Review of Three-Dimensional Liquid Chromatography Platforms for Bottom-Up Proteomics



Abstract
:1. Introduction
2. Recent Developments and Applications of 3D-LC
2.1. Offline 3D-LC Systems
2.2. Online 3D-LC Systems
2.3. Combined Offline-Online 3D-LC Systems
2.4. Comparison of Offline and Online Setups
3. The Power of 3D-LC Compared to 1D and 2D-LC
4. Author’s Outlook and Concluding Remarks
- –
- Should we perform an MD-LC method? If yes, is a 2D-LC or a 3D-LC method suitable?
- –
- Which LC modes should be combined?
- –
- If it is a 3D-LC system, should it be offline, online, or combined?
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PTMs | Post-translational modifications |
| MD | Multidimensional |
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| MudPIT | Multidimensional protein identification technology |
| SCX | Strong cation exchange |
| RPLC | Reversed phase liquid chromatography |
| SAX | Strong anion exchange |
| SEC | Size exclusion chromatography |
| HILIC | Hydrophilic interaction chromatography |
| CZE | Capillary zone electrophoresis |
| GELFrEE | Gel-eluted liquid fraction entrapment electrophoresis |
| LPIEF | Liquid-phase isoelectric focusing |
| ERLIC | Electrostatic repulsion hydrophilic interaction chromatography |
| ZIC | Zwitterion–ion |
| StageTips | Stop and go extraction tips |
| iST | in-StageTip |
| 3D-SISPROT | 3D peptide fractionation technology |
| SILAC | Stable isotope labeling by amino acids in cell culture |
| TMT | Tandem mass tags |
| iTRAQ | Isobaric tags for relative and absolute quantitation |
| DEEP SEQ | DEep Efficient Peptide SEquencing and Quantification |
| PASEF | Parallel accumulation–serial fragmentation |
| SWATH | Sequential window acquisition of all theoretical spectra |
| DIA | Data-independent acquisition |
References
- Tyers, M.; Mann, M. From genomics to proteomics. Nature 2003, 422, 193–197. [Google Scholar] [CrossRef]
- Larance, M.; Lamond, A.I. Multidimensional proteomics for cell biology. Nat. Rev. Mol. Cell Biol. 2015, 16, 269–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youngblood, H.; Robinson, R.; Sharma, A.; Sharma, S. Proteomic Biomarkers of Retinal Inflammation in Diabetic Retinopathy. Int. J. Mol. Sci. 2019, 20, 4755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalita-de Croft, P.; Straube, J.; Lim, M.; Al-Ejeh, F.; Lakhani, S.R.; Saunus, J.M. Proteomic Analysis of the Breast Cancer Brain Metastasis Microenvironment. Int. J. Mol. Sci. 2019, 20, 2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, S.A.; Gillett, D.A.; D’Alton, S.V.; Hamm, M.J.; Abisambra, J.F. Proteomic Techniques to Examine Neuronal Translational Dynamics. Int. J. Mol. Sci. 2019, 20, 3524. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Yuan, H.; Zhang, L.; Zhang, Y. Recent advances on multidimensional liquid chromatography–mass spectrometry for proteomics: From qualitative to quantitative analysis—A review. Anal. Chim. Acta 2012, 731, 1–10. [Google Scholar] [CrossRef]
- Toby, T.K.; Fornelli, L.; Kelleher, N.L. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Tucholski, T.M.; Gregorich, Z.R.; Ge, Y. Top-down Proteomics: Technology Advancements and Applications to Heart Diseases. Expert Rev. Proteom. 2016, 13, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Gregorich, Z.R.; Ge, Y. Top-down proteomics in health and disease: Challenges and opportunities. Proteomics 2014, 14, 1195–1210. [Google Scholar] [CrossRef]
- Gillet, L.C.; Leitner, A.; Aebersold, R. Mass Spectrometry Applied to Bottom-Up Proteomics: Entering the High-Throughput Era for Hypothesis Testing. Annu. Rev. Anal. Chem. 2016, 9, 449–472. [Google Scholar] [CrossRef]
- Mayne, J.; Ning, Z.; Zhang, X.; Starr, A.E.; Chen, R.; Deeke, S.; Chiang, C.-K.; Xu, B.; Wen, M.; Cheng, K.; et al. Bottom-Up Proteomics (2013–2015): Keeping up in the Era of Systems Biology. Anal. Chem. 2016, 88, 95–121. [Google Scholar] [CrossRef] [PubMed]
