Hypercapnia Impairs Na,K-ATPase Function by Inducing Endoplasmic Reticulum Retention of the β-Subunit of the Enzyme in Alveolar Epithelial Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Hypercapnia Increases the Endoplasmic Reticulum Fraction of the Na,K-ATPase β-Subunit
2.2. Elevated CO2 Levels Decrease Na,K-ATPase Plasma Membrane Abundance and Function
2.3. Hypercapnia Induces Endoplasmic Reticulum Retention of the Na,K-ATPase β-Subunit
2.4. Hypercapnia Attenuates Na,K-ATPase α:β Complex Formation
2.5. Elevated CO2 Levels Alter the Oxidizing Environment of the Endoplasmic Reticulum and Promote Oxidation of the Na,K-ATPase β-Subunit
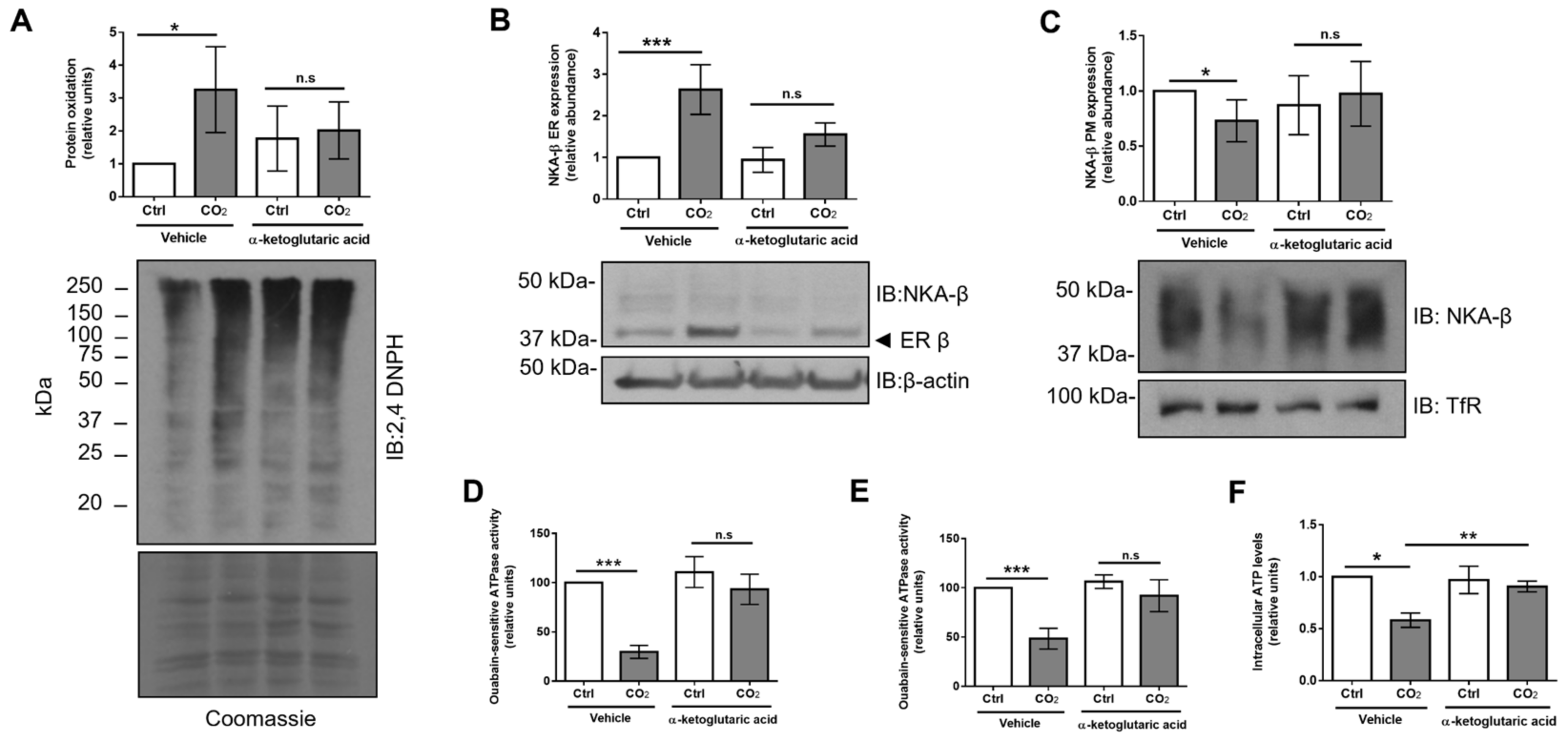
2.6. Treatment with α-Ketoglutaric Acid Ameliorates Hypercapnia-Induced ER Dysfunction and Restores Na,K-ATPase Function
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. CO2 Exposure
4.3. Western Blot Analysis
4.4. Cell Surface Biotinylation
4.5. Co-Immunoprecipitation
4.6. Antibodies and Chemical Compounds
4.7. Immunofluorescent Microscopy
4.8. Isolation of Total, Cytosolic Endoplasmic Reticulum and Golgi Cellular Fractions
4.9. Isolation of Soluble Plasma Membrane Proteins
4.10. Measurement of Na,K-ATPase Enzymatic Activity
4.11. Detection of the Protein Oxidation
4.12. Measurement of Intracellular ATP Levels
4.13. Measurement of Cellular Viability
4.14. Statistics
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α-KG | α-ketoglutaric acid |
| AEC | alveolar epithelial cell |
| ALI | acute lung injury |
| ARDS | acute respiratory distress syndrome |
| ATII | alveolar epithelial type II |
| BiP | binding immunoglobulin protein |
| CO2 | carbon dioxide |
| ENaC | epithelial sodium channel |
| ER | endoplasmic reticulum |
| GFP | green fluorescent protein |
| IDH2 | isocitrate dehydrogenase 2 |
| NKA | Na,K-ATPase |
| PM | plasma membrane |
| TCA | tricarboxylic acid |
| TfR | transferrin receptor |
References
- Clausen, M.V.; Hilbers, F.; Poulsen, H. The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front. Physiol. 2017, 8, 371. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.H. Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 2002, 71, 511–535. [Google Scholar] [CrossRef] [PubMed]
- Tokhtaeva, E.; Sun, H.; Deiss-Yehiely, N.; Wen, Y.; Soni, P.N.; Gabrielli, N.M.; Marcus, E.A.; Ridge, K.M.; Sachs, G.; Vazquez-Levin, M.; et al. The O-glycosylated ectodomain of FXYD5 impairs adhesion by disrupting cell-cell trans-dimerization of Na,K-ATPase beta1 subunits. J. Cell Sci. 2016, 129, 2394–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wujak, L.A.; Blume, A.; Baloglu, E.; Wygrecka, M.; Wygowski, J.; Herold, S.; Mayer, K.; Vadasz, I.; Besuch, P.; Mairbaurl, H.; et al. FXYD1 negatively regulates Na(+)/K(+)-ATPase activity in lung alveolar epithelial cells. Respir. Physiol. Neurobiol. 2016, 220, 54–61. [Google Scholar] [CrossRef]
- Flodby, P.; Kim, Y.H.; Beard, L.L.; Gao, D.; Ji, Y.; Kage, H.; Liebler, J.M.; Minoo, P.; Kim, K.J.; Borok, Z.; et al. Knockout Mice Reveal a Major Role for Alveolar Epithelial Type I Cells in Alveolar Fluid Clearance. Am. J. Respir. Cell Mol. Biol. 2016, 55, 395–406. [Google Scholar] [CrossRef] [Green Version]
