Autoantigen Treatment in Type 1 Diabetes: Unsolved Questions on How to Select Autoantigen and Administration Route

Abstract
:1. Introduction
2. Different Forms of Immune Interventions
3. Autoantigen Treatment
3.1. Possible Mechanisms
3.2. Autoantigen Treatment in Other Autoimmune Diseases than T1D
3.3. What Autoantigen to Use in T1D?
3.4. Peptides in Autoantigen Treatment
3.5. GAD as Autoantigen
3.6. Different Routes for Administration of Autoantigen.
3.7. Intra-lymphatic Administration of Autoantigen
3.8. Adjuvant Vitamin D
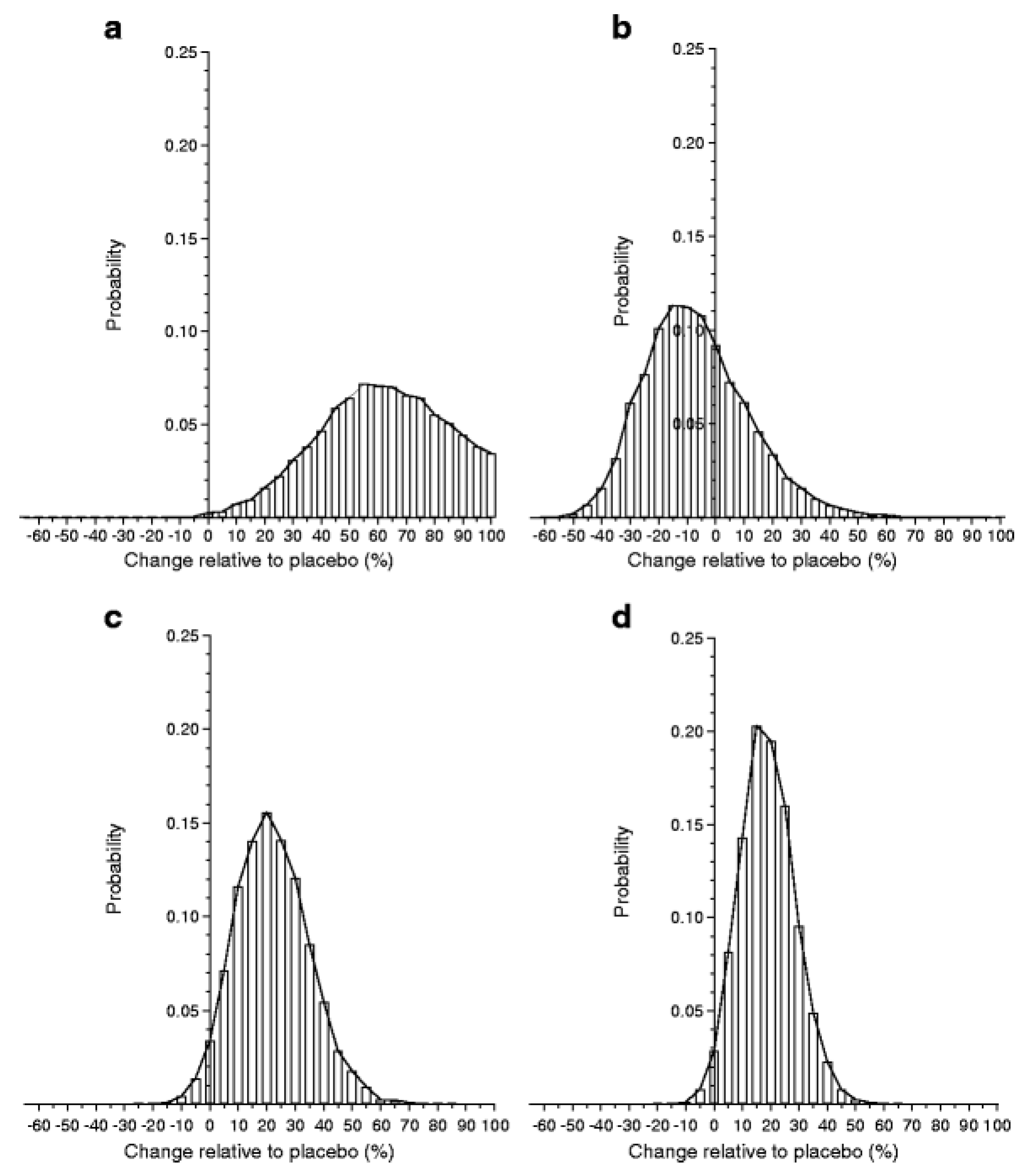
3.9. Experience from Intra-Lymphatic GAD-alum Treatment
- Change in C-peptide (Area Under the Curve [AUC mean 0–120 min) during a mixed-meal tolerance test (MMTT) between baseline to 15 months.
- Proportion of patients with a stimulated maximum C-peptide level above 0.2 nmol/L (0.6 ng/mL) at 15 months.
- Proportion of patients with a stimulated 90min C-peptide level above 0.2 nmol/L (0.6 ng/mL) at 15 months.
- Change in maximum C-peptide during MMTT between baseline and 15 months.
- C-peptide measured at 30, 60, 90, and 120 min during MMTT at 15 months.
- Change in fasting C-peptide between baseline and 15 months.
- Change in HbA1c between baseline and 15 months.
- Change in daily exogenous insulin consumption between baseline and 15 months.
- Change in insulin-dose-adjusted HbA1c (IDAA1c) between baseline and 15 months.
- Proportion of patients with by IDAA1c ≤ 9 at 15 months.
- Number of self-reported episodes of severe hypoglycemia (Severe hypoglycemia defined as needing help from others and/or seizures and/or unconscious) between baseline and 15 months.
