Disparity between Inter-Patient Molecular Heterogeneity and Repertoires of Target Drugs Used for Different Types of Cancer in Clinical Oncology

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Results
2.1. Biosample Sets
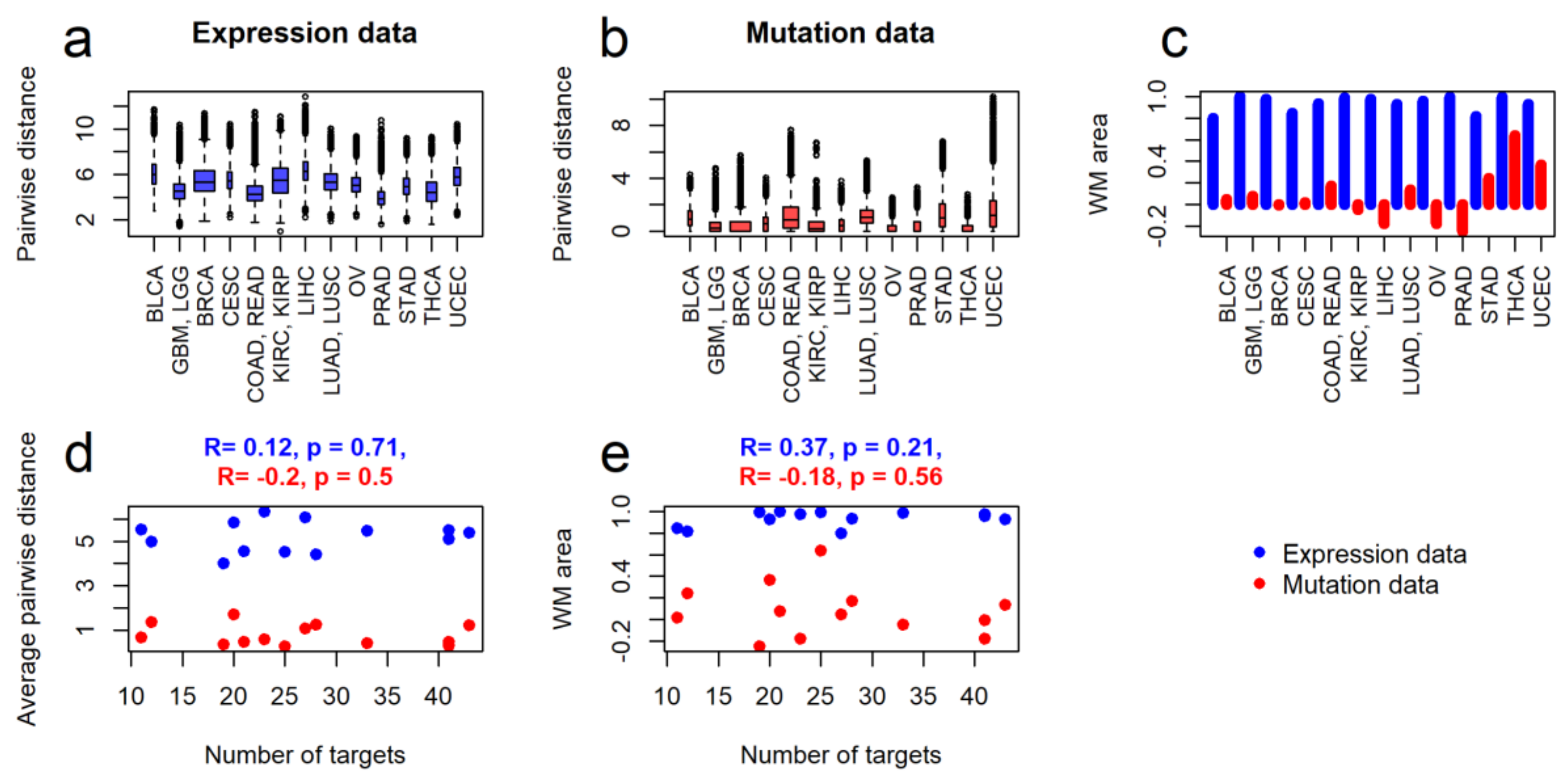
2.2. Intertumoral Heterogeneities
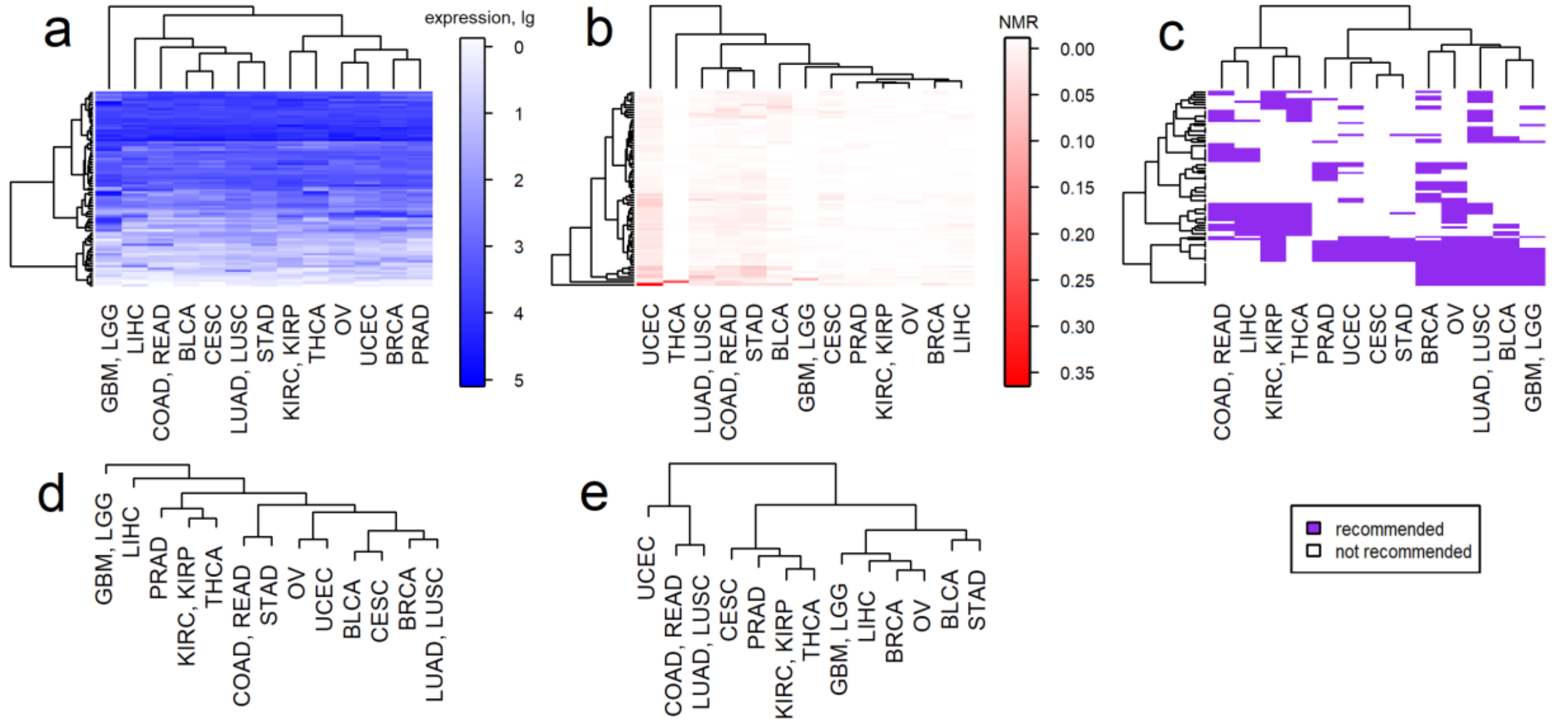
2.3. Clustering of Cancer Types in Relation with Recommended Targeted Therapeutics and Molecular Data
3. Discussion
4. Materials and Methods
4.1. Tumor Samples
4.2. Mutation Data
4.3. Gene Expression Data
4.4. Clinical Utility of Drugs and Molecular Targets
4.5. Clustering Parameters and Quality
4.6. Data Presentation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FDA | US Food and Drug Administration |
| NMRNCCN | Normalized mutation rateNational Comprehensive Cancer Network |
| TCGA | The Cancer Genome Atlas |
| WM method | Watermelon Multisection method |
References
- Bedard, P.L.; Hansen, A.R.; Ratain, M.J.; Siu, L.L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Jamal-Hanjani, M.; Quezada, S.A.; Larkin, J.; Swanton, C. Translational Implications of Tumor Heterogeneity. Clin. Cancer Res. 2015, 21, 1258. [Google Scholar] [CrossRef] [Green Version]
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, T.; Yaguchi, T.; Kawakami, Y. Inter-patient and Intra-tumor Heterogeneity in the Sensitivity to Tumor-targeted Immunity in Colorectal Cancer. Japanese J. Clin. Immunol. 2017, 40, 54–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusnir, M.; Cavalcante, L. Inter-tumor heterogeneity. Hum. Vaccin. Immunother. 