- Geering, K. Functional roles of Na,K-ATPase subunits. Curr. Opin. Nephrol. Hypertens. 2008, 17, 526–532. [Google Scholar] [CrossRef]
- Tokhtaeva, E.; Sachs, G.; Souda, P.; Bassilian, S.; Whitelegge, J.P.; Shoshani, L.; Vagin, O. Epithelial junctions depend on intercellular trans-interactions between the Na,K-ATPase beta(1) subunits. J. Biol. Chem. 2011, 286, 25801–25812. [Google Scholar] [CrossRef] [Green Version]
- Rajasekaran, S.A.; Palmer, L.G.; Quan, K.; Harper, J.F.; Ball, W.J., Jr.; Bander, N.H.; Peralta Soler, A.; Rajasekaran, A.K. Na,K-ATPase beta-subunit is required for epithelial polarization, suppression of invasion, and cell motility. Mol. Biol. Cell 2001, 12, 279–295. [Google Scholar] [CrossRef]
- Rajasekaran, S.A.; Barwe, S.P.; Rajasekaran, A.K. Multiple functions of Na,K-ATPase in epithelial cells. Semin. Nephrol. 2005, 25, 328–334. [Google Scholar] [CrossRef]
- Vagin, O.; Tokhtaeva, E.; Yakubov, I.; Shevchenko, E.; Sachs, G. Inverse correlation between the extent of N-glycan branching and intercellular adhesion in epithelia. Contribution of the Na,K-ATPase beta1 subunit. J. Biol. Chem. 2008, 283, 2192–2202. [Google Scholar] [CrossRef] [Green Version]
- Cummins, E.P.; Strowitzki, M.J.; Taylor, C.T. Mechanisms and consequences of oxygen- and carbon dioxide-sensing in mammals. Physiol. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Vadasz, I.; Hubmayr, R.D.; Nin, N.; Sporn, P.H.; Sznajder, J.I. Hypercapnia: A nonpermissive environment for the lung. Am. J. Respir Cell Mol. Biol. 2012, 46, 417–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nin, N.; Angulo, M.; Briva, A. Effects of hypercapnia in acute respiratory distress syndrome. Ann. Transl. Med. 2018, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Vadasz, I.; Dada, L.A.; Briva, A.; Trejo, H.E.; Welch, L.C.; Chen, J.; Toth, P.T.; Lecuona, E.; Witters, L.A.; Schumacker, P.T.; et al. AMP-activated protein kinase regulates CO2-induced alveolar epithelial dysfunction in rats and human cells by promoting Na,K-ATPase endocytosis. J. Clin. Investig. 2008, 118, 752–762. [Google Scholar] [CrossRef]
- Vadasz, I.; Dada, L.A.; Briva, A.; Helenius, I.T.; Sharabi, K.; Welch, L.C.; Kelly, A.M.; Grzesik, B.A.; Budinger, G.R.; Liu, J.; et al. Evolutionary conserved role of c-Jun-N-terminal kinase in CO2-induced epithelial dysfunction. PLoS ONE 2012, 7, e46696. [Google Scholar] [CrossRef]
- Dada, L.A.; Trejo Bittar, H.E.; Welch, L.C.; Vagin, O.; Deiss-Yehiely, N.; Kelly, A.M.; Baker, M.R.; Capri, J.; Cohn, W.; Whitelegge, J.P.; et al. High CO2 Leads to Na,K-ATPase Endocytosis via c-Jun Amino-Terminal Kinase-Induced LMO7b Phosphorylation. Mol. Cell Biol. 2015, 35, 3962–3973. [Google Scholar] [CrossRef] [Green Version]
- Matthay, M.A.; Folkesson, H.G.; Clerici, C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol. Rev. 2002, 82, 569–600. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, J.L.; Skach, W.R. Protein folding and quality control in the endoplasmic reticulum: Recent lessons from yeast and mammalian cell systems. Curr. Opin. Cell Biol. 2011, 23, 464–475. [Google Scholar] [CrossRef] [Green Version]