- Change in rate of hypoglycemic events between baseline and 15 months.
- Number of patients having at least 1 severe hypoglycemic event between baseline and 15 months.
- Change in glycemic variability/fluctuations (evaluated from data from continuous glucose monitoring FreeStyle LibrePro, Flash Glucose Monitoring [FGM]) over 14-day period between screening and 15 months.
- CInjection site reactions.
- Occurrence of adverse evernts (AEs).
- Laboratory measurements (hematology and clinical chemistry).
- Urine analysis (microalbuminuria, creatinine).
- Physical examinations, including neurological assessments.
- GAD65A titer.
- Vital signs (blood pressure).
- Concentrations of serum autoantibodies toward GAD65 and IA 2.
- Concentrations of serum autoantibody isotypes toward GAD65.
- Total serum Immunoglobulin E (IgE).
- Secretion of interleukin (IL)-1, IL-2, IL-5, IL-13, IL-10, IL-17, interferon (IFN)γ, and tumor necrosis factor (TNF)α by peripheral blood mononuclear cells (PBMCs) on stimulation with GAD65.
- Proliferation of PBMCs on stimulation with GAD65.
- Flow cytometric analysis of PBMC subsets.
- Further exploratory immunological characterization.
- Change in QoL as measured by questionnaire EQ-5D-5L between baseline and month 15.
- Quality-adjusted life years (QALYs) based on the EQ-5D-5L questionnaire.
4. Challenges and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Korsgren, O.; Skyler, J.S.; Skog, O.; Sundberg, F.; Forsander, G.; Ludvigsson, J. Imagining a better future for all people with type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 30. [Google Scholar] [CrossRef] [PubMed]
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Bojestig, M.; Arnqvist, H.J.; Hermansson, G.; Karlberg, B.E.; Ludvigsson, J. Declining incidence of nephropathy in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1994, 330, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Lind, M.; Svensson, A.M.; Rosengren, A. Glycemic control and excess mortality in type 1 diabetes. N. Engl. J. Med. 2015, 372, 880–881. [Google Scholar] [CrossRef] [PubMed]
- Rawshani, A.; Sattar, S.; Franzen, A.T.; Hattersley, A.M.; Svensson, B.; Eliasson, S. Gudbjornsdottir Excess mortality and cardiovascular disease in young adults with type 1 diabetes in relation to age at onset: A nationwide, register-based cohort study. Lancet 2018, 392, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Ludvigsson, J. Time to leave rigid traditions in Type 1 diabetes research. Immunotherapy 2017, 9, 619–621. [Google Scholar] [CrossRef]
- Keenan, H.A.; Sun, J.K.; Levine, J.; Doria, A.; Aiello, L.P.; Eisenbarth, G.; Bonner-Weir, S.; King, G.L. Residual insulin production and pancreatic ß-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes 2010, 59, 2846–2853. [Google Scholar] [CrossRef] [Green Version]
- Madsbad, S.; Alberti, K.G.; Binder, C.; Burrin, J.M.; Faber, O.K.; Krarup, T.; Regeur, L. Role of residual insulin secretion in protecting against ketoacidosis in insulin-dependent diabetes. Br. Med. J. 1979, 2, 1257–1259. [Google Scholar] [CrossRef] [Green Version]
- Steffes, M.W.; Sibley, S.; Jackson, M.; Thomas, W. Beta-cell function and the development of diabetes-related complications in the Diabetes Control and Complications Trial. Diabetes Care 2003, 26, 832–836. [Google Scholar] [CrossRef] [Green Version]
- Wahren, J.; Ekberg, K.; Jörnvall, H. C-peptide is a bioactive peptide. Diabetologia 2007, 50, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Fierabracci, A. Peptide immunotherapies in Type 1 diabetes: Lessons from animal models. Curr. Med. Chem. 2011, 18, 577–586. [Google Scholar] [CrossRef]
- Ludvigsson, J.; Heding, L.; Liedén, G.; Marner, B.; Lernmark, A. Plasmapheresis in the initial treatment of insulin-dependent diabetes mellitus in children. Br. Med. J. 1983, 286, 176–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiller, C.R.; Laupacis, A.; Dupre, J.; Jenner, M.R.; Keown, P.A.; Rodger, W.; Wolfe, B.M. Cyclosporine for treatment of early type I diabetes: Preliminary results. N. Engl. J. Med. 1983, 308, 1226–1227. [Google Scholar] [PubMed]
- Herold, K.C.; Hagopian, W.; Auger, J.A.; Poumian-Ruiz, E.; Taylor, L.; Donaldson, D.; Gitelman, S.E.; Harlan, D.M.; Xu, D.; Zivin, R.A.; et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N. Engl. J. Med. 2002, 346, 1692–1698. [Google Scholar] [CrossRef] [Green Version]
