Transcriptome Analysis Reveals Differences in Key Genes and Pathways Regulating Carbon and Nitrogen Metabolism in Cotton Genotypes under N Starvation and Resupply

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Summary of RNA Sequencing Results
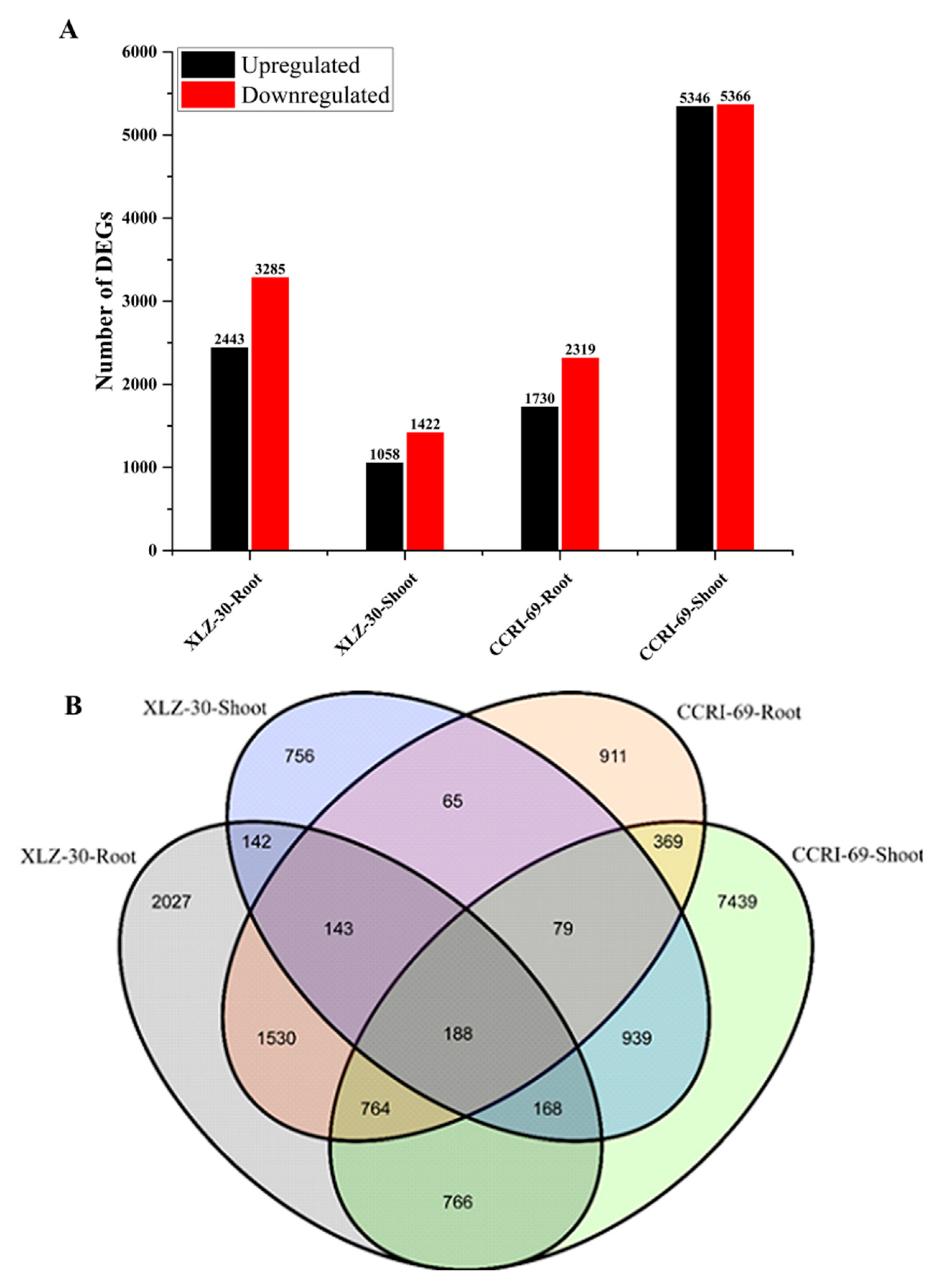
2.2. Differentially Expressed Gene Analysis
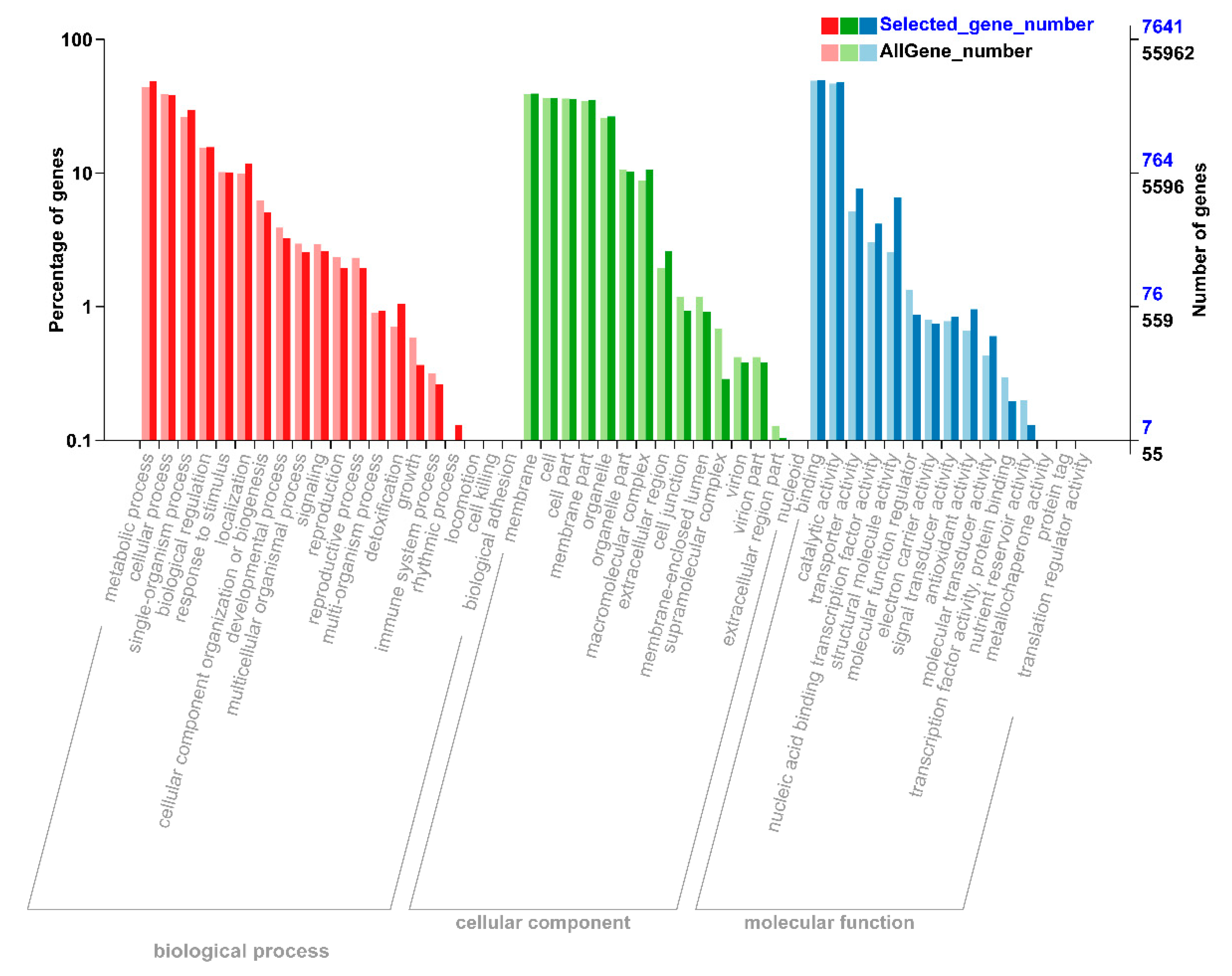
2.3. Gene Ontology (GO) Enrichment Analysis of DEGs
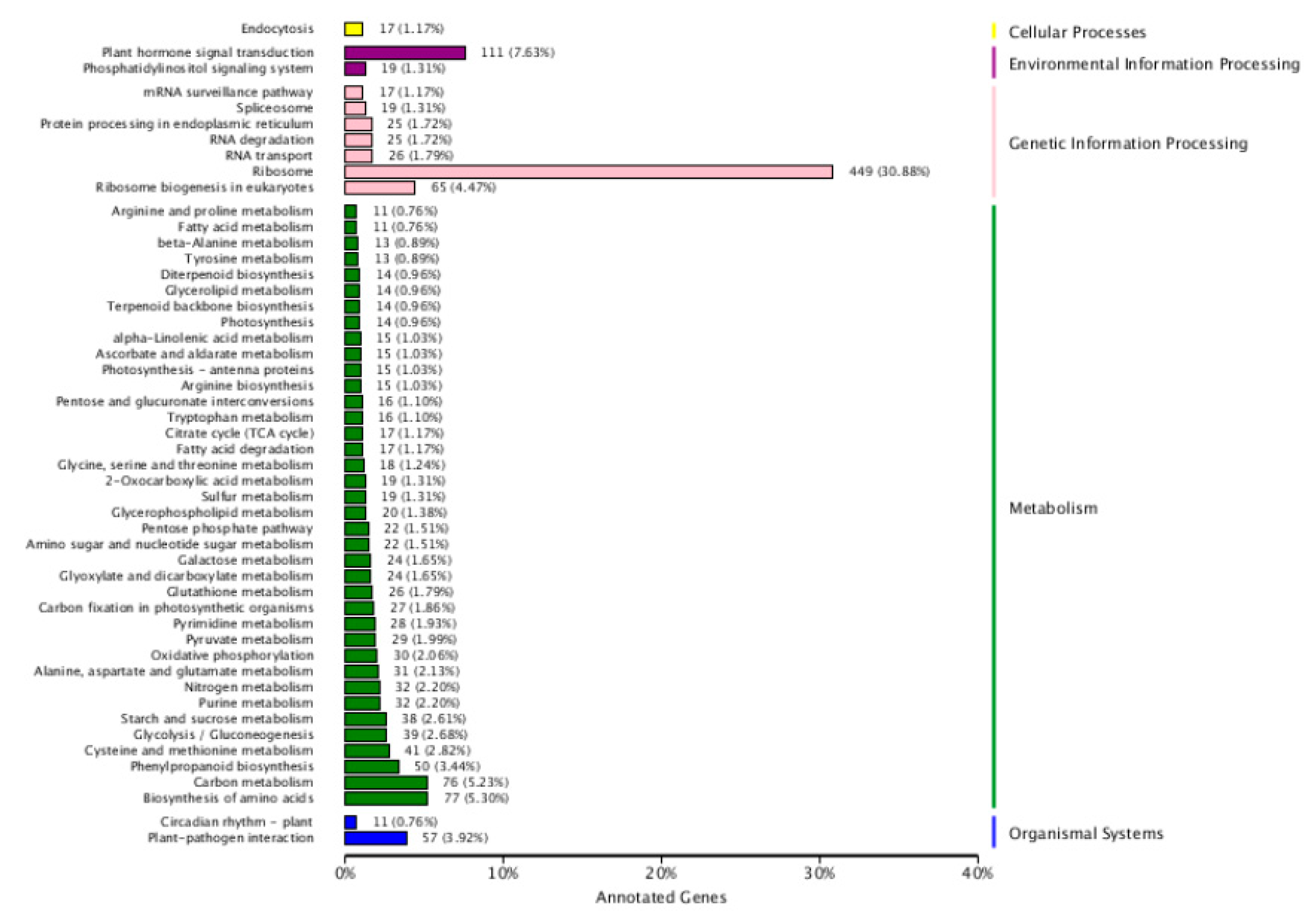
2.4. Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis of DEGs
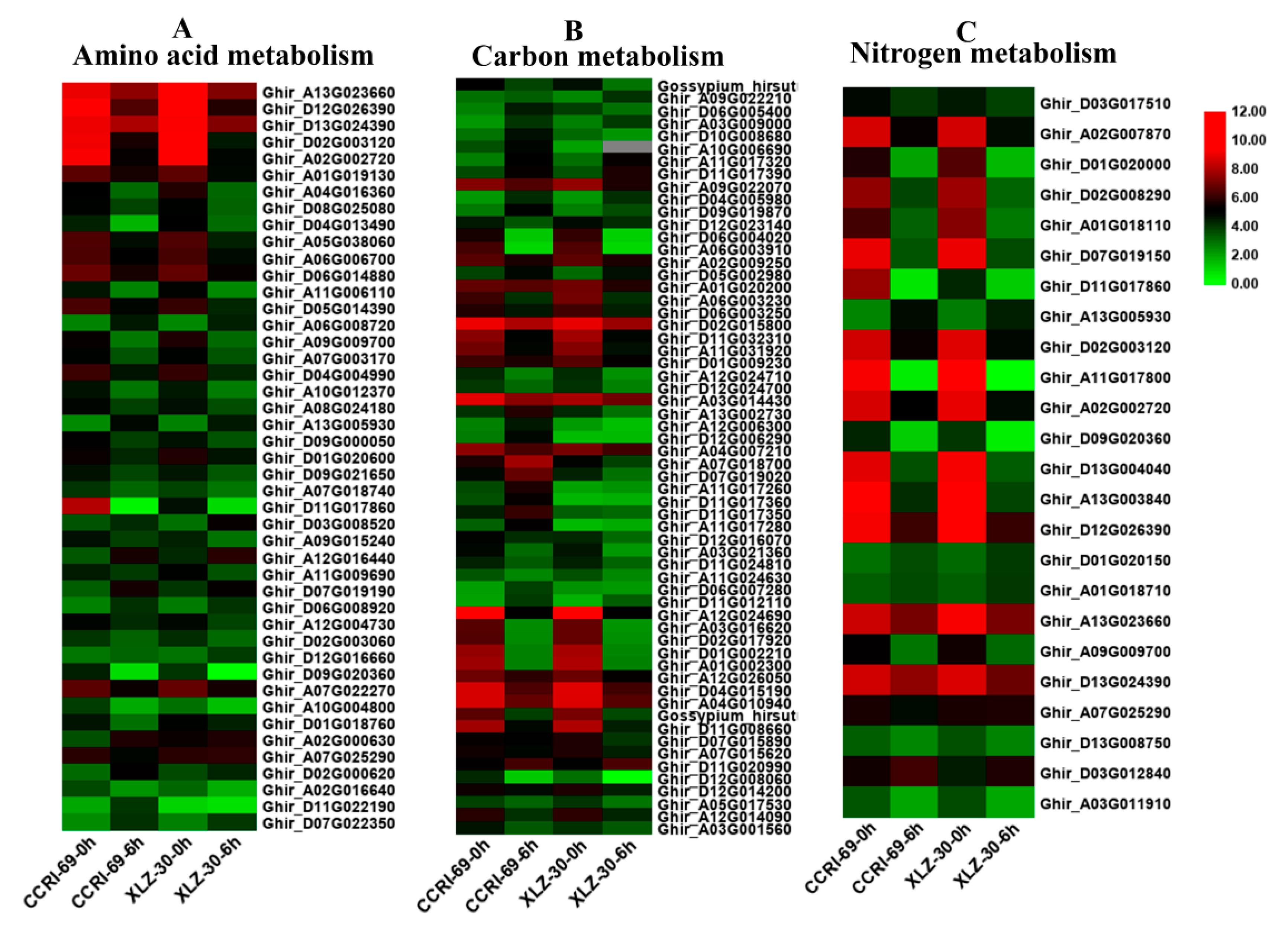
2.5. DEGs Involved in Root Amino Acid, Carbon, and Nitrogen Metabolism
2.6. DEGs Involved in Shoot Amino Acid, Carbon, and Nitrogen Metabolism
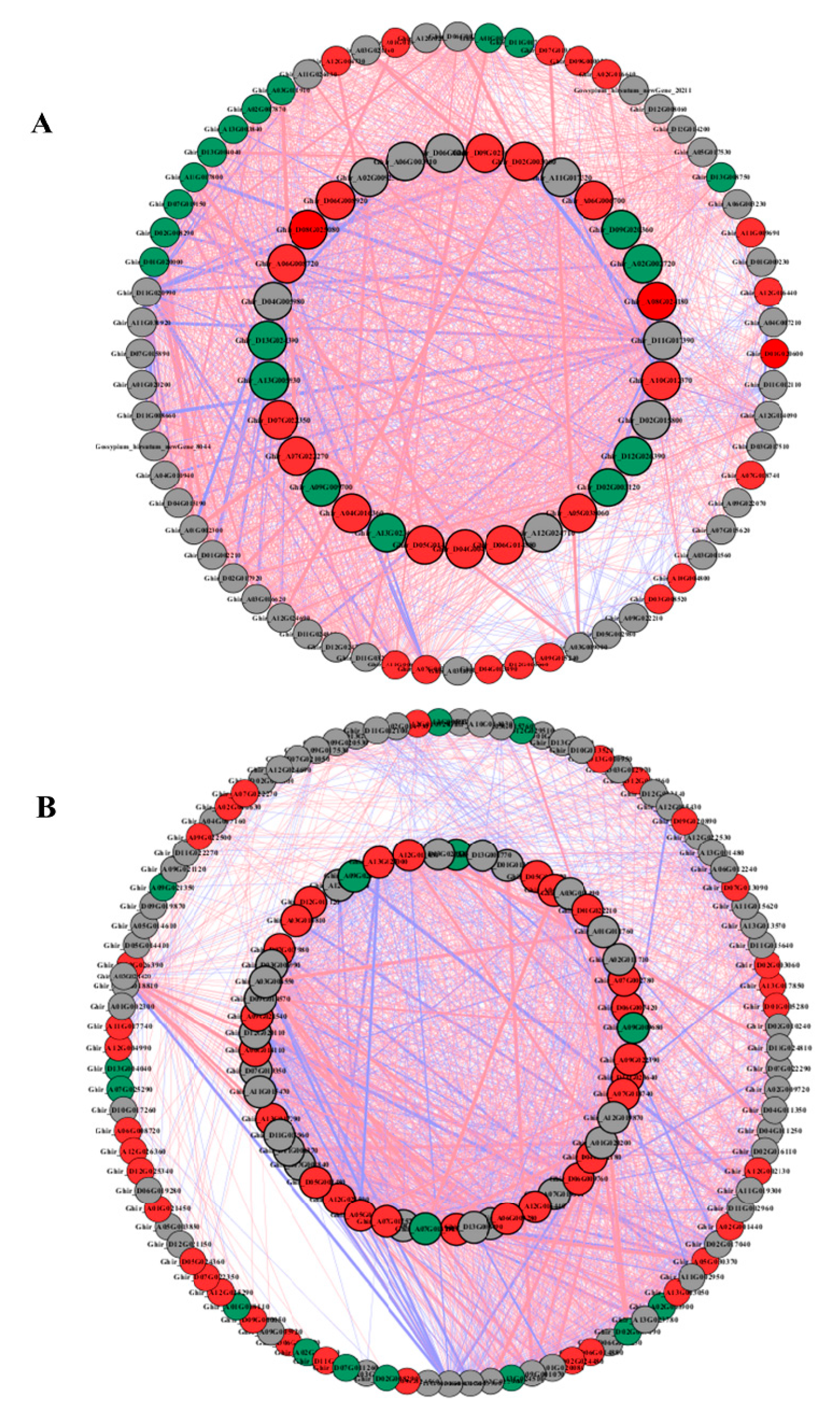
2.7. Coexpression Networks Reveal a Differential Regulatory Network of Amino Acid, Carbon, and Nitrogen Metabolism under N Starvation and N Resupply
2.8. Activities of the Key N Assimilation Enzymes
2.9. Validation of the Expression Patterns of Selected DEGs by qRT-PCR
3. Discussion
3.1. Abundance of Transcripts in the Major Pathways Related to Amino Acid, Carbon, and Nitrogen Metabolism
3.2. Nitrogen Metabolic Networks in Response to Nitrogen Starvation and Resupply Treatments
3.3. Molecular Mechanism and Regulation of Carbon and Nitrogen Metabolism
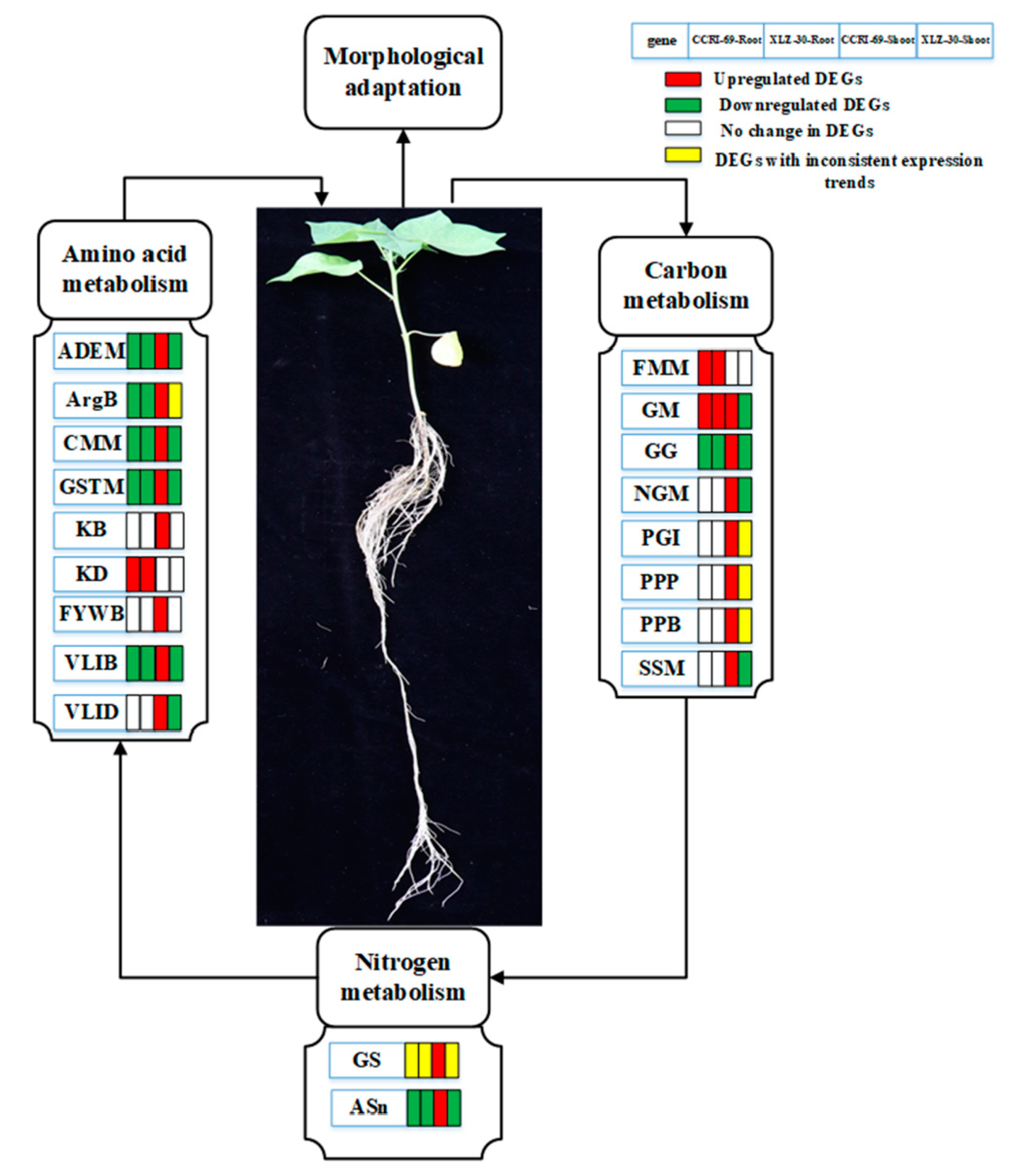
3.4. Hypothesis
4. Materials and Methods
4.1. Plant cultivation and Nitrogen Treatment
4.2. Measurement of Key Enzymes Activities in N Metabolism
4.3. RNA-Seq Sampling, RNA Extraction, and mRNA-Seq Library Construction for Illumina Sequencing
4.4. Data Filtering, Mapping of Reads, and Functional Annotation
4.5. Coexpression Network Analysis of Genes Related to Amino Acid, Carbon and Nitrogen Metabolism
4.6. Validation of RNA-Seq Analysis by qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Iqbal, A.; Qiang, D.; Alamzeb, M.; Xiangru, W.; Huiping, G.; Hengheng, Z.; Nianchang, P.; Xiling, Z.; Meizhen, S. Untangling the molecular mechanisms and functions of nitrate to improve nitrogen use efficiency. J. Sci. Food Agric. 2019, 100, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Crawford, N.M.; Forde, B.G. Molecular and developmental biology of inorganic nitrogen nutrition. Arab. Book Am. Soc. Plant Biol. 2002, 1, e0011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.L.; Clode, P.L.; Kilburn, M.R.; Stockdale, E.A.; Murphy, D. V Competition between plant and bacterial cells at the microscale regulates the dynamics of nitrogen acquisition in wheat (Triticum aestivum). New Phytol. 2013, 200, 796–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, J.; Mitchell, D.C.; Barker, D.W.; Miguez, F.; Sawyer, J.E.; Pantoja, J.; Castellano, M.J. Does nitrogen fertilizer application rate to corn affect nitrous oxide emissions from the rotated soybean crop? J. Environ. Qual. 2015, 44, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, A.D.M. Nitrogen use efficiency of crop plants: Physiological constraints upon nitrogen absorption. CRC Crit. Rev. Plant Sci. 2003, 22, 453–470. [Google Scholar] [CrossRef]