- Tokhtaeva, E.; Sachs, G.; Vagin, O. Assembly with the Na,K-ATPase alpha(1) subunit is required for export of beta(1) and beta(2) subunits from the endoplasmic reticulum. Biochemistry 2009, 48, 11421–11431. [Google Scholar] [CrossRef] [Green Version]
- Tokhtaeva, E.; Sachs, G.; Vagin, O. Diverse pathways for maturation of the Na,K-ATPase beta1 and beta2 subunits in the endoplasmic reticulum of Madin-Darby canine kidney cells. J. Biol. Chem. 2010, 285, 39289–39302. [Google Scholar] [CrossRef] [Green Version]
- Tokhtaeva, E.; Munson, K.; Sachs, G.; Vagin, O. N-glycan-dependent quality control of the Na,K-ATPase beta(2) subunit. Biochemistry 2010, 49, 3116–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Nagata, K. Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 2011, 3, a007526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vohwinkel, C.U.; Lecuona, E.; Sun, H.; Sommer, N.; Vadasz, I.; Chandel, N.S.; Sznajder, J.I. Elevated CO(2) levels cause mitochondrial dysfunction and impair cell proliferation. J. Biol. Chem. 2011, 286, 37067–37076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fergie, N.; Todd, N.; McClements, L.; McAuley, D.; O’Kane, C.; Krasnodembskaya, A. Hypercapnic acidosis induces mitochondrial dysfunction and impairs the ability of mesenchymal stem cells to promote distal lung epithelial repair. FASEB J. 2019, 33, 5585–5598. [Google Scholar] [CrossRef] [Green Version]
- Schutt, F.; Aretz, S.; Auffarth, G.U.; Kopitz, J. Moderately reduced ATP levels promote oxidative stress and debilitate autophagic and phagocytic capacities in human RPE cells. Invest. Ophthalmol. Vis. Sci. 2012, 53, 5354–5361. [Google Scholar] [CrossRef] [Green Version]
- Esposito, L.A.; Melov, S.; Panov, A.; Cottrell, B.A.; Wallace, D.C. Mitochondrial disease in mouse results in increased oxidative stress. Proc. Natl. Acad. Sci. USA 1999, 96, 4820–4825. [Google Scholar] [CrossRef] [Green Version]
- Huppert, L.A.; Matthay, M.A. Alveolar Fluid Clearance in Pathologically Relevant Conditions: In Vitro and In Vivo Models of Acute Respiratory Distress Syndrome. Front. Immunol. 2017, 8, 371. [Google Scholar] [CrossRef]
- Matalon, S.; Bartoszewski, R.; Collawn, J.F. Role of epithelial sodium channels in the regulation of lung fluid homeostasis. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L1229–L1238. [Google Scholar] [CrossRef] [Green Version]
- Sznajder, J.I.; Factor, P.; Ingbar, D.H. Invited review: Lung edema clearance: Role of Na(+)-K(+)-ATPase. J. Appl. Physiol. (1985) 2002, 93, 1860–1866. [Google Scholar] [CrossRef]
- Sznajder, J.I. Alveolar edema must be cleared for the acute respiratory distress syndrome patient to survive. Am. J. Respir Crit. Care Med. 2001, 163, 1293–1294. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, J.; Matthay, M.A. Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu. Rev. Physiol. 2013, 75, 593–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, M.; Masterson, C.; Laffey, J.G. Permissive hypercapnia: What to remember. Curr. Opin. Anaesthesiol 2015, 28, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Nin, N.; Muriel, A.; Penuelas, O.; Brochard, L.; Lorente, J.A.; Ferguson, N.D.; Raymondos, K.; Rios, F.; Violi, D.A.; Thille, A.W.; et al. Severe hypercapnia and outcome of mechanically ventilated patients with moderate or severe acute respiratory distress syndrome. Intensive Care Med. 2017, 43, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Casalino-Matsuda, S.M.; Nair, A.; Beitel, G.J.; Gates, K.L.; Sporn, P.H. Hypercapnia Inhibits Autophagy and Bacterial Killing in Human Macrophages by Increasing Expression of Bcl-2 and Bcl-xL. J. Immunol. 