2012, 8, 1143–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Merajver, S.D.; Li, J.Z. Cancer classification in the genomic era: Five contemporary problems. Hum. Genomics 2015, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Harada, S.; Arend, R.; Dai, Q.; Levesque, J.A.; Winokur, T.S.; Guo, R.; Heslin, M.J.; Nabell, L.; Nabors, L.B.; Limdi, N.A.; et al. Implementation and utilization of the molecular tumor board to guide precision medicine. Oncotarget 2017, 8, 57845–57854. [Google Scholar] [CrossRef] [Green Version]
- Shabani Azim, F.; Houri, H.; Ghalavand, Z.; Nikmanesh, B. Next Generation Sequencing in Clinical Oncology: Applications, Challenges and Promises: A Review Article. Iran. J. Public Health 2018, 47, 1453–1457. [Google Scholar]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. (Poznan, Poland) 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- International Cancer Genome Consortium, T.I.C.G.; Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Suntsova, M.; Gaifullin, N.; Allina, D.; Reshetun, A.; Li, X.; Mendeleeva, L.; Surin, V.; Sergeeva, A.; Spirin, P.; Prassolov, V.; et al. Atlas of RNA sequencing profiles for normal human tissues. Sci. Data 2019, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Padma, V.V. An overview of targeted cancer therapy. BioMedicine 2015, 5, 19. [Google Scholar] [CrossRef]