- Sarasketa, A.; González-Moro, M.B.; González-Murua, C.; Marino, D. Exploring ammonium tolerance in a large panel of Arabidopsis thaliana natural accessions. J. Exp. Bot. 2014, 65, 6023–6033. [Google Scholar] [CrossRef] [Green Version]
- Good, A.G.; Shrawat, A.K.; Muench, D.G. Can less yield more? Is reducing nutrient input into the environment compatible with maintaining crop production? Trends Plant. Sci. USA 2004, 9, 597–605. [Google Scholar] [CrossRef]
- Socolow, R.H. Nitrogen management and the future of food: Lessons from the management of energy and carbon. Proc. Natl. Acad. Sci. USA 1999, 96, 6001–6008. [Google Scholar] [CrossRef] [Green Version]
- Asif, I.; Dong, Q.; Wang, Z.; Wang, X.; Gui, H.; Zhang, H.; Pang, N.; Zhang, X.; Song, M. Growth and nitrogen metabolism are associated with nitrogen-use efficiency in cotton genotypes. Plant Physiol. Biochem. 2020, 149, 61–74. [Google Scholar]
- Hou, Z.; Li, P.; Li, B.; Gong, J.; Wang, Y. Effects of fertigation scheme on N uptake and N use efficiency in cotton. Plant Soil 2007, 290, 115–126. [Google Scholar] [CrossRef]
- Den Herder, G.; Van Isterdael, G.; Beeckman, T.; De Smet, I. The roots of a new green revolution. Trends Plant Sci. 2010, 15, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Bi, Y.-M.; Rothstein, S.J. Understanding plant response to nitrogen limitation for the improvement of crop nitrogen use efficiency. J. Exp. Bot. 2010, 62, 1499–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgerton, M.D. Increasing crop productivity to meet global needs for feed, food, and fuel. Plant Physiol. 2009, 149, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chardon, F.; Noël, V.; Masclaux-Daubresse, C. Exploring NUE in crops and in Arabidopsis ideotypes to improve yield and seed quality. J. Exp. Bot. 2012, 63, 3401–3412. [Google Scholar] [CrossRef] [Green Version]
- Le Gouis, J.; Béghin, D.; Heumez, E.; Pluchard, P. Genetic differences for nitrogen uptake and nitrogen utilisation efficiencies in winter wheat. Eur. J. Agron. 2000, 12, 163–173. [Google Scholar] [CrossRef]
- Namai, S.; Toriyama, K.; Fukuta, Y. Genetic variations in dry matter production and physiological nitrogen use efficiency in rice (Oryza sativa L.) varieties. Breed. Sci. 2009, 59, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Presterl, T.; Seitz, G.; Landbeck, M.; Thiemt, E.M.; Schmidt, W.; Geiger, H.H. Improving nitrogen-use efficiency in european maize. Crop. Sci. 2003, 43, 1259–1265. [Google Scholar] [CrossRef]