- Keymeulen, B.; Vandemeulebroucke, E.; Ziegler, A.G.; Mathieu, C.; Kaufman, L.; Hale, G.; Gorus, F.; Goldman, M.; Walter, M.; Candon, S.; et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N. Engl. J. Med. 2005, 352, 2598–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, N.; Hagopian, W.; Ludvigsson, J.; Jain, S.M.; Wahlen, J.; Ferry, R.J., Jr.; Bode, B.; Aronoff, S.; Holland, C.; Carlin, D.; et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet 2011, 378, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Hagopian, W.; Ferry, R.J., Jr.; Sherry, N.; Carlin, D.; Bonvini, E.; Johnson, S.; Stein, K.E.; Koenig, S.; Daifotis, A.G.; Herold, K.C.; et al. Teplizumab preservesC-peptide in recent-onset type 1 diabetes: Two-year results from the randomized, placebo-controlled Protégé trial. Diabetes 2013, 62, 3901–3908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Korsgren, O.; Skog, O.; Ludvigsson, J. Teplizumab in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 1879–1880. [Google Scholar] [CrossRef]
- Mastrandrea, L.; Yu, J.; Behrens, T.; Buchlis, J.; Albini, C.; Fourtner, S.; Quattrin, T. Etanercept treatment in children with new-onset type 1 diabetes: Pilot randomized, placebo-controlled, double-blind study. Diabetes Care 2009, 32, 1244–1249. [Google Scholar] [CrossRef] [Green Version]
- Haller, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Michels, A.W.; Rosenthal, S.M.; Shuster, J.J.; Zou, B.; Brusko, T.M.; Hulme, M.A.; Wasserfall, C.H.; et al. Anti-thymocyte globulin/G-CSF treatment preserves b cell function in patients with established type 1 diabetes. J. Clin. Investig. 2015, 125, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Rigby, M.R.; Harris, K.M.; Pinckney, A.; DiMeglio, L.A.; Rendell, M.S.; Felner, E.I.; Dostou, J.M.; Gitelman, S.E.; Griffin, K.J.; Tsalikian, E.; et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J. Clin. Investig. 2015, 125, 3285–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pescovitz, M.D.; Greenbaum, C.J.; Krause-Steinrauf, H.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; McGee, P.F.; Moran, A.M.; et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N. Engl. J. Med. 2009, 361, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Robert, S.; Korf, H.; Gysemans, C.; Mathieu, C. Antigen-based vs. systemic immunomodulation in type 1 diabetes: The pros and cons. Islets 2013, 5, 53–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roep, B.O.; Wheeler, D.C.S.; Peakman, M. Antigen-based immune modulation therapy for type 1 diabetes: The era of precision medicine. Lancet Diabetes Endocrinol. 2019, 7, 65–74. [Google Scholar] [CrossRef]
- Shoda, L.K.; Young, D.L.; Ramanujan, S.; Whiting, C.C.; Atkinson, M.A.; Bluestone, J.A.; Eisenbarth, G.S.; Mathis, D.; Rossini, A.A.; Campbell, S.E.; et al. A comprehensive review of interventions in the NOD mouse and implications for translation. Immunity 2005, 23, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Boettler, T.; von Herrath, M. Type 1 diabetes vaccine development: Animal models vs. humans. Hum. Vaccines 2011, 7, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Staeva-Vieira, T.; Peakman, M.; von Herrath, M. Translational mini-review series on type 1 diabetes: Immune-based therapeutic approaches for type 1 diabetes. Clin. Exp. Immunol. 2007, 148, 17–31. [Google Scholar] [CrossRef]
- Quinn, A.; McInerney, B.; Reich, E.P.; Kim, O.; Jensen, K.P.; Sercarz, E.E. Regulatory and effector CD4 T cells in nonobese diabetic mice recognize overlapping determinants on glutamic acid decarboxylase and use distinct V beta genes. J. Immunol. 2001, 166, 2982–2991. [Google Scholar] [CrossRef] [Green Version]
- Homann, D.; Holz, A.; Bot, A.; Coon, B.; Wolfe, T.; Petersen, J.; Dyrberg, T.P.; Grusby, M.J.; von Herrath, M.G. Autoreactive CD4+ T cells protect from autoimmune diabetes via bystander suppression using the IL-4/Stat6 pathway. Immunity 1999, 11, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Peakman, M.; von Herrath, M. Antigen-specific immunotherapy for type 1 diabetes: Maximizing the potential. Diabetes 2010, 59, 2087–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tisch, R.; Wang, B.; Serreze, D.V. Induction of glutamic acid decarboxylase 65-specific Th2 cells and suppression of autoimmune diabetes at late stages of disease is epitope dependent. J. Immunol. 1999, 163, 1178–1187. [Google Scholar]
- Cabello-Kindelan, C.; Mackey, S.; Sands, A.; Rodriguez, J.; Vazquez, C.; Pugliese, A.; Bayer, A.L. Immunomodulation Followed By Antigen-Specific Treg Infusion Controls Islet Autoimmunity. Diabetes 2019, 11, 190061. [Google Scholar] [CrossRef] [PubMed]
- Umeshappa, C.S.; Singha, S.; Blanco, J.; Shao, K.; Nanjundappa, R.H.; Yamanouchi, J.; Parés, A.; Serra, P.; Yang, Y.; Santamaria, P. Suppression of a broad spectrum of liver autoimmune pathologies by single peptide-MHC-based nanomedicines. Nat. Commun. 2019, 10, 2150. [Google Scholar] [CrossRef] [PubMed]
- Ludvigsson, J. Adequate doses of autoantigen administered using the appropriate route may create tolerance and stop autoimmunity. Diabetologia 2009, 52, 175–176. [Google Scholar] [CrossRef] [Green Version]