- Yan, L.; Rosen, N.; Arteaga, C. Targeted cancer therapies. Chin. J. Cancer 2011, 30, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Joo, W.D.; Visintin, I.; Mor, G. Targeted cancer therapy--are the days of systemic chemotherapy numbered? Maturitas 2013, 76, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Nahta, R.; Esteva, F.J. Trastuzumab: Triumphs and tribulations. Oncogene 2007, 26, 3637–3643. [Google Scholar] [CrossRef] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Gridelli, C.; De Marinis, F.; Di Maio, M.; Cortinovis, D.; Cappuzzo, F.; Mok, T. Gefitinib as first-line treatment for patients with advanced non-small-cell lung cancer with activating epidermal growth factor receptor mutation: Review of the evidence. Lung Cancer 2011, 71, 249–257. [Google Scholar] [CrossRef]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and Safety of a Specific Inhibitor of the BCR-ABL Tyrosine Kinase in Chronic Myeloid Leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef] [Green Version]
- Gerber, D.E. Targeted therapies: A new generation of cancer treatments. Am. Fam. Physician 2008, 77, 311–319. [Google Scholar]
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef]
- Tan, C.; Du, X. KRAS mutation testing in metastatic colorectal cancer. World J. Gastroenterol. 2012, 18, 5171–5180. [Google Scholar]
- U S Food and Drug Administration Home Page. Available online: https://www.fda.gov/ (accessed on 15 October 2018).
- Principles for Codevelopment of an In Vitro Companion Diagnostic Device with a Therapeutic Product. Available online: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/de (accessed on 22 July 2019).