- Blein-Nicolas, M.; Zivy, M. Thousand and one ways to quantify and compare protein abundances in label-free bottom-up proteomics. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2016, 1864, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by Mass Spectrometry: Approaches, Advances, and Applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, N.M.; Coon, J.J. The Role of Electron Transfer Dissociation in Modern Proteomics. Anal. Chem. 2018, 90, 40–64. [Google Scholar] [CrossRef]
- Andrews, G.L.; Simons, B.L.; Young, J.B.; Hawkridge, A.M.; Muddiman, D.C. Performance Characteristics of a New Hybrid Quadrupole Time-of-Flight Tandem Mass Spectrometer (TripleTOF 5600). Anal. Chem. 2011, 83, 5442–5446. [Google Scholar] [CrossRef] [Green Version]
- Muntel, J.; Xuan, Y.; Berger, S.T.; Reiter, L.; Bachur, R.; Kentsis, A.; Steen, H. Advancing Urinary Protein Biomarker Discovery by Data-Independent Acquisition on a Quadrupole-Orbitrap Mass Spectrometer. J. Proteome Res. 2015, 14, 4752–4762. [Google Scholar] [CrossRef] [Green Version]
- Bayat, P.; Lesage, D.; Cole, R.B. Low-energy collision-induced dissociation (low-energy CID), collision-induced dissociation (CID), and higher energy collision dissociation (HCD) mass spectrometry for structural elucidation of saccharides and clarification of their dissolution mechanism in DMAc/LiCl. J. Mass Spectrom. 2018, 53, 705–716. [Google Scholar] [CrossRef]
- Di Palma, S.; Hennrich, M.L.; Heck, A.J.R.; Mohammed, S. Recent advances in peptide separation by multidimensional liquid chromatography for proteome analysis. J. Proteom. 2012, 75, 3791–3813. [Google Scholar] [CrossRef]
- Ranjbar, L.; Foley, J.P.; Breadmore, M.C. Multidimensional liquid-phase separations combining both chromatography and electrophoresis—A review. Anal. Chim. Acta 2017, 950, 7–31. [Google Scholar] [CrossRef]
- Vivó-Truyols, G.; van der Wal, S.; Schoenmakers, P.J. Comprehensive Study on the Optimization of Online Two-Dimensional Liquid Chromatographic Systems Considering Losses in Theoretical Peak Capacity in First- and Second-Dimensions: A Pareto-Optimality Approach. Anal. Chem. 2010, 82, 8525–8536. [Google Scholar] [CrossRef]
- Li, X.; Stoll, D.R.; Carr, P.W. Equation for Peak Capacity Estimation in Two-Dimensional Liquid Chromatography. Anal. Chem. 2009, 81, 845–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Washburn, M.P.; Wolters, D.; Yates, J.R. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001, 19, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Tao, D.; Qiao, X.; Sun, L.; Hou, C.; Gao, L.; Zhang, L.; Shan, Y.; Liang, Z.; Zhang, Y. Development of a Highly Efficient 2-D System with a Serially Coupled Long Column and Its Application in Identification of Rat Brain Integral Membrane Proteins with Ionic Liquids-Assisted Solubilization and Digestion. J. Proteome Res. 2011, 10, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Millioni, R.; Tolin, S.; Puricelli, L.; Sbrignadello, S.; Fadini, G.P.; Tessari, P.; Arrigoni, G. High Abundance Proteins Depletion vs Low Abundance Proteins Enrichment: Comparison of Methods to Reduce the Plasma Proteome Complexity. PLoS ONE 2011, 6, e19603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.-Z.; Li, N.; Wang, Y.-T.; Liu, N.; Guo, M.-Q.; Sun, B.-q.; Zhou, H.; Liu, L.; Wu, J.-L. Acid/Salt/pH Gradient Improved Resolution and Sensitivity in Proteomics Study Using 2D SCX-RP LC–MS. J. Proteome Res. 2017, 16, 3470–3475. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, S.; Zhang, Y.; Chen, S.; Liu, P.; Liu, B. An off-line high pH reversed-phase fractionation and nano-liquid chromatography–mass spectrometry method for global proteomic profiling of cell lines. J. Chromatogr. B 2015, 974, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Vonk, R.J.; Gargano, A.F.G.; Davydova, E.; Dekker, H.L.; Eeltink, S.; de Koning, L.J.; Schoenmakers, P.J. Comprehensive Two-Dimensional Liquid Chromatography with Stationary-Phase-Assisted Modulation Coupled to High-Resolution Mass Spectrometry Applied to Proteome Analysis of Saccharomyces cerevisiae. Anal. Chem. 2015, 87, 5387–5394. [Google Scholar] [CrossRef]