- Anbessa, Y.; Juskiw, P.; Good, A.; Nyachiro, J.; Helm, J. Genetic variability in nitrogen use efficiency of spring barley. Crop. Sci. 2009, 49, 1259–1269. [Google Scholar] [CrossRef]
- Bouchet, A.-S.; Laperche, A.; Bissuel-Belaygue, C.; Snowdon, R.; Nesi, N.; Stahl, A. Nitrogen use efficiency in rapeseed. A review. Agron. Sustain. Dev. 2016, 36, 38. [Google Scholar] [CrossRef]
- Zhang, H.; Xiaoqiong, F.U.; Xiangru, W.; Huiping, G.U.I.; Qiang, D.; Nianchang, P.; Zhun, W.; Zhang, X.; Meizhen, S. Identification and screening of nitrogen-efficient cotton genotypes under low and normal nitrogen environments at the seedling stage. J. Cott. Res. 2018, 1, 6. [Google Scholar] [CrossRef] [Green Version]
- Ranathunge, K.; El-kereamy, A.; Gidda, S.; Bi, Y.-M.; Rothstein, S.J. AMT1; 1 transgenic rice plants with enhanced NH4+ permeability show superior growth and higher yield under optimal and suboptimal NH4+ conditions. J. Exp. Bot. 2014, 65, 965–979. [Google Scholar] [CrossRef] [PubMed]
- Coruzzi, G.M.; Zhou, L. Carbon and nitrogen sensing and signaling in plants: Emerging ‘matrix effects’. Curr. Opin. Plant Biol. 2001, 4, 247–253. [Google Scholar] [CrossRef]
- Zheng, Z.-L. Carbon and nitrogen nutrient balance signaling in plants. Plant Signal. Behav. 2009, 4, 584–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krapp, A.; Traong, H.-N. Regulation of C/N interaction in model plant species. J. Crop. Improv. 2006, 15, 127–173. [Google Scholar] [CrossRef]
- Bao, A.; Zhao, Z.; Ding, G.; Shi, L.; Xu, F.; Cai, H. Accumulated expression level of cytosolic glutamine synthetase 1 gene (OsGS1; 1 or OsGS1; 2) alter plant development and the carbon-nitrogen metabolic status in rice. PLoS ONE 2014, 9, e95581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reich, P.B.; Hobbie, S.E.; Lee, T.; Ellsworth, D.S.; West, J.B.; Tilman, D.; Knops, J.M.H.; Naeem, S.; Trost, J. Nitrogen limitation constrains sustainability of ecosystem response to CO2. Nature 2006, 440, 922–925. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Nesi, A.; Fernie, A.R.; Stitt, M. Metabolic and signaling aspects underpinning the regulation of plant carbon nitrogen interactions. Mol. Plant 2010, 3, 973–996. [Google Scholar] [CrossRef]
- Foyer, C.H.; Parry, M.; Noctor, G. Markers and signals associated with nitrogen assimilation in higher plants. J. Exp. Bot. 2003, 54, 585–593. [Google Scholar] [CrossRef]