- Martens, H.; Goxe, B.; Geenen, V. The thymic repertoire of neuroendocrine-related self-antigens: Physiological implications in T-cell life and death. Immunol. Today 1996, 17, 312–317. [Google Scholar] [CrossRef]
- Kyewski, B.; Klein, L. A central role for central tolerance. Annu. Rev. Immunol. 2006, 24, 571–606. [Google Scholar] [CrossRef] [PubMed]
- Funda, D.P.; Palová-Jelínková, L.; Goliá, J.; Kroulíková, Z.; Fajstová, A.; Hudcovic, T.; Špíšek, R. Optimal Tolerogenic Dendritic Cells in Type 1 Diabetes (T1D) Therapy: What Can We Learn From Non-obese Diabetic (NOD) Mouse Models? Front. Immunol. 2019, 10, 967. [Google Scholar] [CrossRef]
- Wraith, D.C.; Smilek, D.E.; Mitchell, D.J.; Steinman, L.; McDevitt, H.O. Antigen recognition in autoimmune encephalomyelitis and the potential for peptide-mediated immunotherapy. Cell 1989, 59, 247–255. [Google Scholar] [CrossRef]
- Sharma, S.D.; Nag, B.; Su, X.M.; Green, D.; Spack, E.; Clark, B.R.; Sriram, S. Antigen-specific therapy of experimental allergic encephalomyelitis by soluble class II major histocompatibility complex-peptide complexes. Proc. Natl Acad. Sci. USA 1991, 88, 11465–11469. [Google Scholar] [CrossRef] [Green Version]
- Bielekova, B.; Goodwin, B.; Richert, N.; Cortese, I.; Kondo, T.; Afshar, G.; Gran, B.; Eaton, J.; Antel, J.; Frank, J.A.; et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: Results of a phase II clinical trial with an altered peptide ligand. Nat. Med. 2000, 6, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Comi, G.; Panitch, H.; Oger, J.; Antel, J.; Conlon, P.; Steinman, L. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. Nat. Med. 2000, 6, 1176–1182. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.G.; Catz, I.; Ferenczi, L.Z.; Krantz, M.J. Intravenous synthetic peptide MBP8298 delayed disease progression in an HLA class II-defined cohort of patients with progressive multiple sclerosis: Results of a 24-month double-blind placebo-controlled clinical trial and 5 years of follow-up treatment. Eur. J. Neurol. 2006, 13, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, A.; Vollmer, T.; Antel, J.; Arnold, D.L.; Bodner, C.A.; Campagnolo, D.; Gianettoni, J.; Jalili, F.; Kachuck, N.; Lapierre, Y.; et al. Induction of antigen-specific tolerance in multiple sclerosis after immunization with DNA encoding myelin basic protein in a randomized, placebo-controlled phase 1/2 trial. Arch. Neurol. 2007, 64, 1407–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garren, H.; Robinson, W.H.; Krasulová, E.; Havrdová, E.; Nadj, C.; Selmaj, K.; Losy, J.; Nadj, I.; Radue, E.W.; Kidd, B.A.; et al. Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis. Ann. Neurol. 2008, 63, 611–620. [Google Scholar] [CrossRef]
- Walczak, A.; Siger, M.; Ciach, A.; Szczepanik, M.; Selmaj, K. Transdermal application of myelin peptides in multiple sclerosis treatment. JAMA Neurol. 2013, 70, 1105–1109. [Google Scholar] [CrossRef]
- Juryńczyk, M.; Walczak, A.; Jurewicz, A.; Jesionek-Kupnicka, D.; Szczepanik, M.; Selmaj, K. Immune regulation of multiple sclerosis by transdermally applied myelin peptides. Ann. Neurol. 2010, 68, 593–601. [Google Scholar] [CrossRef]
- Freedman, M.S.; Bar-Or, A.; Oger, J.; Traboulsee, A.; Patry, D.; Young, C.; Olsson, T.; Li, D.; Hartung, H.P.; Krantz, M.; et al. A phase III study evaluating the efficacy and safety of MBP8298 in secondary progressive MS. Neurology 2011, 77, 1551–1560. [Google Scholar] [CrossRef]
- Spack, E.G.; McCutcheon, M.; Corbelletta, N.; Nag, B.; Passmore, D.; Sharma, S.D. Induction of tolerance in experimental autoimmune myasthenia gravis with solubilized MHC class II: Acetylcholine receptor peptide complexes. J. Autoimmun. 1995, 8, 787–807. [Google Scholar] [CrossRef]
- Koffeman, E.C.; Genovese, M.; Amox, D.; Keogh, E.; Santana, E.; Matteson, E.L.; Kavanaugh, A.; Molitor, J.A.; Schiff, M.H.; Posever, J.O.; et al. Epitope-specific immunotherapy of rheumatoid arthritis: Clinical responsiveness occurs with immune deviation and relies on the expression of a cluster of molecules associated with T cell tolerance in a double-blind, placebo-controlled, pilot phase II trial. Arthritis Rheumatol. 2009, 60, 3207–3216. [Google Scholar] [CrossRef]
- Pozsgay, J.; Szekanecz, Z.; Sármay, G. Antigen-specific immunotherapies in rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Goel, G.; King, T.; Daveson, A.J.; Andrews, J.M.; Krishnarajah, J.; Krause, R.; Brown, G.J.E.; Fogel, R.; Barish, C.F.; Epstein, R.; et al. Epitope-specific immunotherapy targeting CD4-positive T cells in coeliac disease; two randomised, double-blind, placebo-controlled phase 1 studies. Lancet Gastroenterol. Hepatol. 2017, 2, 479–493. [Google Scholar] [CrossRef]