- Zolotovskaia, M.A.; Sorokin, M.I.; Emelianova, A.A.; Borisov, N.M.; Kuzmin, D.V.; Borger, P.; Garazha, A.V.; Buzdin, A.A. Pathway Based Analysis of Mutation Data Is Efficient for Scoring Target Cancer Drugs. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artemov, A.; Aliper, A.; Korzinkin, M.; Lezhnina, K.; Jellen, L.; Zhukov, N.; Roumiantsev, S.; Gaifullin, N.; Zhavoronkov, A.; Borisov, N.; et al. A method for predicting target drug efficiency in cancer based on the analysis of signaling pathway activation. Oncotarget 2015, 6, 29347–29356. [Google Scholar] [CrossRef]
- Buzdin, A.A.; Zhavoronkov, A.A.; Korzinkin, M.B.; Venkova, L.S.; Zenin, A.A.; Smirnov, P.Y.; Borisov, N.M. Oncofinder, a new method for the analysis of intracellular signaling pathway activation using transcriptomic data. Front. Genet. 2014, 5, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzdin, A.; Sorokin, M.; Poddubskaya, E.; Borisov, N. High-Throughput Mutation Data Now Complement Transcriptomic Profiling: Advances in Molecular Pathway Activation Analysis Approach in Cancer Biology. Cancer Inform. 2019, 18, 1176935119838844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poddubskaya, E.V.; Baranova, M.P.; Allina, D.O.; Sekacheva, M.I.; Makovskaia, L.A.; Kamashev, D.E.; Suntsova, M.V.; Barbara, V.S.; Kochergina-Nikitskaya, I.N.; Aleshin, A.A. Personalized prescription of imatinib in recurrent granulosa cell tumor of the ovary: Case report. Mol. Case Stud. 2019, 5, a003434. [Google Scholar] [CrossRef]
- Koh, W.-J.; Abu-Rustum, N.R.; Bean, S.; Bradley, K.; Campos, S.M.; Cho, K.R.; Chon, H.S.; Chu, C.; Clark, R.; Cohn, D.; et al. Cervical Cancer, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 64–84. [Google Scholar] [CrossRef] [Green Version]
- Mohler, J.L.; Antonarakis, E.S.; Armstrong, A.J.; D’Amico, A.V.; Davis, B.J.; Dorff, T.; Eastham, J.A.; Enke, C.A.; Farrington, T.A.; Higano, C.S.; et al. Prostate Cancer, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 479–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Cederquist, L.; Chen, Y.-J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Engstrom, P.F.; et al. NCCN Guidelines Insights: Colon Cancer, Version 2.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettinger, D.S.; Aisner, D.L.; Wood, D.E.; Akerley, W.; Bauman, J.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; Dilling, T.J.; Dobelbower, M.; et al. NCCN Guidelines Insights: Non–Small Cell Lung Cancer, Version 5.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 807–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, S.H.; Elias, A.D.; Gradishar, W.J. NCCN Guidelines Updates: Breast Cancer. J. Natl. Compr. Cancer Netw. 2018, 16, 605–610. [Google Scholar] [CrossRef] [Green Version]
- Nabors, L.B.; Portnow, J.; Ammirati, M.; Baehring, J.; Brem, H.; Butowski, N.; Fenstermaker, R.A.; Forsyth, P.; Hattangadi-Gluth, J.; Holdhoff, M.; et al. NCCN Guidelines Insights: Central Nervous System Cancers, Version 1.2017. J. Natl. Compr. Cancer Netw. 2017, 15, 1331–1345. [Google Scholar] [CrossRef]
- Koh, W.-J.; Abu-Rustum, N.R.; Bean, S.; Bradley, K.; Campos, S.M.; Cho, K.R.; Chon, H.S.; Chu, C.; Cohn, D.; Crispens, M.A.; et al. Uterine Neoplasms, Version 1.2018, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2018, 16, 170–199. [Google Scholar] [CrossRef] [Green Version]
- Jonasch, E. NCCN Guidelines Updates: Management of Metastatic Kidney Cancer. J. Natl. Compr. Canc. Netw. 2019, 17, 587–589. [Google Scholar]
- Morgan, R.J.; Armstrong, D.K.; Alvarez, R.D.; Bakkum-Gamez, J.N.; Behbakht, K.; Chen, L.-M.; Copeland, L.; Crispens, M.A.; DeRosa, M.; Dorigo, O.; et al. Ovarian Cancer, Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2016, 14, 1134–1163. [Google Scholar] [CrossRef]