- Mooshammer, M.; Wanek, W.; Hämmerle, I.; Fuchslueger, L.; Hofhansl, F.; Knoltsch, A.; Schnecker, J.; Takriti, M.; Watzka, M.; Wild, B. Adjustment of microbial nitrogen use efficiency to carbon: Nitrogen imbalances regulates soil nitrogen cycling. Nat. Commun. 2014, 5, 3694. [Google Scholar] [CrossRef]
- Krouk, G.; Lacombe, B.; Bielach, A.; Perrine-Walker, F.; Malinska, K.; Mounier, E.; Hoyerova, K.; Tillard, P.; Leon, S.; Ljung, K. Nitrate-regulated auxin transport by NRT1. 1 defines a mechanism for nutrient sensing in plants. Dev. Cell 2010, 18, 927–937. [Google Scholar] [CrossRef]
- Patterson, K.; Cakmak, T.; Cooper, A.; Lager, I.D.A.; Rasmusson, A.G.; Escobar, M.A. Distinct signalling pathways and transcriptome response signatures differentiate ammonium-and nitrate-supplied plants. Plant Cell Environ. 2010, 33, 1486–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Qu, J.; Liu, H. Effect of liquid property on adsorption and catalytic reduction of nitrate over hydrotalcite-supported Pd-Cu catalyst. J. Mol. Catal. A Chem. 2007, 272, 31–37. [Google Scholar] [CrossRef]
- Gifford, M.L.; Dean, A.; Gutierrez, R.A.; Coruzzi, G.M.; Birnbaum, K.D. Cell-specific nitrogen responses mediate developmental plasticity. Proc. Natl. Acad. Sci. USA 2008, 105, 803–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.J.; Smith, C.J.; Chen, D. Insight into the active organic nitrogen pool estimated by isotopic equilibrium approaches. Soil Sci. Soc. Am. J. 2003, 67, 1773–1780. [Google Scholar] [CrossRef]
- Vidal, E.A.; Gutiérrez, R.A. A systems view of nitrogen nutrient and metabolite responses in Arabidopsis. Curr. Opin. Plant Biol. 2008, 11, 521–529. [Google Scholar] [CrossRef]
- Alvarez, J.M.; Vidal, E.A.; Gutiérrez, R.A. Integration of local and systemic signaling pathways for plant N responses. Curr. Opin. Plant Biol. 2012, 15, 185–191. [Google Scholar] [CrossRef]
- Scheible, W.-R.; Morcuende, R.; Czechowski, T.; Fritz, C.; Osuna, D.; Palacios-Rojas, N.; Schindelasch, D.; Thimm, O.; Udvardi, M.K.; Stitt, M. Genome-wide reprogramming of primary and secondary metabolism, protein synthesis, cellular growth processes, and the regulatory infrastructure of Arabidopsis in response to nitrogen. Plant Physiol. 2004, 136, 2483–2499. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Van den Ende, W.; Tillberg, J.-E. Fructan accumulation induced by nitrogen deficiency in barley leaves correlates with the level of sucrose: Fructan 6-fructosyltransferase mRNA. Planta 2000, 211, 701–707. [Google Scholar] [CrossRef]