- Chen, W.; Wang, S.; Adhikari, S.; Deng, Z.; Wang, L.; Chen, L.; Ke, M.; Yang, P.; Tian, R. Simple and Integrated Spintip-Based Technology Applied for Deep Proteome Profiling. Anal. Chem. 2016, 88, 4864–4871. [Google Scholar] [CrossRef]
- Zhou, H.; Hou, W.; Lambert, J.-P.; Tian, R.; Figeys, D. Analysis of low-abundance proteins using the proteomic reactor with pH fractionation. Talanta 2010, 80, 1526–1531. [Google Scholar] [CrossRef]
- Lee, H.-J.; Na, K.; Kwon, M.-S.; Park, T.; Kim, K.S.; Kim, H.; Paik, Y.-K. A new versatile peptide-based size exclusion chromatography platform for global profiling and quantitation of candidate biomarkers in hepatocellular carcinoma specimens. Proteomics 2011, 11, 1976–1984. [Google Scholar] [CrossRef]
- Garbis, S.D.; Roumeliotis, T.I.; Tyritzis, S.I.; Zorpas, K.M.; Pavlakis, K.; Constantinides, C.A. A Novel Multidimensional Protein Identification Technology Approach Combining Protein Size Exclusion Prefractionation, Peptide Zwitterion−Ion Hydrophilic Interaction Chromatography, and Nano-Ultraperformance RP Chromatography/nESI-MS2 for the in-Depth Analysis of the Serum Proteome and Phosphoproteome: Application to Clinical Sera Derived from Humans with Benign Prostate Hyperplasia. Anal. Chem. 2011, 83, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Di Palma, S.; Stange, D.; van de Wetering, M.; Clevers, H.; Heck, A.J.R.; Mohammed, S. Highly Sensitive Proteome Analysis of FACS-Sorted Adult Colon Stem Cells. J. Proteome Res. 2011, 10, 3814–3819. [Google Scholar] [CrossRef] [PubMed]
- Di Palma, S.; Boersema, P.J.; Heck, A.J.R.; Mohammed, S. Zwitterionic Hydrophilic Interaction Liquid Chromatography (ZIC-HILIC and ZIC-cHILIC) Provide High Resolution Separation and Increase Sensitivity in Proteome Analysis. Anal. Chem. 2011, 83, 3440–3447. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.-L.; Wang, S.-S.; Hu, H.; He, C.-W.; Wan, J.-B.; Su, H.-X.; Wang, Y.-T.; Li, P. Online comprehensive two-dimensional hydrophilic interaction chromatography×reversed-phase liquid chromatography coupled with hybrid linear ion trap Orbitrap mass spectrometry for the analysis of phenolic acids in Salvia miltiorrhiza. J. Chromatogr. A 2018, 1536, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Wang, X.; Han, J.; Gao, F.; Xu, L.; Xiao, Z.; Bai, P.; Wang, Q.; Zhang, B. Two dimensional separations of human urinary protein digest using a droplet-interfaced platform. Anal. Chim. Acta 2015, 863, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Ye, M.; Han, G.; Jiang, X.; Wang, F.; Yu, Z.; Chen, R.; Zou, H. Reversed-Phase-Reversed-Phase Liquid Chromatography Approach with High Orthogonality for Multidimensional Separation of Phosphopeptides. Anal. Chem. 2010, 82, 53–56. [Google Scholar] [CrossRef]
- Donato, P.; Cacciola, F.; Sommella, E.; Fanali, C.; Dugo, L.; Dachà, M.; Campiglia, P.; Novellino, E.; Dugo, P.; Mondello, L. Online Comprehensive RPLC × RPLC with Mass Spectrometry Detection for the Analysis of Proteome Samples. Anal. Chem. 2011, 83, 2485–2491. [Google Scholar] [CrossRef]
- Dou, M.; Tsai, C.-F.; Piehowski, P.D.; Wang, Y.; Fillmore, T.L.; Zhao, R.; Moore, R.J.; Zhang, P.; Qian, W.-J.; Smith, R.D.; et al. Automated Nanoflow Two-Dimensional Reversed-Phase Liquid Chromatography System Enables In-Depth Proteome and Phosphoproteome Profiling of Nanoscale Samples. Anal. Chem. 2019, 91, 9707–9715. [Google Scholar] [CrossRef]
