection | [42] |
| Sivarajah et al. | 2006 | Wistar | 50 | 15 min | Opening of mitochondrial KATP channels | Single IV injection | [31] |
| Zhu et al. | 2007 | Wistar | 14 | 7 day | Elevation of H2S concentrations | Daily IP injection | [30] |
| Zhu et al. | 2008 | Sprague–Dawley | 2.8 and 14 | 20 min | Inhibition of apoptosis | Single IV injection | [44] |
| Zhuo et al. | 2009 | Wistar | 14 | 6 days | Inhibition of apoptosis | Daily IP injection | [32] |
| Sivarajah et al. | 2009 | Wistar | 50 | 15 min | Inhibition of apoptosis and inflammation | Single IV injection | [24] |
| Pan et al. | 2009 | Sprague–Dawley | 0.1, 1, 3, 10 and 30 † | 1 day | Activation of protein kinase C | Single IP injection | [45] |
| Yao et al. | 2010 | Sprague–Dawley | 1, 10, 30, 100, and 300 ‡ | 10 min | Inhibition of apoptosis | Single IV injection | [40] |
| Yao et al. | 2012 | Wistar | 14 | 7 days | Inhibition of apoptosis | Daily IP injection | [29] |
| Issa et al. | 2013 | Wistar | 3.57 | 10 min | Inhibition of inflammation and iNOS expression and activation of Akt/eNOS pathway | Single IV injection | [18] |
| Li et al. | 2015 | Sprague–Dawley | 1.4, 2.8, and 14 | 10 min | Inhibition of endo/sarcoplasmic reticulum stress | Single IV injection | [46] |
| Kang et al. | 2017 | Sprague–Dawley | 30 | 30 min | Inhibition of apoptosis | Single IP injection | [28] |
| Primer Name | Gene bank Accession No. | Primer Sequence (5′→3’) |
|---|---|---|
| CSE | NM_017074.1 | Forward: TTGTATACAGCCGCTCTGGA Reverse: CGAGCGAAGGTCAAACAGTG |
| CBS | NM_012522.2 | Forward: TGGTGACTCTCGGGAACATG Reverse: AGGTGGATCGGCTTGAACTG |
| 3-MST | NM_138843.1 | Forward: GGCATCGAACCTGGACACATC Reverse: ACTGGCGTTGGATCTCCTCTG |
| iNOS | NM_012611 | Forward: ACCATGGAGCATCCCAAGTA Reverse: CAGCGCATACCACTTCAGC |
| eNOS | NM_021838.2 | Forward: TGACCCTCACCGATACAACA Reverse: CGGGTGTCTAGATCCATGC |
| nNOS | NM_052799.1 | Forward: AATCTCAGGTCGGCCATCAC Reverse: ATCCCCCAAGGTAGAGCCAT |
| RKIP | NM_017236.1 | Forward: ACTTCCTGGTGGTCAACATGAA Reverse: TCCGGAGCCCACGTATTC |
| NF-κB p50 | NM_001276711.1 | Forward: AGAGGATGTGGGGTTTCAGG Reverse: GCTGAGCATGAAGGTGGATG |
| ß-actin | NM_031144.3 | Forward: GCGTCCACCCGCGAGTACAAC Reverse: CGACGACGAGCGCAGCGATA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeddi, S.; Gheibi, S.; Kashfi, K.; Carlström, M.; Ghasemi, A. Dose-Dependent Effects of Long-Term Administration of Hydrogen Sulfide on Myocardial Ischemia–Reperfusion Injury in Male Wistar Rats: Modulation of RKIP, NF-κB, and Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 1415. https://doi.org/10.3390/ijms21041415
Jeddi S, Gheibi S, Kashfi K, Carlström M, Ghasemi A. Dose-Dependent Effects of Long-Term Administration of Hydrogen Sulfide on Myocardial Ischemia–Reperfusion Injury in Male Wistar Rats: Modulation of RKIP, NF-κB, and Oxidative Stress. International Journal of Molecular Sciences. 2020; 21(4):1415. https://doi.org/10.3390/ijms21041415
Chicago/Turabian StyleJeddi, Sajad, Sevda Gheibi, Khosrow Kashfi, Mattias Carlström, and Asghar Ghasemi. 2020. "Dose-Dependent Effects of Long-Term Administration of Hydrogen Sulfide on Myocardial Ischemia–Reperfusion Injury in Male Wistar Rats: Modulation of RKIP, NF-κB, and Oxidative Stress" International Journal of Molecular Sciences 21, no. 4: 1415. https://doi.org/10.3390/ijms21041415
APA StyleJeddi, S., Gheibi, S., Kashfi, K., Carlström, M., & Ghasemi, A. (2020). Dose-Dependent Effects of Long-Term Administration of Hydrogen Sulfide on Myocardial Ischemia–Reperfusion Injury in Male Wistar Rats: Modulation of RKIP, NF-κB, and Oxidative Stress. International Journal of Molecular Sciences, 21(4), 1415. https://doi.org/10.3390/ijms21041415