- Ajani, J.A.; D’Amico, T.A.; Almhanna, K.; Bentrem, D.J.; Chao, J.; Das, P.; Denlinger, C.S.; Fanta, P.; Farjah, F.; Fuchs, C.S.; et al. Gastric Cancer, Version 3.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2016, 14, 1286–1312. [Google Scholar] [CrossRef]
- Haddad, R.I.; Nasr, C.; Bischoff, L.; Busaidy, N.L.; Byrd, D.; Callender, G.; Dickson, P.; Duh, Q.-Y.; Ehya, H.; Goldner, W.; et al. NCCN Guidelines Insights: Thyroid Carcinoma, Version 2.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 1429–1440. [Google Scholar] [CrossRef] [Green Version]
- Flaig, T.W.; Spiess, P.E.; Agarwal, N.; Bangs, R.; Boorjian, S.A.; Buyyounouski, M.K.; Downs, T.M.; Efstathiou, J.A.; Friedlander, T.; Greenberg, R.E.; et al. NCCN Guidelines Insights: Bladder Cancer, Version 5.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 1041–1053. [Google Scholar] [CrossRef] [Green Version]
- Benson, A.B.; D’Angelica, M.I.; Abbott, D.E.; Abrams, T.A.; Alberts, S.R.; Saenz, D.A.; Are, C.; Brown, D.B.; Chang, D.T.; Covey, A.M.; et al. NCCN Guidelines Insights: Hepatobiliary Cancers, Version 1.2017. J. Natl. Compr. Canc. Netw. 2017, 15, 563–573. [Google Scholar] [CrossRef]
- Nagpal, S.; Thomas, L.; Recht, S. Advances in the management of glioblastoma: The role of temozolomide and MGMT testing. Clin. Pharmacol. Adv. Appl. 2012, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Zolotovskaia, M.A.; Sorokin, M.I.; Roumiantsev, S.A.; Borisov, N.M.; Buzdin, A.A. Pathway Instability Is an Effective New Mutation-Based Type of Cancer Biomarkers. Front. Oncol. 2019, 8. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Forbes, S.A.; Bhamra, G.; Bamford, S.; Dawson, E.; Kok, C.; Clements, J.; Menzies, A.; Teague, J.W.; Futreal, P.A.; Stratton, M.R. The Catalogue of Somatic Mutations in Cancer (COSMIC). In Current Protocols in Human Genetics; Haines, J.L., Korf, B.R., Morton, C.C., Seidman, C.E., Seidman, J.G., Smith, D.R., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; ISBN 978-0-471-14290-4. [Google Scholar]
- Efron, B.; Tibshirani, R. On testing the significance of sets of genes. Ann. Appl. Stat. 2007, 1, 107–129. [Google Scholar] [CrossRef] [Green Version]
- Brock, G.; Pihur, V.; Datta, S.; Datta, S. ClValid: An R package for cluster validation. J. Stat. Softw. 2008, 25, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Handl, J.; Knowles, J.; Kell, D.B. Computational cluster validation in post-genomic data analysis. Bioinformatics 2005, 21, 3201–3212. [Google Scholar] [CrossRef] [Green Version]
- Dunn, J.C. Well-Separated Clusters and Optimal Fuzzy Partitions. J. Cybern. 1974, 4, 95–104. [Google Scholar] [CrossRef]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Qiu, P.; Ji, Y. TCGA-Assembler: Open-source software for retrieving and processing TCGA data. Nat. Methods 2014, 11, 599–600. [Google Scholar] [CrossRef] [PubMed]
- Clifford, H.; Wessely, F.; Pendurthi, S.; Emes, R.D. Comparison of clustering methods for investigation of genome-wide methylation array data. Front. Genet. 2011, 2, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, J.R. Induction of decision trees. Mach. Learn. 1986, 1, 81–106. [Google Scholar] [CrossRef] [Green Version]
- van Kessel, K.E.M.; Zuiverloon, T.C.M.; Alberts, A.R.; Boormans, J.L.; Zwarthoff, E.C. Targeted therapies in bladder cancer: An overview of in vivo research. Nat. Rev. Urol. 2015, 12, 681–694. [Google Scholar] [CrossRef]