- Chandran, A.K.N.; Priatama, R.A.; Kumar, V.; Xuan, Y.; Je, B.I.l.; Kim, C.M.; Jung, K.-H.; Han, C. Genome-wide transcriptome analysis of expression in rice seedling roots in response to supplemental nitrogen. J. Plant Physiol. 2016, 200, 62–75. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Garvin, D.F.; Kochian, L. V Nitrate-induced genes in tomato roots. Array analysis reveals novel genes that may play a role in nitrogen nutrition. Plant Physiol. 2001, 127, 345–359. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.W.; Cothren, J.T. Cotton: ORIGIN, History, Technology, and Production; John Wiley & Sons: Hoboken, NJ, USA, 1999. [Google Scholar]
- Krouk, G.; Mirowski, P.; LeCun, Y.; Shasha, D.E.; Coruzzi, G.M. Predictive network modeling of the high-resolution dynamic plant transcriptome in response to nitrate. Genome Biol. 2010, 11, R123. [Google Scholar] [CrossRef] [Green Version]
- Xin, W.; Zhang, L.; Zhang, W.; Gao, J.; Yi, J.; Zhen, X.; Li, Z.; Zhao, Y.; Peng, C.; Zhao, C. An Integrated Analysis of the Rice Transcriptome and Metabolome Reveals Differential Regulation of Carbon and Nitrogen Metabolism in Response to Nitrogen Availability. Int. J. Mol. Sci. 2019, 20, 2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Li, X.; Chen, Q.; Wang, L.; Qi, K.; Yin, H.; Qiao, X.; Wang, P.; Zhang, S.; Wu, J. Dynamic transcriptome analysis of root nitrate starvation and re-supply provides insights into nitrogen metabolism in pear (Pyrus bretschneideri). Plant Sci. 2018, 277, 322–333. [Google Scholar] [CrossRef]
- Hockin, N.L.; Mock, T.; Mulholland, F.; Kopriva, S.; Malin, G. The response of diatom central carbon metabolism to nitrogen starvation is different from that of green algae and higher plants. Plant Physiol. 2012, 158, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Quan, X.; Zeng, J.; Ye, L.; Chen, G.; Han, Z.; Shah, J.M.; Zhang, G. Transcriptome profiling analysis for two Tibetan wild barley genotypes in responses to low nitrogen. BMC Plant Biol. 2016, 16, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelli, M.; Duo, Y.; Konda, A.R.; Zhang, C.; Holding, D.; Dweikat, I. Identification of differentially expressed genes between sorghum genotypes with contrasting nitrogen stress tolerance by genome-wide transcriptional profiling. BMC Genom. 2014, 15, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeedipour, S.; Moradi, F. Comparison of the drought stress responses of tolerant and sensitive wheat cultivars during grain filling: Impact of invertase activity on carbon metabolism during kernel development. J. Agric. Sci. 2011, 3, 32. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Misra, A.; Nargund, S.; Coleman, G.D.; Sriram, G. Concurrent isotope-assisted metabolic flux analysis and transcriptome profiling reveal responses of poplar cells to altered nitrogen and carbon supply. Plant J. 2018, 93, 472–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, R.A.; Paiva, N.L. Stress-induced phenylpropanoid metabolism. Plant Cell 1995, 7, 1085. [Google Scholar] [CrossRef]