2015, 194, 5388–5396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gates, K.L.; Howell, H.A.; Nair, A.; Vohwinkel, C.U.; Welch, L.C.; Beitel, G.J.; Hauser, A.R.; Sznajder, J.I.; Sporn, P.H. Hypercapnia impairs lung neutrophil function and increases mortality in murine pseudomonas pneumonia. Am. J. Respir. Cell Mol. Biol. 2013, 49, 821–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Gates, K.L.; Trejo, H.; Favoreto, S., Jr.; Schleimer, R.P.; Sznajder, J.I.; Beitel, G.J.; Sporn, P.H. Elevated CO2 selectively inhibits interleukin-6 and tumor necrosis factor expression and decreases phagocytosis in the macrophage. FASEB J. 2010, 24, 2178–2190. [Google Scholar] [CrossRef] [Green Version]
- Gwozdzinska, P.; Buchbinder, B.A.; Mayer, K.; Herold, S.; Morty, R.E.; Seeger, W.; Vadasz, I. Hypercapnia Impairs ENaC Cell Surface Stability by Promoting Phosphorylation, Polyubiquitination and Endocytosis of beta-ENaC in a Human Alveolar Epithelial Cell Line. Front. Immunol. 2017, 8, 591. [Google Scholar] [CrossRef] [Green Version]
- Vagin, O.; Kraut, J.A.; Sachs, G. Role of N-glycosylation in trafficking of apical membrane proteins in epithelia. Am. J. Physiol. Renal. Physiol. 2009, 296, F459–F469. [Google Scholar] [CrossRef] [Green Version]
- Beggah, A.; Mathews, P.; Beguin, P.; Geering, K. Degradation and endoplasmic reticulum retention of unassembled alpha- and beta-subunits of Na,K-ATPase correlate with interaction of BiP. J. Biol. Chem. 1996, 271, 20895–20902. [Google Scholar] [CrossRef] [Green Version]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Jager, R.; Bertrand, M.J.; Gorman, A.M.; Vandenabeele, P.; Samali, A. The unfolded protein response at the crossroads of cellular life and death during endoplasmic reticulum stress. Biol. Cell 2012, 104, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Hebert, D.N. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect Biol. 2013, 5, a013201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid. Med. Cell Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Marcocci, L.; Liu, L.; Suzuki, Y.J. Cell signaling by protein carbonylation and decarbonylation. Antioxid Redox Signal. 2010, 12, 393–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalle-Donne, I.; Aldini, G.; Carini, M.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell Mol. Med. 2006, 10, 389–406. [Google Scholar] [CrossRef]
- Suzuki, Y.J.; Carini, M.; Butterfield, D.A. Protein carbonylation. Antioxid Redox Signal. 2010, 12, 323–325. [Google Scholar] [CrossRef]
- Rasmussen, H.H.; Hamilton, E.J.; Liu, C.C.; Figtree, G.A. Reversible oxidative modification: Implications for cardiovascular physiology and pathophysiology. Trends Cardiovasc Med. 2010, 20, 85–90. [Google Scholar] [CrossRef]
- England, K.; Cotter, T. Identification of carbonylated proteins by MALDI-TOF mass spectroscopy reveals susceptibility of ER. Biochem Biophys Res. Commun. 2004, 320, 123–130. [Google Scholar] [CrossRef]