- Daveson, A.J.M.; Ee, H.C.; Andrews, J.M.; King, T.; Goldstein, K.E.; Dzuris, J.L.; MacDougall, J.A.; Williams, L.J.; Treohan, A.; Cooreman, M.P.; et al. Epitope-specific immunotherapy targeting CD4-positive T cells in celiac disease: Safety, pharmacokinetics, and effects on intestinal histology and plasma cytokines with escalating dose regimens of Nexvax2 in a randomized, double-blind, placebo-controlled phase 1 study. EBioMedicine 2017, 26, 78–90. [Google Scholar] [PubMed] [Green Version]
- Purcell, A.W.; Sechi, S.; DiLorenzo, T.P. The Evolving Landscape of Autoantigen Discovery and Characterization in Type 1 Diabetes. Diabetes 2019, 68, 879–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, P.A.; Delong, T.; Baker, R.L.; Fitzgerald-Miller, L.; Wagner, R.; Cook, G.; Rewers, M.R.; Michels, A.; Haskins, K. Chromogranin A is a T Cell Antigen in Human Type 1 Diabetes. J. Autoimmun. 2014, 50, 38–41. [Google Scholar] [CrossRef] [Green Version]
- Delong, T.; Wiles, T.A.; Baker, R.L.; Bradley, B.; Barbour, G.; Reisdorph, R.; Armstrong, M.; Powell, R.L.; Reisdorph, N.; Kumar, N.; et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 2016, 351, 711–714. [Google Scholar] [CrossRef] [Green Version]
- McGinty, J.W.; Chow, I.T.; Greenbaum, C.; Odegard, J.; Kwok, W.W.; James, E.A. Recognition of posttranslationally modified GAD65 epitopes in subjects with type 1 diabetes. Diabetes 2014, 63, 3033–3040. [Google Scholar] [CrossRef] [Green Version]
- Stadinski, B.D.; Delong, T.; Reisdorph, N.; Reisdorph, R.; Powell, R.L.; Armstrong, M.; Piganelli, J.D.; Barbour, G.; Bradley, B.; Crawford, F.; et al. Chromogranin A is an autoantigen in type diabetes. Nat. Immunol. 2010, 11, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Marré, M.L.; James, E.A.; Piganelli, J.D. β cell ER stress and the implications for immunogenicity in type 1 diabetes. Front. Cell Dev. Biol. 2015, 3, 67. [Google Scholar] [CrossRef] [Green Version]
- Strollo, R.; Vinci, C.; Napoli, N.; Pozzilli, P.; Ludvigsson, J.; Nissim, A. Antibodies to post-translationally modified insulin as a novel biomarker for prediction of type 1 diabetes in children. Diabetologia 2017, 60, 1467–1474. [Google Scholar] [CrossRef]
- Strollo, R.; Vinci, C.; Napoli, N.; Fioriti, E.; Maddaloni, E.; Åkerman, L.; Casas, R.; Pozzilli, P.; Ludvigsson, J.; Nissim, A. Antibodies to oxidized insulin improve prediction of type 1 diabetes in children with positive standard islet autoantibodies. Diabetes Metab. Res. Rev. 2019, 35, e3132. [Google Scholar] [CrossRef] [PubMed]
- Jarchum, I.; Nichol, L.; Trucco, M.; Santamaria, T.P. Identification of Novel IGRP Epitopes Targeted in Type 1 Diabetes Patients. Clin. Immunol. 2008, 127, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, R.; Omidian, Z.; Giwa, A.; Cornwell, B.; Majety, N.; Bell, D.R.; Lee, S.; Zhang, H.; Michels, A.; Desiderio, S.; et al. A Public BCR Present in a Unique Dual-Receptor-Expressing Lymphocyte from Type 1 Diabetes Patients Encodes a Potent T Cell Autoantigen. Cell 2019, 177, 1583–1599. [Google Scholar] [CrossRef] [PubMed]
- Hannelius, U.; Beam, C.; Ludvigsson, J. HLA haplotypes associate with clinical response to GAD-specific immunotherapy. Abstract IDS, Peking. 2020. [Google Scholar]
- Smith, E.L.; Peakman, M. Peptide Immunotherapy for Type 1 Diabetes—Clinical Advances. Front. Immunol. 2018, 9, 392. [Google Scholar] [CrossRef] [PubMed]
- Siljander, H.T.; Simell, S.; Hekkala, A.; Lähde, J.; Simell, T.; Vähäsalo, P.; Veijola, R.; Ilonen, J.; Simell, O.; Knip, M. Predictive characteristics of diabetes-associated autoantibodies among children with HLA-conferred disease susceptibility in the general population. Diabetes 2009, 58, 2835–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; James, E.A.; Sanda, S.; Greenbaum, C.; Kwok, W.W. CD4+ T cells recognize diverse epitopes within GAD65: Implications for repertoire development and diabetes monitoring. Immunology 2013, 138, 269–279. [Google Scholar] [CrossRef]
- Harrison, L.C.; Wentworth, J.M.; Zhang, Y.; Bandala-Sanchez, E.; Böhmer, R.M.; Neale, A.M.; Stone, N.L.; Naselli, G.; Bosco, J.J.; Auyeung, P.; et al. Antigen-based vaccination and prevention of type 1 diabetes. Curr. Diabetes Rep. 2013, 13, 616–623. [Google Scholar] [CrossRef]
- Diabetes Prevention Trial-Type 1 Diabetes Study Group. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N. Engl. J. Med. 2002, 345, 1685–1691. [Google Scholar]
- Skyler, J.S.; Krischer, J.P.; Wolfsdorf, J.; Cowie, C.; Palmer, J.P.; Greenbaum, C.; Cuthbertson, D.; Rafkin-Mervis, L.E.; Chase, H.P.; Leschek, E. Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial-Type 1. Diabetes Care 2005, 28, 1068–1076. [Google Scholar]
- Every, A.L.; Kramer, D.R.; Mannering, S.I.; Lew, A.M.; Harrison, L.C. Intranasal vaccination with proinsulin DNA induces regulatory CD4+ T cells that prevent experimental autoimmune diabetes. J. Immunol. 2006, 176, 4608–4615. [Google Scholar] [CrossRef] [Green Version]