- Dou, M.; Zhu, Y.; Liyu, A.; Liang, Y.; Chen, J.; Piehowski, P.D.; Xu, K.; Zhao, R.; Moore, R.J.; Atkinson, M.A.; et al. Nanowell-mediated two-dimensional liquid chromatography enables deep proteome profiling of <1000 mammalian cells. Chem. Sci. 2018, 9, 6944–6951. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Mun, D.-G.; So, J.E.; Bae, J.; Kim, H.; Masselon, C.; Lee, S.-W. Efficient Exploitation of Separation Space in Two-Dimensional Liquid Chromatography System for Comprehensive and Efficient Proteomic Analyses. Anal. Chem. 2016, 88, 11734–11741. [Google Scholar] [CrossRef]
- Horvatovich, P.; Hoekman, B.; Govorukhina, N.; Bischoff, R. Multidimensional chromatography coupled to mass spectrometry in analysing complex proteomics samples. J. Sep. Sci. 2010, 33, 1421–1437. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Jiang, B.; Zhao, B.; Zhang, L.; Zhang, Y. Recent Advances in Multidimensional Separation for Proteome Analysis. Anal. Chem. 2019, 91, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Malerod, H.; Lundanes, E.; Greibrokk, T. Recent advances in on-line multidimensional liquid chromatography. Anal. Methods 2010, 2, 110–122. [Google Scholar] [CrossRef]
- Zhang, X.; Fang, A.; Riley, C.P.; Wang, M.; Regnier, F.E.; Buck, C. Multi-dimensional liquid chromatography in proteomics—A review. Anal. Chim. Acta 2010, 664, 101–113. [Google Scholar] [CrossRef] [Green Version]
- van de Meent, M.H.M.; de Jong, G.J. Novel liquid-chromatography columns for proteomics research. TrAC Trends Anal. Chem. 2011, 30, 1809–1818. [Google Scholar] [CrossRef]
- Makarov, A.; Scigelova, M. Coupling liquid chromatography to Orbitrap mass spectrometry. J. Chromatogr. A 2010, 1217, 3938–3945. [Google Scholar] [CrossRef] [Green Version]
- Tao, D.; Zhang, L.; Shan, Y.; Liang, Z.; Zhang, Y. Recent advances in micro-scale and nano-scale high-performance liquid-phase chromatography for proteome research. Anal. Bioanal. Chem. 2011, 399, 229–241. [Google Scholar] [CrossRef]
- Motoyama, A.; Xu, T.; Ruse, C.I.; Wohlschlegel, J.A.; Yates, J.R. Anion and Cation Mixed-Bed Ion Exchange for Enhanced Multidimensional Separations of Peptides and Phosphopeptides. Anal. Chem. 2007, 79, 3623–3634. [Google Scholar] [CrossRef] [Green Version]
- Wolters, D.A.; Washburn, M.P.; Yates, J.R. An Automated Multidimensional Protein Identification Technology for Shotgun Proteomics. Anal. Chem. 2001, 73, 5683–5690. [Google Scholar] [CrossRef]
- Dai, J.; Jin, W.-H.; Sheng, Q.-H.; Shieh, C.-H.; Wu, J.-R.; Zeng, R. Protein Phosphorylation and Expression Profiling by Yin-Yang Multidimensional Liquid Chromatography (Yin-Yang MDLC) Mass Spectrometry. J. Proteome Res. 2007, 6, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Shen, X.; Sun, L. Strong cation exchange-reversed phase liquid chromatography-capillary zone electrophoresis-tandem mass spectrometry platform with high peak capacity for deep bottom-up proteomics. Anal. Chim. Acta 2018, 1012, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lorne Burke, T.W.; Mant, C.T.; Black, J.A.; Hodges, R.S. Strong cation-exchange high-performance liquid chromatography of peptides: Effect of non-specific hydrophobic interactions and linearization of peptide retention behaviour. J. Chromatogr. A 1989, 476, 377–389. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, H.; Smith, K.; Wu, S. Two-dimensional separation using high-pH and low-pH reversed phase liquid chromatography for top-down proteomics. Int. J. Mass Spectrom. 2018, 427, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Hood, B.L.; Zhou, M.; Chan, K.C.; Lucas, D.A.; Kim, G.J.; Issaq, H.J.; Veenstra, T.D.; Conrads, T.P. Investigation of the Mouse Serum Proteome. J. Proteome Res. 2005, 4, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, X.; Gao, M.; Yang, P.; Zhang, X. Comparison of 2-D LC and 3-D LC with post- and pre-tryptic-digestion SEC fractionation for proteome analysis of normal human liver tissue. Proteomics 2007, 7, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Gilar, M.; Olivova, P.; Daly, A.E.; Gebler, J.C. Orthogonality of Separation in Two-Dimensional Liquid Chromatography. Anal. Chem. 2005, 77, 6426–6434. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Want, E.J.; Chen, W.; Keating, W.; Nussbaumer, W.; Moore, R.; Gentle, T.M.; Siuzdak, G. Sepsis Plasma Protein Profiling with Immunodepletion, Three-Dimensional Liquid Chromatography Tandem Mass Spectrometry, and Spectrum Counting. J. Proteome Res. 2006, 5, 3154–3160. [Google Scholar] [CrossRef]