- Sanchez-Ballesta, M.T.; Lafuente, M.T.; Zacarias, L.; Granell, A. Involvement of phenylalanine ammonia-lyase in the response of Fortune mandarin fruits to cold temperature. Physiol. Plant. 2000, 108, 382–389. [Google Scholar] [CrossRef]
- van der Werf, A.; Kooijman, A.; Welschen, R.; Lambers, H. Respiratory energy costs for the maintenance of biomass, for growth and for ion uptake in roots of Carex diandra and Carex acutiformis. Physiol. Plant. 1988, 72, 483–491. [Google Scholar] [CrossRef]
- Pratelli, R.; Pilot, G. Regulation of amino acid metabolic enzymes and transporters in plants. J. Exp. Bot. 2014, 65, 5535–5556. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Okamoto, M.; Xing, X.; Crawford, N.M. Microarray analysis of the nitrate response in Arabidopsis roots and shoots reveals over 1000 rapidly responding genes and new linkages to glucose, trehalose-6-phosphate, iron, and sulfate metabolism. Plant Physiol. 2003, 132, 556–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-Y.; Hsu, P.-K.; Tsay, Y.-F. Uptake, allocation and signaling of nitrate. Trends Plant Sci. 2012, 17, 458–467. [Google Scholar] [CrossRef]
- Scheible, W.-R.; Gonzalez-Fontes, A.; Lauerer, M.; Muller-Rober, B.; Caboche, M.; Stitt, M. Nitrate acts as a signal to induce organic acid metabolism and repress starch metabolism in tobacco. Plant Cell 1997, 9, 783–798. [Google Scholar] [CrossRef]
- Chen, B.-M.; Wang, Z.-H.; Li, S.-X.; Wang, G.-X.; Song, H.-X.; Wang, X.-N. Effects of nitrate supply on plant growth, nitrate accumulation, metabolic nitrate concentration and nitrate reductase activity in three leafy vegetables. Plant Sci. 2004, 167, 635–643. [Google Scholar] [CrossRef]
- Hakeem, K.R.; Ahmad, A.; Iqbal, M.; Gucel, S.; Ozturk, M. Nitrogen-efficient rice cultivars can reduce nitrate pollution. Environ. Sci. Pollut. Res. 2011, 18, 1184–1193. [Google Scholar] [CrossRef]
- Ye, X.; Hong, J.; Shi, L.; Xu, F. Adaptability mechanism of nitrogen-efficient germplasm of natural variation to low nitrogen stress in Brassica napus. J. Plant Nutr. 2010, 33, 2028–2040. [Google Scholar] [CrossRef]
- Lemaître, T.; Gaufichon, L.; Boutet-Mercey, S.; Christ, A.; Masclaux-Daubresse, C. Enzymatic and metabolic diagnostic of nitrogen deficiency in Arabidopsis thaliana Wassileskija accession. Plant Cell Physiol. 2008, 49, 1056–1065. [Google Scholar] [CrossRef]