- Näntö-Salonen, K.; Kupila, A.; Simell, S.; Siljander, H.; Salonsaari, T.; Hekkala, A.; Korhonen, S.; Erkkola, R.; Sipilä, J.I.; Haavisto, L.; et al. Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: A double-blind, randomised controlled trial. Lancet 2008, 372, 1746–1755. [Google Scholar] [CrossRef]
- Achenbach, P.; Barker, J.; Bonifacio, E. Pre-POINT Study Group Modulating the natural history of type 1 diabetes in children at high genetic risk by mucosal insulin immunization. Curr. Diabetes Rep. 2008, 8, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Elding Larsson, H.; Lundgren, M.; Jonsdottir, B.; Cuthbertson, D.; Krischer, J.; DiAPREV-IT Study Group. Combined therapy with GABA and proinsulin/alum acts synergistically to restore long-term normoglycemia by modulating T-cell autoimmunity and promoting β-cell replication in newly diabetic NOD mice. Pediatr. Diabetes 2018, 19, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Alleva, D.G.; Maki, R.A.; Putnam, A.L.; Robinson, J.M.; Kipnes, M.S.; Dandona, P.; Marks, J.B.; Simmons, D.L.; Greenbaum, C.J.; Jimenez, R.G.; et al. Immunomodulation in type 1 diabetes by NBI-6024, an altered peptide ligand of the insulin (B9-23) epitope. Scand J. Immunol 2006, 63, 59–69. [Google Scholar] [CrossRef]
- Walter, M.; Philotheou, A.; Bonnici, F.; Ziegler, A.G.; Jimenez, R. No effect of the altered peptide ligand NBI-6024 β-cell residual function and insulin needs in new-onset type 1 diabetes. Diabetes Care 2009, 32, 2036–2040. [Google Scholar] [CrossRef] [Green Version]
- Thrower, S.L.; James, L.; Hall, W.; Green, K.M.; Arif, S.; Allen, J.S.; Van-Krinks, C.; Lozanoska-Ochser, B.; Marquesini, L.; Brown, S.; et al. Proinsulin peptide immunotherapy in type 1 diabetes: Report of a first-in man Phase I safety study. Clin. Exp. Immunol. 2009, 155, 156–165. [Google Scholar] [CrossRef]
- Alhadj Ali, M.; Liu, Y.F.; Arif, S.; Tatovic, D.; Shariff, H.; Gibson, V.B.; Yusuf, N.; Baptista, R.; Eichmann, M.; Petrov, N.; et al. Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes. Sci. Transl. Med. 2017, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Baekkeskov, S.; Nielsen, J.H.; Marner, B.; Ludvigsson, J.; Lernmark, A. Autoantibodies in newly diagnosed diabetic children immunoprecipitate human pancreatic islet cell proteins. Nature 1982, 298, 167–169. [Google Scholar] [CrossRef]
- Franklin, I.K.; Wollheim, C.B. GABA in the endocrine pancreas: Its putative role as an islet cell paracrine-signalling molecule. J. Gen. Physiol. 2004, 123, 185–190. [Google Scholar] [CrossRef]
- Tian, J.; Dang, H.; Nguyen, A.V.; Chen, Z.; Kaufman, D.L. Combined therapy with GABA and proinsulin/alum acts synergistically to restore long-term normoglycemia by modulating T-cell autoimmunity and promoting β-cell replication in newly diabetic NOD mice. Diabetes 2014, 63, 3128–3134. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, D.L.; Clare-Salzler, M.; Tian, J.; Forsthuber, T.; Ting, G.S.; Robinson, P.; Atkinson, M.A.; Sercarz, E.E.; Tobin, A.J.; Lehmann, P.V. Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature 1993, 366, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Tisch, R.; Yang, X.D.; Singer, S.M.; Liblau, R.S.; Fugger, L.; McDevitt, H.O. Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic mice. Nature 1993, 366, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Clare-Salzler, M.; Herschenfeld, A.; Middleton, B.; Newman, D.; Mueller, R.; Arita, S.; Evans, C.; Atkinson, M.A.; Mullen, Y.; et al. Modulating autoimmune responses to GAD inhibits disease progression and prolongs islet graft survival in diabetes-prone mice. Nat. Med. 1996, 2, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Agardh, C.D.; Cilio, C.M.; Lethagen, A.; Lynch, K.; Leslie, R.D.; Palmér, M.; Harris, R.A.; Robertson, J.A.; Lernmark, A. Clinical evidence for the safety of GAD65 immunomodulation in adult-onset autoimmune diabetes. J. Diabetes Complicat. 2005, 19, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Agardh, C.-D.; Lynch, K.; Palmér, M.; Lernmark, A. GAD65 vaccination significantly reduces insulin dependence at five years follow-up in a dose escalating study in adult-onset autoimmune diabetes patients. Diabetologia 2008, 51 (Suppl. 1), S230. [Google Scholar]
- Ludvigsson, J.; Faresjo, M.; Hjorth, M.; Axelsson, S.; Chéramy, M.; Pihl, M.; Vaarala, O.; Forsander, G.; Ivarsson, S.; Johansson, C.; et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N. Engl. J. Med. 2008, 359, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Greenbaum, C.J.; Mandrup-Poulsen, T.; McGee, P.F.; Battelino, T.; Haastert, B.; Ludvigsson, J.; Pozzilli, P.; Lachin, J.M.; Kolb, H.; Type 1 Diabetes Trial Net Research Group; et al. Mixed-meal tolerance test versus glucagon stimulation test for the assessment of beta-cell function in therapeutic trials in type diabetes. Diabetes Care 2008, 31, 1966–1971. [Google Scholar] [CrossRef] [Green Version]