- Betancourt, L.H.; De Bock, P.-J.; Staes, A.; Timmerman, E.; Perez-Riverol, Y.; Sanchez, A.; Besada, V.; Gonzalez, L.J.; Vandekerckhove, J.; Gevaert, K. SCX charge state selective separation of tryptic peptides combined with 2D-RP-HPLC allows for detailed proteome mapping. J. Proteom. 2013, 91, 164–171. [Google Scholar] [CrossRef]
- Zhao, P.; Schulz, T.C.; Sherrer, E.S.; Weatherly, D.B.; Robins, A.J.; Wells, L. The human embryonic stem cell proteome revealed by multidimensional fractionation followed by tandem mass spectrometry. Proteomics 2015, 15, 554–566. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Li, M.; Yang, Y.; Guo, Z.; Sun, Y.; Shao, C.; Li, M.; Sun, W.; Gao, Y. A comprehensive analysis and annotation of human normal urinary proteome. Sci. Rep. 2017, 7, 3024. [Google Scholar] [CrossRef]
- Spicer, V.; Ezzati, P.; Neustaeter, H.; Beavis, R.C.; Wilkins, J.A.; Krokhin, O.V. 3D HPLC-MS with Reversed-Phase Separation Functionality in All Three Dimensions for Large-Scale Bottom-Up Proteomics and Peptide Retention Data Collection. Anal. Chem. 2016, 88, 2847–2855. [Google Scholar] [CrossRef] [PubMed]
- Boichenko, A.P.; Govorukhina, N.; van der Zee, A.G.J.; Bischoff, R. Multidimensional separation of tryptic peptides from human serum proteins using reversed-phase, strong cation exchange, weak anion exchange, and fused-core fluorinated stationary phases. J. Sep. Sci. 2013, 36, 3463–3470. [Google Scholar] [CrossRef] [PubMed]
- Loroch, S.; Schommartz, T.; Brune, W.; Zahedi, R.P.; Sickmann, A. Multidimensional electrostatic repulsion–hydrophilic interaction chromatography (ERLIC) for quantitative analysis of the proteome and phosphoproteome in clinical and biomedical research. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2015, 1854, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Bandhakavi, S.; Stone, M.D.; Onsongo, G.; Van Riper, S.K.; Griffin, T.J. A Dynamic Range Compression and Three-Dimensional Peptide Fractionation Analysis Platform Expands Proteome Coverage and the Diagnostic Potential of Whole Saliva. J. Proteome Res. 2009, 8, 5590–5600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millioni, R.; Tolin, S.; Fadini, G.P.; Falda, M.; van Breukelen, B.; Tessari, P.; Arrigoni, G. High confidence and sensitivity four-dimensional fractionation for human plasma proteome analysis. Amino Acids 2012, 43, 2199–2202. [Google Scholar] [CrossRef]
- Alpert, A.J. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. J. Chromatogr. A 1990, 499, 177–196. [Google Scholar] [CrossRef]
- Zhou, H.; Di Palma, S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.R.; Mohammed, S. Toward a Comprehensive Characterization of a Human Cancer Cell Phosphoproteome. J. Proteome Res. 2013, 12, 260–271. [Google Scholar] [CrossRef]
- Pernikářová, V.; Sedláček, V.; Potěšil, D.; Procházková, I.; Zdráhal, Z.; Bouchal, P.; Kučera, I. Proteomic responses to a methyl viologen-induced oxidative stress in the wild type and FerB mutant strains of Paracoccus denitrificans. J. Proteom. 2015, 125, 68–75. [Google Scholar] [CrossRef]
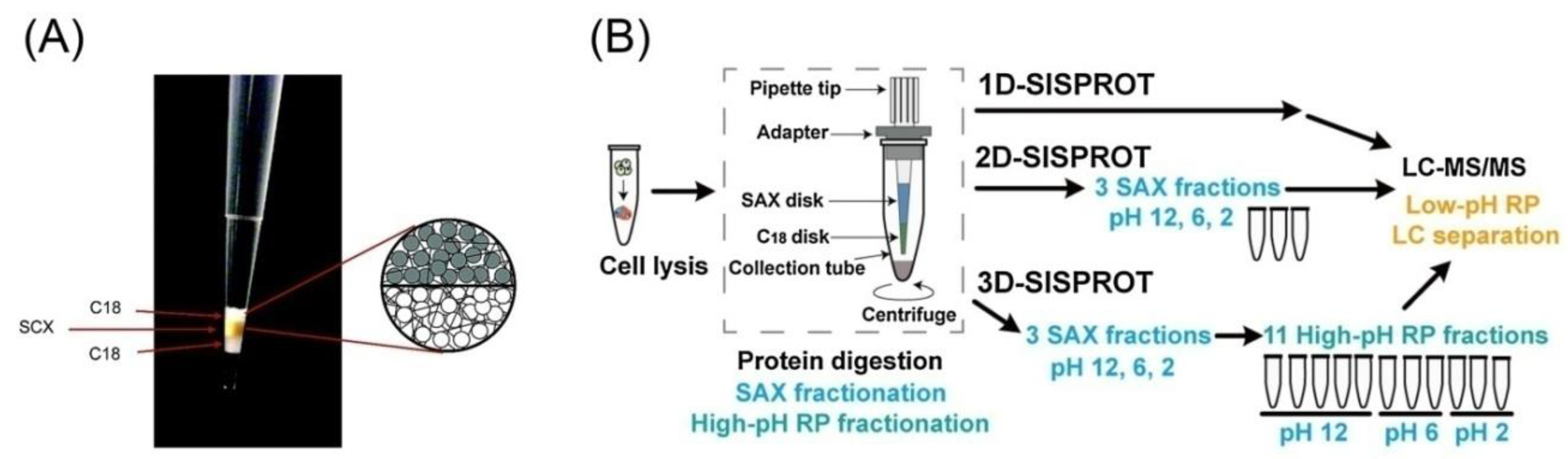
- Chen, W.; Adhikari, S.; Chen, L.; Lin, L.; Li, H.; Luo, S.; Yang, P.; Tian, R. 3D-SISPROT: A simple and integrated spintip-based protein digestion and three-dimensional peptide fractionation technology for deep proteome profiling. J. Chromatogr. A 2017, 1498, 207–214. [Google Scholar] [CrossRef]