- Britto, D.T.; Kronzucker, H.J. NH4+ toxicity in higher plants: A critical review. J. Plant Physiol. 2002, 159, 567–584. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Chen, G.X.; Cheng, J.P.; Nevo, E.; Gutterman, Y. Phenological and phenotypic differences and correlations among genotypes of Hordeum spontaneum originating from different locations in Israel. Genet. Resour. Crop. Evol. 2008, 55, 995–1005. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, S.; Su, Y.; Lin, Z.; Guo, J.; Li, M.; Wang, Z.; Que, Y.; Xu, L. Transcripts and low nitrogen tolerance: Regulatory and metabolic pathways in sugarcane under low nitrogen stress. Environ. Exp. Bot. 2019, 163, 97–111. [Google Scholar] [CrossRef]
- Bi, Y.-M.; Wang, R.-L.; Zhu, T.; Rothstein, S.J. Global transcription profiling reveals differential responses to chronic nitrogen stress and putative nitrogen regulatory components in Arabidopsis. BMC Genom. 2007, 8, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Q.N.; Zhou, X.A.; Ai, H.S.; Wang, C.; Zhou, R.; Chen, S.L. Identification of genes associated with nitrogen-use efficiency by genome-wide transcriptional analysis of two soybean genotypes. BMC Genom. 2011, 12, 525. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, A.; Gui, H.; Zhang, H.; Wang, X.; Pang, N.; Dong, Q.; Song, M. Genotypic Variation in Cotton Genotypes for Phosphorus-Use Efficiency. Agronomy 2019, 9, 689. [Google Scholar] [CrossRef] [Green Version]
- Silveira, J.A.G.; Matos, J.C.S.; Cecatto, V.M.; Viegas, R.A.; Oliveira, J.T.A. Nitrate reductase activity, distribution, and response to nitrate in two contrasting Phaseolus species inoculated with Rhizobium spp. Environ. Exp. Bot. 2001, 46, 37–46. [Google Scholar] [CrossRef]
- Wang, G.; Ding, G.; Li, L.; Cai, H.; Ye, X.; Zou, J.; Xu, F. Identification and characterization of improved nitrogen efficiency in interspecific hybridized new-type Brassica napus. Ann. Bot. 2014, 114, 549–559. [Google Scholar] [CrossRef]
- Groat, R.G.; Vance, C.P. Root nodule enzymes of ammonia assimilation in alfalfa (Medicago sativa L.): Developmental patterns and response to applied nitrogen. Plant Physiol. 1981, 67, 1198–1203. [Google Scholar] [CrossRef] [Green Version]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; He, X.; Wu, D.; Zhu, B.; Cai, S.; Nadira, U.A.; Jabeen, Z.; Zhang, G. Comparative transcriptome profiling of two Tibetan wild barley genotypes in responses to low potassium. PLoS ONE 2014, 9, e100567. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Chen, J.; Zhou, X.; Chen, X.; Li, Q.; Tan, H.; Dong, X.; Xiao, Y.; Chen, L.; Chen, W. Dynamic metabolic and transcriptomic profiling of methyl jasmonate-treated hairy roots reveals synthetic characters and regulators of lignan biosynthesis in Isatis indigotica Fort. Plant Biotechnol. J. 2016, 14, 2217–2227. [Google Scholar] [CrossRef] [Green Version]