- Ludvigsson, J.; Hjorth, M.; Chéramy, M.; Axelsson, S.; Pihl, M.; Forsander, G.; Nilsson, N.Ö.; Samuelsson, B.O.; Wood, T.; Aman, J.; et al. Extended evaluation of the safety and efficacy of GAD treatment of children and adolescents with recent-onset type 1 diabetes: A randomised controlled trial. Diabetologia 2011, 54, 634–640. [Google Scholar] [CrossRef] [Green Version]
- Wherrett, D.K.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Marks, J.B.; et al. Antigen-based therapy with glutamicacid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: A randomised double-blind trial. Lancet 2011, 378, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Ludvigsson, J.; Krisky, D.; Casas, R.; Battelino, T.; Castaño, L.; Greening, J.; Kordonouri, O.; Otonkoski, T.; Pozzilli, P.; Robert, J.J.; et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N. Engl. J. Med. 2012, 366, 433–442. [Google Scholar] [CrossRef]
- Tavira, B.; Cheramy, M.; Axelsson, S.; Åkerman, L.; Ludvigsson, J.; Casas, R. Effect of simultaneous vaccination with H1N1 and GAD-alum on GAD65-induced immune response. Diabetologia 2017, 60, 1276–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludvigsson, J.; Chéramy, M.; Axelsson, S.; Pihl, M.; Akerman, L.; Casas, R. GAD-treatment of children and adolescents with recent-onset Type 1 diabetes preserves residual insulin secretion after 30 months. for the clinical GAD-study group in Sweden. Diabetes Metab. Res. Rev. 2014, 3, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Beam, C.A.; MacCallum, C.; Herold, K.C.; Wherrett, D.K.; Palmer, J.; Ludvigsson, J. GAD vaccine reduces insulin loss in recently diagnosed type 1 diabetes: Findings from a Bayesian meta-analysis. Diabetologia 2017, 60, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Writing Committee for the Type 1 Diabetes TrialNet Oral Insulin Study Group; Krischer, J.P.; Schatz, D.A.; Bundy, B.; Skyler, J.S.; Greenbaum, C.J. Effect of Oral Insulin on Prevention of Diabetes in Relatives of Patients With Type 1 Diabetes: A Randomized Clinical Trial. JAMA 2017, 318, 1891–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonifacio, E.; Ziegler, A.G.; Klingensmith, G.; Schober, E.; Bingley, P.J.; Rottenkolber, M.; Theil, A.; Eugster, A.; Puff, R.; Peplow, C.; et al. Effects of high-dose oral insulin on immune responses in children at high risk for type 1 diabetes: The Pre-POINT randomized clinical trial. J. Am. Med. Assoc. 2015, 313, 1541–1549. [Google Scholar] [CrossRef]
- Ziegler, A.G.; Danne, T.; Dunger, D.B.; Berner, R.; Puff, R.; Kiess, W.; Agiostratidou, G.; Todd, J.A.; Bonifacio, E. Primary prevention of beta-cell autoimmunity and type 1 diabetes-The Global Platform for the Prevention of Autoimmune Diabetes (GPPAD) perspectives. Mol. Metab. 2016, 5, 255–262. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, A.G.; Achenbach, P.; Berner, R.; Casteels, K.; Danne, T.; Gündert, M.; Hasford, J.; Hoffmann, V.S.; Kordonouri, O.; Lange, K.; et al. Oral insulin therapy for primary prevention of type 1 diabetes in infants with high genetic risk: The GPPAD-POInT (global platform for the prevention of autoimmune diabetes primary oral insulin trial) study protocol. BMJ Open 2019, 9, e028578. [Google Scholar] [CrossRef] [Green Version]
- Pozzilli, P.; Pitocco, D.; Visalli, N.; Cavallo, M.G.; Buzzetti, R.; Crinò, A.; Spera, S.; Suraci, C.; Multari, G.; Cervoni, M.; et al. No effect of oral insulin on residual beta-cell function in recent-onset type I diabetes (the IMDIAB VII). Diabetologia 2000, 43, 1000–1004. [Google Scholar] [CrossRef] [Green Version]
- Chaillous, L.; Lefèvre, H.; Thivolet, C.; Boitard, C.; Lahlou, N.; Atlan-Gepner, C.; Bouhanick, B.; Mogenet, A.; Nicolino, M.; Carel, J.C.; et al. Oral insulin administration and residual beta-cell function in recent-onset type 1 diabetes: A multicentre randomised controlled trial. Diabète Insuline Orale group. Lancet 2000, 356, 545–549. [Google Scholar] [CrossRef]
- Takiishi, T.; Korf, H.; Van Belle, T.L.; Robert, S.; Grieco, F.A.; Caluwaerts, S.; Galleri, L.; Spagnuolo, I.; Steidler, L.; Van Huynegem, K.; et al. Reversal of autoimmune diabetes by restoration of antigen-specific tolerance using genetically modified Lactococcus lactis in mice. J. Clin. Investig. 2012, 122, 1717–1725. [Google Scholar] [CrossRef]
- Robert, S.; Van Huynegem, K.; Gysemans, C.; Mathieu, C.; Rottiers, P.; Steidler, L. Trimming of two major type 1 diabetes driving antigens, GAD65 and IA-2, allows for successful expression in Lactococcus lactis. Benef. Microbes 2015, 6, 591–601. [Google Scholar] [CrossRef]