- Zolotovskaia, M.A.; Tkachev, V.S.; Seryakov, A.P.; Kuzmin, D.V.; Kamashev, D.E.; Sorokin, M.I.; Roumiantsev, S.A.; Buzdin, A.A. Mutation enrichment and transcriptomic activation signatures of 419 molecular pathways in cancer. Cancers (Basel). 2020, 12, 271. [Google Scholar] [CrossRef] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Buzdin, A.; Sorokin, M.; Garazha, A.; Sekacheva, M.; Kim, E.; Zhukov, N.; Wang, Y.; Li, X.; Kar, S.; Hartmann, C.; et al. Molecular pathway activation - New type of biomarkers for tumor morphology and personalized selection of target drugs. Semin. Cancer Biol. 2018, 53, 110–124. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Crispo, F.; Notarangelo, T.; Pietrafesa, M.; Lettini, G.; Storto, G.; Sgambato, A.; Maddalena, F.; Landriscina, M. Braf inhibitors in thyroid cancer: Clinical impact, mechanisms of resistance and future perspectives. Cancers (Basel). 2019, 11, 1388. [Google Scholar] [CrossRef] [Green Version]
- Jeske, Y.W.; Ali, S.; Byron, S.A.; Gao, F.; Mannel, R.S.; Ghebre, R.G.; DiSilvestro, P.A.; Lele, S.B.; Pearl, M.L.; Schmidt, A.P.; et al. FGFR2 mutations are associated with poor outcomes in endometrioid endometrial cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2017, 145, 366–373. [Google Scholar] [CrossRef] [Green Version]
- Westphal, M.; Maire, C.L.; Lamszus, K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 2017, 31, 723–735. [Google Scholar] [CrossRef] [Green Version]
- Couffignal, C.; Desgrandchamps, F.; Mongiat-Artus, P.; Ravery, V.; Ouzaid, I.; Roupret, M.; Phe, V.; Ciofu, C.; Tubach, F.; Mentre, F.; et al. The Diagnostic and Prognostic Performance of Urinary FGFR3 Mutation Analysis in Bladder Cancer Surveillance: A Prospective Multicenter Study. Urology 2015, 86, 1185–1191. [Google Scholar] [CrossRef]
- Rolfo, C.; Raez, L. New targets bring hope in squamous cell lung cancer: Neurotrophic tyrosine kinase gene fusions. Lab. Investig. 2017, 97, 1268–1270. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tang, P.M.K.; Zhou, Y.; Cheng, A.S.L.; Yu, J.; Kang, W.; To, K.F. Targeting the Oncogenic FGF-FGFR Axis in Gastric Carcinogenesis. Cells 2019, 8, 637. [Google Scholar] [CrossRef] [Green Version]
- Buzdin, A.; Sorokin, M.; Garazha, A.; Glusker, A.; Aleshin, A.; Poddubskaya, E.; Sekacheva, M.; Kim, E.; Gaifullin, N.; Giese, A.; et al. RNA sequencing for research and diagnostics in clinical oncology. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Rodon, J.; Soria, J.C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Braña, I.; et al. Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial. Nat. Med. 2019, 25, 751–758. [Google Scholar] [CrossRef]
- Poddubskaya, E.V.; Baranova, M.P.; Allina, D.O.; Smirnov, P.Y.; Albert, E.A.; Kirilchev, A.P.; Aleshin, A.A.; Sekacheva, M.I.; Suntsova, M.V. Personalized prescription of tyrosine kinase inhibitors in unresectable metastatic cholangiocarcinoma. Exp. Hematol. Oncol. 2018, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, M.; Poddubskaya, E.; Baranova, M.; Glusker, A.; Kogoniya, L.; Markarova, E.; Allina, D.; Suntsova, M.; Tkachev, V.; Garazha, A.; et al. RNA sequencing profiles and diagnostic signatures linked with response to ramucirumab in gastric cancer. Cold Spring Harb. Mol. case Stud. 2020. mcs.a004945. [Google Scholar] [CrossRef]
- Poddubskaya, E.; Bondarenko, A.; Boroda, A.; Zotova, E.; Glusker, A.; Sletina, S.; Makovskaia, L.; Kopylov, P.; Sekacheva, M.; Moisseev, A.; et al. Transcriptomics-Guided Personalized Prescription of Targeted Therapeutics for Metastatic ALK-Positive Lung Cancer Case Following Recurrence on ALK Inhibitors. Front. Oncol. 2019, 9, 1026. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Jensen, M.A.; Zenklusen, J.C. A practical guide to The Cancer Genome Atlas (TCGA). In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2016; Vol. 1418, pp. 111–141. [Google Scholar]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416. [Google Scholar] [CrossRef] [Green Version]
- Selleckem.com. Available online: https://www.selleckchem.com (accessed on 22 July 2019).