- Xue, L.; Lin, L.; Zhou, W.; Chen, W.; Tang, J.; Sun, X.; Huang, P.; Tian, R. Mixed-mode ion exchange-based integrated proteomics technology for fast and deep plasma proteome profiling. J. Chromatogr. A 2018, 1564, 76–84. [Google Scholar] [CrossRef]
- Ishihama, Y.; Rappsilber, J.; Mann, M. Modular Stop and Go Extraction Tips with Stacked Disks for Parallel and Multidimensional Peptide Fractionation in Proteomics. J. Proteome Res. 2006, 5, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Adachi, J.; Hashiguchi, K.; Nagano, M.; Sato, M.; Sato, A.; Fukamizu, K.; Ishihama, Y.; Tomonaga, T. Improved Proteome and Phosphoproteome Analysis on a Cation Exchanger by a Combined Acid and Salt Gradient. Anal. Chem. 2016, 88, 7899–7903. [Google Scholar] [CrossRef] [PubMed]
- McDonald, W.H.; Ohi, R.; Miyamoto, D.T.; Mitchison, T.J.; Yates, J.R. Comparison of three directly coupled HPLC MS/MS strategies for identification of proteins from complex mixtures: Single-dimension LC-MS/MS, 2-phase MudPIT, and 3-phase MudPIT. Int. J. Mass Spectrom. 2002, 219, 245–251. [Google Scholar] [CrossRef]
- Wei, J.; Sun, J.; Yu, W.; Jones, A.; Oeller, P.; Keller, M.; Woodnutt, G.; Short, J.M. Global Proteome Discovery Using an Online Three-Dimensional LC−MS/MS. J. Proteome Res. 2005, 4, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chen, R.; Zhu, J.; Sun, D.; Song, C.; Wu, Y.; Ye, M.; Wang, L.; Zou, H. A Fully Automated System with Online Sample Loading, Isotope Dimethyl Labeling and Multidimensional Separation for High-Throughput Quantitative Proteome Analysis. Anal. Chem. 2010, 82, 3007–3015. [Google Scholar] [CrossRef]
- Song, C.; Wang, F.; Ye, M.; Cheng, K.; Chen, R.; Zhu, J.; Tan, Y.; Wang, H.; Figeys, D.; Zou, H. Improvement of the Quantification Accuracy and Throughput for Phosphoproteome Analysis by a Pseudo Triplex Stable Isotope Dimethyl Labeling Approach. Anal. Chem. 2011, 83, 7755–7762. [Google Scholar] [CrossRef]
- Xu, B.; Wang, F.; Song, C.; Sun, Z.; Cheng, K.; Tan, Y.; Wang, H.; Zou, H. Large-Scale Proteome Quantification of Hepatocellular Carcinoma Tissues by a Three-Dimensional Liquid Chromatography Strategy Integrated with Sample Preparation. J. Proteome Res. 2014, 13, 3645–3654. [Google Scholar] [CrossRef]
- Zhou, F.; Sikorski, T.W.; Ficarro, S.B.; Webber, J.T.; Marto, J.A. Online Nanoflow Reversed Phase-Strong Anion Exchange-Reversed Phase Liquid Chromatography–Tandem Mass Spectrometry Platform for Efficient and In-Depth Proteome Sequence Analysis of Complex Organisms. Anal. Chem. 2011, 83, 6996–7005. [Google Scholar] [CrossRef] [Green Version]
- Ficarro, S.B.; Zhang, Y.; Carrasco-Alfonso, M.J.; Garg, B.; Adelmant, G.; Webber, J.T.; Luckey, C.J.; Marto, J.A. Online Nanoflow Multidimensional Fractionation for High Efficiency Phosphopeptide Analysis. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Lu, Y.; Ficarro, S.B.; Adelmant, G.; Jiang, W.; Luckey, C.J.; Marto, J.A. Genome-scale proteome quantification by DEEP SEQ mass spectrometry. Nat. Commun. 2013, 4, 2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, H.C.H.; Kong, R.P.W.; Szeto, S.S.W.; Zhao, Y.; Zhang, Z.; Wang, Y.; Li, G.; Quan, Q.; Lee, S.M.Y.; Lam, H.C.; et al. A versatile reversed phase-strong cation exchange-reversed phase (RP–SCX–RP) multidimensional liquid chromatography platform for qualitative and quantitative shotgun proteomics. Analyst 2015, 140, 1237–1252. [Google Scholar] [CrossRef] [PubMed]
- Fournier, M.L.; Gilmore, J.M.; Martin-Brown, S.A.; Washburn, M.P. Multidimensional Separations-Based Shotgun Proteomics. Chem. Rev. 2007, 107, 3654–3686. [Google Scholar] [CrossRef]
- Motoyama, A.; Yates, J.R. Multidimensional LC Separations in Shotgun Proteomics. Anal. Chem. 2008, 80, 7187–7193. [Google Scholar] [CrossRef] [Green Version]