- Pujana, M.A.; Han, J.-D.J.; Starita, L.M.; Stevens, K.N.; Tewari, M.; Ahn, J.S.; Rennert, G.; Moreno, V.; Kirchhoff, T.; Gold, B. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat. Genet. 2007, 39, 1338. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Reads | Mapped Reads | Uniquely Mapped Reads | Clean Reads | Multiple Map Reads | GC (%) | Q30 (%) |
|---|---|---|---|---|---|---|---|
| T1 | 58,570,080 | 53,610,649 (91.37%) | 51,091,336 (87.10%) | 29,285,040 | 2,519,313 (4.31%) | 45.19 | 95.22 |
| T2 | 45,719,979 | 44,065,333 (96.38%) | 41,797,242 (91.42%) | 22,859,989 | 2,268,090 (4.96%) | 44.99 | 95.66 |
| T3 | 46,445,903 | 42,395,883 (91.28%) | 40,346,255 (86.87%) | 23,222,951 | 2,049,628 (4.41%) | 45.02 | 95.44 |
| T4 | 46,768,972 | 44,190,679 (94.60%) | 41,918,645 (89.75%) | 23,584,486 | 2,275,367 (4.84%) | 45.12 | 95.44 |
| T5 | 44,412,336 | 58,095,133 (93.23%) | 39,466,925 (88.80%) | 22,206,156 | 1,961,540 (4.43%) | 44.88 | 95.42 |
| T6 | 43,632,818 | 39,893,880 (91.41%) | 37,631,996 (86.55%) | 21,816,409 | 2,261,883 (5.03%) | 45.90 | 95.71 |
| T7 | 45,474,475 | 40,764,984 (89.65%) | 38,554,429 (84.79%) | 22,737,237 | 2,210,555 (4.86%) | 45.06 | 95.70 |
| T8 | 47,407,099 | 44,979,691 (94.88%) | 42,692,137 (90.05%) | 23,703,549 | 2,287,554 (4.83%) | 45.17 | 95.50 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iqbal, A.; Dong, Q.; Wang, X.; Gui, H.; Zhang, H.; Zhang, X.; Song, M. Transcriptome Analysis Reveals Differences in Key Genes and Pathways Regulating Carbon and Nitrogen Metabolism in Cotton Genotypes under N Starvation and Resupply. Int. J. Mol. Sci. 2020, 21, 1500. https://doi.org/10.3390/ijms21041500
Iqbal A, Dong Q, Wang X, Gui H, Zhang H, Zhang X, Song M. Transcriptome Analysis Reveals Differences in Key Genes and Pathways Regulating Carbon and Nitrogen Metabolism in Cotton Genotypes under N Starvation and Resupply. International Journal of Molecular Sciences. 2020; 21(4):1500. https://doi.org/10.3390/ijms21041500
Chicago/Turabian StyleIqbal, Asif, Qiang Dong, Xiangru Wang, Huiping Gui, Hengheng Zhang, Xiling Zhang, and Meizhen Song. 2020. "Transcriptome Analysis Reveals Differences in Key Genes and Pathways Regulating Carbon and Nitrogen Metabolism in Cotton Genotypes under N Starvation and Resupply" International Journal of Molecular Sciences 21, no. 4: 1500. https://doi.org/10.3390/ijms21041500
APA StyleIqbal, A., Dong, Q., Wang, X., Gui, H., Zhang, H., Zhang, X., & Song, M. (2020). Transcriptome Analysis Reveals Differences in Key Genes and Pathways Regulating Carbon and Nitrogen Metabolism in Cotton Genotypes under N Starvation and Resupply. International Journal of Molecular Sciences, 21(4), 1500. https://doi.org/10.3390/ijms21041500