- Dukan, S.; Farewell, A.; Ballesteros, M.; Taddei, F.; Radman, M.; Nystrom, T. Protein oxidation in response to increased transcriptional or translational errors. Proc. Natl. Acad. Sci. USA 2000, 97, 5746–5749. [Google Scholar] [CrossRef] [Green Version]
- Zdzisinska, B.; Zurek, A.; Kandefer-Szerszen, M. Alpha-Ketoglutarate as a Molecule with Pleiotropic Activity: Well-Known and Novel Possibilities of Therapeutic Use. Arch. Immunol. Ther. Exp. (Warsz) 2017, 65, 21–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; He, L.; Yao, K. The Antioxidative Function of Alpha-Ketoglutarate and Its Applications. Biomed. Res. Int. 2018, 2018, 3408467. [Google Scholar] [CrossRef] [PubMed]
- Chin, R.M.; Fu, X.; Pai, M.Y.; Vergnes, L.; Hwang, H.; Deng, G.; Diep, S.; Lomenick, B.; Meli, V.S.; Monsalve, G.C.; et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014, 510, 397–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, C.D.; Wong, D.S.; Bozidis, P.; Zhang, A.; Colberg-Poley, A.M. Isolation of Endoplasmic Reticulum, Mitochondria, and Mitochondria-Associated Membrane and Detergent Resistant Membrane Fractions from Transfected Cells and from Human Cytomegalovirus-Infected Primary Fibroblasts. Curr. Protoc. Cell Biol. 2007, 37, 3–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnani, N.D.; Dada, L.A.; Queisser, M.A.; Brazee, P.L.; Welch, L.C.; Anekalla, K.R.; Zhou, G.; Vagin, O.; Misharin, A.V.; Budinger, G.R.S.; et al. HIF and HOIL-1L-mediated PKCzeta degradation stabilizes plasma membrane Na,K-ATPase to protect against hypoxia-induced lung injury. Proc. Natl. Acad. Sci. USA 2017, 114, E10178–E10186. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kryvenko, V.; Wessendorf, M.; Morty, R.E.; Herold, S.; Seeger, W.; Vagin, O.; Dada, L.A.; Sznajder, J.I.; Vadász, I. Hypercapnia Impairs Na,K-ATPase Function by Inducing Endoplasmic Reticulum Retention of the β-Subunit of the Enzyme in Alveolar Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 1467. https://doi.org/10.3390/ijms21041467
Kryvenko V, Wessendorf M, Morty RE, Herold S, Seeger W, Vagin O, Dada LA, Sznajder JI, Vadász I. Hypercapnia Impairs Na,K-ATPase Function by Inducing Endoplasmic Reticulum Retention of the β-Subunit of the Enzyme in Alveolar Epithelial Cells. International Journal of Molecular Sciences. 2020; 21(4):1467. https://doi.org/10.3390/ijms21041467
Chicago/Turabian StyleKryvenko, Vitalii, Miriam Wessendorf, Rory E. Morty, Susanne Herold, Werner Seeger, Olga Vagin, Laura A. Dada, Jacob I. Sznajder, and István Vadász. 2020. "Hypercapnia Impairs Na,K-ATPase Function by Inducing Endoplasmic Reticulum Retention of the β-Subunit of the Enzyme in Alveolar Epithelial Cells" International Journal of Molecular Sciences 21, no. 4: 1467. https://doi.org/10.3390/ijms21041467
APA StyleKryvenko, V., Wessendorf, M., Morty, R. E., Herold, S., Seeger, W., Vagin, O., Dada, L. A., Sznajder, J. I., & Vadász, I. (2020). Hypercapnia Impairs Na,K-ATPase Function by Inducing Endoplasmic Reticulum Retention of the β-Subunit of the Enzyme in Alveolar Epithelial Cells. International Journal of Molecular Sciences, 21(4), 1467. https://doi.org/10.3390/ijms21041467