- Guiochon, G.; Marchetti, N.; Mriziq, K.; Shalliker, R.A. Implementations of two-dimensional liquid chromatography. J. Chromatogr. A 2008, 1189, 109–168. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Kelstrup, C.D.; Batth, T.S.; Larsen, S.C.; Haldrup, C.; Bramsen, J.B.; Sørensen, K.D.; Høyer, S.; Ørntoft, T.F.; Andersen, C.L.; et al. An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 2017, 4, 587–599. [Google Scholar] [CrossRef] [Green Version]
- Meier, F.; Brunner, A.-D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation–Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell. Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef] [Green Version]
- Anjo, S.I.; Santa, C.; Manadas, B. SWATH-MS as a tool for biomarker discovery: From basic research to clinical applications. Proteomics 2017, 17, 1600278. [Google Scholar] [CrossRef]
- Kawashima, Y.; Watanabe, E.; Umeyama, T.; Nakajima, D.; Hattori, M.; Honda, K.; Ohara, O. Optimization of Data-Independent Acquisition Mass Spectrometry for Deep and Highly Sensitive Proteomic Analysis. Int. J. Mol. Sci. 2019, 20, 5932. [Google Scholar] [CrossRef] [Green Version]
- Keshishian, H.; Burgess, M.W.; Specht, H.; Wallace, L.; Clauser, K.R.; Gillette, M.A.; Carr, S.A. Quantitative, multiplexed workflow for deep analysis of human blood plasma and biomarker discovery by mass spectrometry. Nat. Protoc. 2017, 12, 1683–1701. [Google Scholar] [CrossRef] [PubMed]
- Mun, D.-G.; Bhin, J.; Kim, S.; Kim, H.; Jung, J.H.; Jung, Y.; Jang, Y.E.; Park, J.M.; Kim, H.; Jung, Y.; et al. Proteogenomic Characterization of Human Early-Onset Gastric Cancer. Cancer Cell 2019, 35, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| 3D-LC setup | Sample (Protein Amount) | MS | Identified Proteins * | Identified Unique Peptides | Fraction Number | MS Time (h) | Year, Reference |
|---|---|---|---|---|---|---|---|
| RP-SCX-RP | Human plasma (500 µg) | Agilent 1100 LC/MSD Trap | 484 | - | 40 | 100 | 2006, [57] |
| SEC-SCX-RP | Human liver (1 mg) | QSTARXL | 636 | 3451 | 120 | 206 | 2007, [55] |
| SEC-HILIC-RP | Human serum (7.4 mg) | Quadrupole ion trap | 1955 | - | 20 | 60 | 2011, [31] |
| ERLIC- RP- RP | Human serum (1.2 mg) | QTOF | 1088 | 208 | 144 | 216 | 2013, [62] |
| SCX-RP-RP | Mouse embryonic fibroblast (1 mg) | LTQ Orbitrap XL | 5051 | 29,843 | 30 | - | 2013, [58] |
| SCX-HILIC-RP | K562 cells (3 mg) | LTQ-Orbitrap Velos | 4708 | 22,148 | 63 | 126 | 2013, [67] |
| SCX-HILIC-RP | HeLa cells (500 µg) | LTQ-Orbitrap Velos | 3424 | 11,980 | 63 | 126 | 2013, [67] |
| SEC-HILIC-RP | Paracoccus denitrificans (8 mg) | Orbitrap Elite | 2627 | - | 36 | 66 | 2015, [68] |
| SCX-RP-RP | Human embryonic stem cell (-) | Thermo Finnigan LTQ | 3184 | ~24,000 | 100 | 117 | 2015, [59] |
| RP- RP- RP | Jurkat cells (720 µg) | TripleTOF 5600 | 14,230 | 251,166 | 126 | 189 | 2016, [61] |
| GELFrEE-RP-RP & LPIEF-RP-RP | Human urine (5 mg) | TripleTOF 5600 | 6085 | 68,151 | 360 | 720 | 2017, [60] |
| SAX-RP-RP | HEK 293T cells (30 µg) | Orbitrap Fusion | 8222 | 74,432 | 11 | 20.4 | 2017, [69] |
| SCX & SAX-RP-RP | Human serum (-) | Orbitrap Fusion | 862 | - | 12 | 12 | 2018, [70] |
| 3D-LC Setup | Sample (Amount) | MS | Identified Proteins * | Identified Unique Peptides | Fraction Number | MS Time (h) | Year, Reference |
|---|---|---|---|---|---|---|---|
| RP-SCX-RP | Saccharomyces cerevisiae (200 µg) | LCQ Deca XP | ~1900 | - | 60 | 140 | 2005, [76] |
| RP-SAX-RP | Saccharomyces cerevisiae (5 µg) | Orbitrap XL | 4004 | 26,468 | 101 | 202 | 2011, [80] |
| 3821 | 25,091 | 51 | 102 | 2011, [80] | |||
| RP-SAX-RP | Murine embryonic stem cells (25 µg) | Orbitrap XL | 11,352 | 211,535 | 20 | 580 | 2013, [82] |
| RP-SCX-RP | PC12 cell (100 µg) | AB Sciex QSTAR XL QTOF | 6345 | 97,309 | 12 | 24 | 2015, [83] |
| Pre-Fractionation | LC Setup | Sample (Amount) | MS | Identified Proteins * | Identified Unique Peptides | Fraction Number | MS Time (h) | Year, Reference |
|---|---|---|---|---|---|---|---|---|