- Murtagh, F.; Legendre, P. Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? J. Classif. 2014, 31, 274–295. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2 : Elegant graphics for data analysis; Springer: New York, NY, USA, 2009; ISBN 9780387981413. [Google Scholar]
- R Core Team (2018). R: A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, Austria. Available online: https://www.r-project.org/ (accessed on 23 July 2019).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Site of Tumor | Number of Samples | TCGA Tumor Abbreviation | Full Name of Tumor, TCGA |
|---|---|---|---|
| Bladder | 136 | BLCA | Bladder Urothelial Carcinoma |
| Brain | 426 | GBM, LGG | Glioblastoma multiforme, Lower Grade Glioma |
| Breast | 797 | BRCA | Breast invasive carcinoma |
| Cervix | 177 | CESC | Cervical squamous cell carcinoma and endocervical adenocarcinoma |
| Colorectal | 603 | COAD, READ | Colon adenocarcinoma, Rectum adenocarcinoma |
| Kidney | 595 | KIRC, KIRP | Kidney renal clear cell carcinoma, Kidney renal papillary cell carcinoma |
| Liver | 185 | LIHC | Liver Hepatocellular carcinoma |
| Lung | 525 | LUAD, LUSC | Lung adenocarcinoma, Lung squamous cell carcinoma |
| Ovary | 315 | OV | Ovarian serous cystadenocarcinoma |
| Prostate | 256 | PRAD | Prostate adenocarcinoma |
| Stomach | 235 | STAD | Stomach adenocarcinoma |
| Thyroid | 393 | THCA | Thyroid carcinoma |
| Uterus | 247 | UCEC | Uterine Corpus Endometrial Carcinoma |
| Cancer Type | All Stages, all Genes | All Stages, Drug Target Genes | ||
|---|---|---|---|---|
| Cancer type / Drug usage cluster | DNA mutation | Gene expression | DNA mutation | Gene expression |
| COAD, READ / Cluster 1 | − | − | − | + |
| LIHC / Cluster 1 | − | − | + | + |
| KIRC, KIRP / Cluster 1 | + | + | + | + |
| THCA / Cluster 1 | + | + | − | + |
| PRAD / Cluster 2 | + | − | + | + |
| UCEC / Cluster 2 | − | + | − | + |
| CESC / Cluster 2 | + | + | + | + |
| STAD / Cluster 2 | − | + | − | + |
| BRCA / Cluster 3 | + | + | + | + |
| OV / Cluster 3 | + | + | + | + |
| LUAD, LUSC / Cluster 3 | − | + | − | + |
| BLCA / Cluster 3 | + | + | + | + |
| GBM, LGG / Cluster 3 | + | − | + | − |
| Stage I, all genes | Stage I, drug target genes | |||
| LIHC / Cluster 1 | + | − | + | + |
| THCA / Cluster 1 | + | + | − | − |
| COAD, READ / Cluster 1 | − | + | − | + |
| KIRC, KIRP / Cluster 1 | + | + | + | + |
| BRCA / Cluster 2 | − | + | − | − |
| LUAD, LUSC / Cluster 2 | − | + | − | − |
| Stage II, all genes | Stage II, drug target genes | |||
| LIHC / Cluster 1 | + | + | + | + |
| THCA / Cluster 1 | + | + | − | + |
| COAD, READ / Cluster 1 | − | − | − | − |
| KIRC, KIRP / Cluster 1 | + | + | + | + |
| BRCA / Cluster 2 | − | + | + | + |
| LUAD, LUSC / Cluster 2 | + | + | + | + |
| STAD / Cluster 2 | + | + | − | + |
| Stage III, all genes | Stage III, drug target genes | |||
| LIHC / Cluster 1 | + | + | + | + |
| THCA / Cluster 1 | + | + | − | − |
| COAD, READ / Cluster 1 | − | − | + | + |