| RP | SCX-RP | Human liver (200 µg) | LTQ-Orbitrap Velos | 2759 | - | 36 | 124 | 2014, [79] |
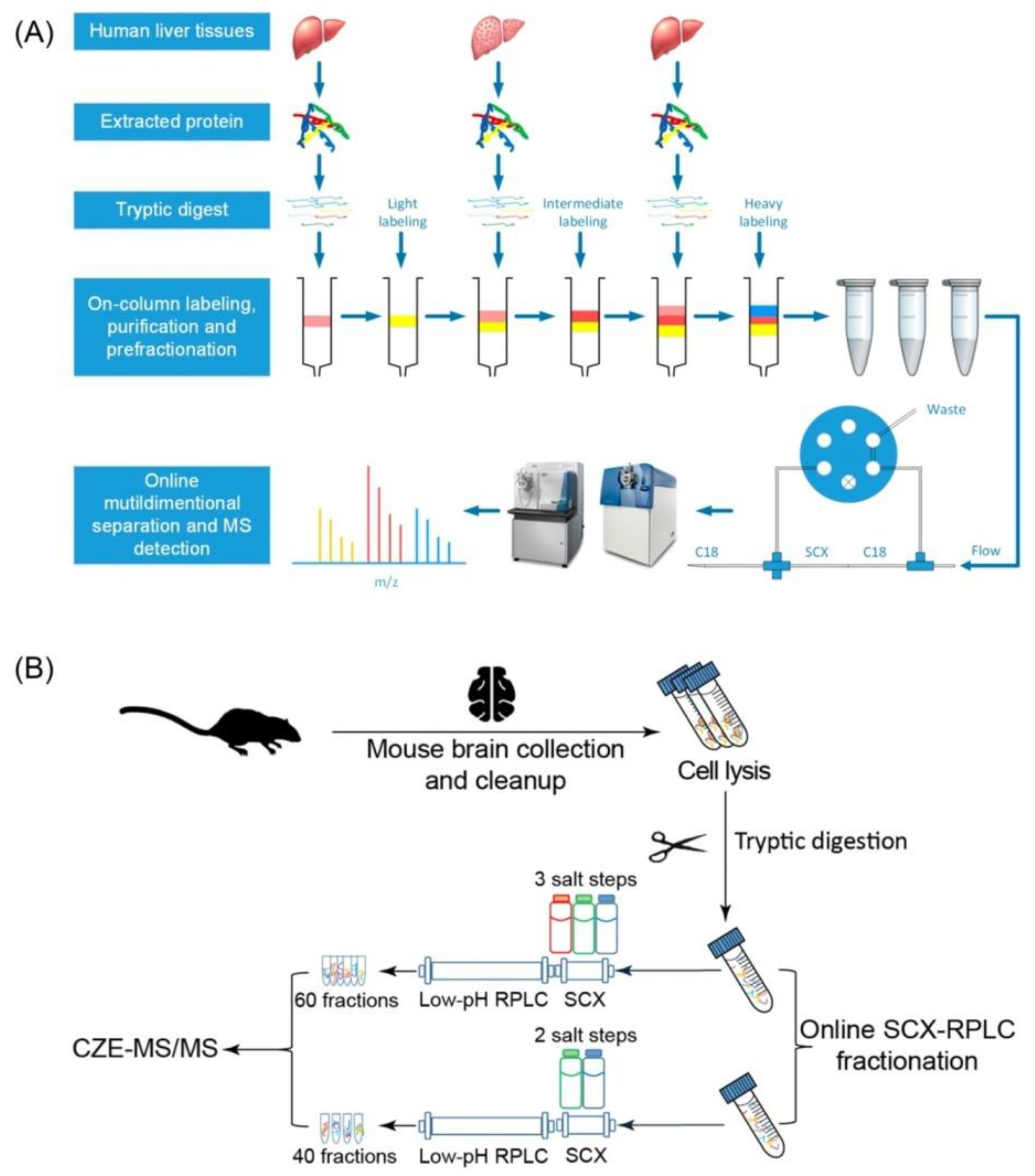
| SCX-RP | CZE | Mouse brain (500 µg) | Q-Exactive HF | 8200 | 65,000 | 60 | 70 | 2018, [51] |
| Feature | Offline 3D-LC | Online 3D-LC |
|---|---|---|
| Setup | Simple | Usually sophisticated |
| Ability to transfer technologies to other laboratories | High | Low-medium |
| Sample amount | High, up to several mg Not suitable for samples with a limited amount | Low, from several µg to 100 µg Allow only low-amount samples |
| Operation | Separated dimensions, simple and flexible to operate | Usually full-automation, well-trained skill required |
| Sample pooling | Allow | Usually not allow |
| Sample handling | High | Low |
| Sample loss | High-medium | Low |
| Sample | MS | Paramater | 1D | 2D | 3D | Reference |
|---|---|---|---|---|---|---|
| HEK 293T cells | Orbitrap Fusion | Sample amount (µg) | 6 | 30 | 30 | [69] |
| Setup | RP | SAX-RP | SAX-RP-RP | |||
| Id. proteins | 4275 ± 26 | 5380 ± 88 | 8222 ± 109 | |||
| Id. unique peptides | 19,768 ± 333 | 34,912 ± 925 | 74,432 ± 996 | |||
| Fraction number | 1 | 3 | 11 | |||
| MS time (h) | 1.4 | 4.2 | 20.4 | |||
| Jurkat cells | TripleTOF 5600 | Sample amount (µg) | 1 | 200 | 720 | [61] |
| Setup | RP | RP-RP | RP-RP-RP | |||
| Id. proteins | 2568 | 8757 | 14,230 | |||
| Id. unique peptides | 11,878 | 109,461 | 251,166 | |||
| Fraction number | 1 | 21 | 126 | |||
| MS time (h) | 1.5 | 31.5 | 189 | |||
| HeLa cells & K562 cells | LTQ-Orbitrap Velos | Sample amount (µg) * | 125 and 750 | 500 and 3000 | 500 | [67] |
| Setup | RP | HILIC-RP | SCX-HILIC-RP | |||
| Unique phos.pro. * | 1570 and 1763 | 3891 and 4132 | 3424 and 4708 | |||
| Unique phos.pep. * | 3726 and 4104 | 16,637 and 16,722 | 11,980 and 22,148 | |||
| Fraction number | 1 | 20 | 63 | |||
| MS time (h) | 2 | 40 | 126 | |||
| Mouse embryonic fibroblast | AB Sciex QSTAR XL QTOF | Sample amount (µg) | 15 | 15 | [83] | |
| Setup | RP-RP | RP-SCX-RP | ||||
| Id. proteins | 2358 | 3114 | ||||
| Id. unique peptides | 13,573 | 25,792 | ||||
| Human urine | TripleTOF 5600 | Sample amount (µg) | 20 | 200 | 5000 | [60] |
| Setup | RP | RP-RP | GELFrEE-RP-RP and LPIEF-RP-RP | |||
| Id. proteins | 808 | 3162 | 6085 | |||
| Id. unique peptides | 13,895 | 25,940 | 68,151 | |||
| Fraction number | 1 | 20 | 360 | |||
| MS time (h) | 2 × 3 | 40 | 720 | |||
| Mouse brain | Q-Exactive HF | Sample amount (µg) | 500 | 500 | [51] | |
| Setup | RP-RP | SCX-RP-CZE | ||||
| Id. proteins | 8900 | 8200 | ||||
| Id. unique peptides | 70,000 | 65,000 | ||||
| Fraction number | 30 | 60 | ||||
| MS time (h) | 60 | 70 | ||||
| E. coli | LCQ Deca XP ion trap | Sample amount (µg) | 20 | 20 | [80] | |
| Setup | RP-RP | RP-SAX-RP | ||||
| Id. proteins | 702 | 923 | ||||
| Id. unique peptides | 2923 | 4254 | ||||
| Fraction number | 40 | 37 | ||||
| MS time (h) | 80 | 74 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duong, V.-A.; Park, J.-M.; Lee, H. Review of Three-Dimensional Liquid Chromatography Platforms for Bottom-Up Proteomics. Int. J. Mol. Sci. 2020, 21, 1524. https://doi.org/10.3390/ijms21041524
Duong V-A, Park J-M, Lee H. Review of Three-Dimensional Liquid Chromatography Platforms for Bottom-Up Proteomics. International Journal of Molecular Sciences. 2020; 21(4):1524. https://doi.org/10.3390/ijms21041524
Chicago/Turabian StyleDuong, Van-An, Jong-Moon Park, and Hookeun Lee. 2020. "Review of Three-Dimensional Liquid Chromatography Platforms for Bottom-Up Proteomics" International Journal of Molecular Sciences 21, no. 4: 1524. https://doi.org/10.3390/ijms21041524
APA StyleDuong, V. -A., Park, J. -M., & Lee, H. (2020). Review of Three-Dimensional Liquid Chromatography Platforms for Bottom-Up Proteomics. International Journal of Molecular Sciences, 21(4), 1524. https://doi.org/10.3390/ijms21041524