| KIRC, KIRP / Cluster 1 | + | + | + | + |
| BRCA / Cluster 2 | + | + | + | − |
| LUAD, LUSC / Cluster 2 | − | + | − | + |
| STAD / Cluster 2 | + | + | − | + |
| BLCA / Cluster 2 | − | + | + | + |
| Stage IV, all genes | Stage IV, drug target genes | |||
| THCA / Cluster 1 | + | + | − | + |
| KIRC, KIRP / Cluster 1 | + | + | − | + |
| BLCA / Cluster 2 | − | + | + | + |
| COAD, READ / Cluster 2 | − | + | + | + |
| Cancer Type | Maximum Possible Score (Max) | Mutation Data, Observed Score (Percentage Share of Max) | Expression Data, Observed Score (Percentage Share of Max) |
|---|---|---|---|
| COAD, READ | 10+ | 2+ (20%) | 6+ (60%) |
| LIHC | 8+ | 7+ (87.5%) | 6+ (75%) |
| KIRC, KIRP | 10+ | 9+ (90%) | 10+ (100%) |
| THCA | 10+ | 5+ (50%) | 8+ (80%) |
| PRAD | 2+ | 2+ (100%) | 1+ (50%) |
| UCEC | 2+ | 0+ (0%) | 2+ (100%) |
| CESC | 2+ | 2+ (100%) | 2+ (100%) |
| STAD | 6+ | 2+ (33%) | 6+ (100%) |
| BRCA | 8+ | 3+ (37.5%) | 6+ (75%) |
| OV | 2+ | 2+ (100%) | 2+ (100%) |
| LUAD, LUSC | 8+ | 0+ (0%) | 7% (87.5%) |
| BLCA | 6+ | 4+ (67%) | 6+ (100%) |
| GBM, LGG | 2+ | 2+ (100%) | 0+ (0%) |
| All cancer types | 76+ | 37+ (49%) | 62+ (82%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zolotovskaia, M.A.; Sorokin, M.I.; Petrov, I.V.; Poddubskaya, E.V.; Moiseev, A.A.; Sekacheva, M.I.; Borisov, N.M.; Tkachev, V.S.; Garazha, A.V.; Kaprin, A.D.; et al. Disparity between Inter-Patient Molecular Heterogeneity and Repertoires of Target Drugs Used for Different Types of Cancer in Clinical Oncology. Int. J. Mol. Sci. 2020, 21, 1580. https://doi.org/10.3390/ijms21051580
Zolotovskaia MA, Sorokin MI, Petrov IV, Poddubskaya EV, Moiseev AA, Sekacheva MI, Borisov NM, Tkachev VS, Garazha AV, Kaprin AD, et al. Disparity between Inter-Patient Molecular Heterogeneity and Repertoires of Target Drugs Used for Different Types of Cancer in Clinical Oncology. International Journal of Molecular Sciences. 2020; 21(5):1580. https://doi.org/10.3390/ijms21051580
Chicago/Turabian StyleZolotovskaia, Marianna A., Maxim I. Sorokin, Ivan V. Petrov, Elena V. Poddubskaya, Alexey A. Moiseev, Marina I. Sekacheva, Nicolas M. Borisov, Victor S. Tkachev, Andrew V. Garazha, Andrey D. Kaprin, and et al. 2020. "Disparity between Inter-Patient Molecular Heterogeneity and Repertoires of Target Drugs Used for Different Types of Cancer in Clinical Oncology" International Journal of Molecular Sciences 21, no. 5: 1580. https://doi.org/10.3390/ijms21051580
APA StyleZolotovskaia, M. A., Sorokin, M. I., Petrov, I. V., Poddubskaya, E. V., Moiseev, A. A., Sekacheva, M. I., Borisov, N. M., Tkachev, V. S., Garazha, A. V., Kaprin, A. D., Shegay, P. V., Giese, A., Kim, E., Roumiantsev, S. A., & Buzdin, A. A. (2020). Disparity between Inter-Patient Molecular Heterogeneity and Repertoires of Target Drugs Used for Different Types of Cancer in Clinical Oncology. International Journal of Molecular Sciences, 21(5), 1580. https://doi.org/10.3390/ijms21051580