- Cook, D.P.; Gysemans, C.; Mathieu, C. Lactococcus lactis As a Versatile Vehicle for Tolerogenic Immunotherapy. Front. Immunol. 2018, 8, 1961. [Google Scholar] [CrossRef] [Green Version]
- Robert, S.; Gysemans, C.; Takiishi, T.; Korf, H.; Spagnuolo, I.; Sebastiani, G.; Van Huynegem, K.; Steidler, L.; Caluwaerts, S.; Demetter, P.; et al. Oral delivery of glutamic acid decarboxylase (GAD)-65 and IL10 by Lactococcus lactis reverses diabetes in recent-onset NOD mice. Diabetes 2014, 63, 2876–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husseiny, M.I.; Rawson, J.; Kaye, A.; Nair, I.; Todorov, I.; Hensel, M.; Kandeel, F.; Ferreri, K. An oral vaccine for type 1 diabetes based on live attenuated Salmonella. Vaccine 2014, 32, 2300–2307. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Yu, J.; Chong, D.K.; Hough, J.; Engen, P.C.; Langridge, W.H. A plant-based cholera toxin B subunit-insulin fusion protein protects against the development of autoimmune diabetes. Nat. Biotechnol. 1998, 16, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Denes, B.; Krausova, V.; Fodor, N.; Timiryasova, T.; Henderson, D.; Hough, J.; Yu, J.; Fodor, I.; Langridge, W.H. Protection of NOD mice from type 1 diabetes after oral inoculation with vaccinia viruses expressing adjuvanted islet autoantigens. J. Immunother. 2005, 28, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Kwon, K.C.; Verma, D.; Singh, N.D.; Herzog, R.; Daniell, H. Oral delivery of human biopharmaceuticals, autoantigens and vaccine antigens bioencapsulated in plant cells. Adv. Drug Deliv. Rev. 2013, 65, 782–799. [Google Scholar] [CrossRef] [Green Version]
- Pujol-Autonell, I.; Serracant-Prat, A.; Cano-Sarabia, M.; Ampudia, R.M.; Rodriguez-Fernandez, S.; Sanchez, A.; Izquierdo, C.; Stratmann, T.; Puig-Domingo, M.; Maspoch, D.; et al. Use of autoantigen-loaded phosphatidylserine-liposomes to arrest autoimmunity in type 1 diabetes. PLoS ONE 2015, 10, e0127057. [Google Scholar] [CrossRef] [Green Version]
- Harrison, L.C.; Honeyman, M.C.; Steele, C.E.; Stone, N.L.; Sarugeri, E.; Bonifacio, E.; Couper, J.J.; Colman, P.G. Pancreatic beta-cell function and immune responses to insulin after administration of intranasal insulin to humans at risk for type 1 diabetes. Diabetes Care 2004, 27, 2348–2355. [Google Scholar] [CrossRef] [Green Version]
- Maloy, K.J.; Erdmann, I.; Basch, V.; Sierro, S.; Kramps, T.A.; Zinkernagel, R.M.; Oehen, S.; Kündig, T.M. Intralymphatic immunization enhances DNA vaccination. Proc. Natl. Acad. Sci. USA 2001, 98, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Johansen, P.A.C.; Häffner, A.C.; Koch, F.; Zepter, K.; Erdmann, I.; Maloy, K.; Simard, J.J.; Storni, T.; Senti, G.; Bot, A.; et al. Direct intralymphatic injection of peptide vaccines enhances immunogenicity. Eur. J. Immunol. 2005, 35, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gomez, J.M.; Johansen, P.; Erdmann, I.; Senti, G.; Crameri, R.; Kündig, T.M. Intralymphatic Injections as a New Administration Route for Allergen-Specific Immunotherapy. Int. Arch. Allergy Immunol. 2009, 150, 59–65. [Google Scholar] [PubMed] [Green Version]
- Von Beust, B.R.; Johansen, P.; Smith, K.A.; Bot, A.; Storni, T.; Kündig, T.M. Improving the therapeutic index for CpG oliogdeoxynucleotides by intralymphatic administration. Eur. J. immunol. 2005, 35, 1869–1876. [Google Scholar] [CrossRef] [PubMed]
- Senti, G.; Prinz Vavricka, B.M.; Erdmann, I.; Diaz, M.I.; Markus, R.; McCormack, S.J.; Simard, J.J.; Wüthrich, B.; Crameri, R.; Graf, N.; et al. Intralymphatic allergen administration renders specific immunotherapy faster and safer: A randomized controlled trial. Proc. Natl. Acad. Sci. USA 2008, 105, 17908–17912. [Google Scholar] [CrossRef] [Green Version]
- Senti, G.; Crameri, R.; Kuster, D.; Johansen, P.; Martinez-Gomez, J.M.; Graf, N.; Steiner, M.; Hothorn, L.A.; Grönlund, H.; Tivig, C.; et al. Intralymphatic immunotherapy for cat allergy induces tolerance after only 3 injections. J. Allergy Clin. Immunol. 2012, 129, 1290–1296. [Google Scholar] [CrossRef]
- Hyllander, T.; Latif, L.; Petersson-Westin, U.; Cardell, L.O. Intralymphatic allergen-specific immunotherapy: An effective and safe alternative treatment route for pollen-induced allergic rhinitis. J. Allergy Clin. Immunol. 2013, 131, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Ludvigsson, J.; Wahlberg, J.; Casas, R. Intralymphatic Injection of Autoantigen in Type 1 Diabetes. N. Engl. J. Med. 2017, 376, 697–699. [Google Scholar] [CrossRef] [Green Version]
- Ludvigsson, J.; Tavira, B.; Casas, R. More on Intralymphatic Injection of Autoantigen in Type 1 Diabetes. N. Engl. J. Med. 2017, 377, 403–405. [Google Scholar] [CrossRef]
- Ludvigsson, J. Combination therapy for preservation of beta-cell function in Type 1 diabetes: New attitudes and strategies are needed! Immunol. Lett. 2014, 159, 30–35. [Google Scholar] [CrossRef]
- Pozzilli, P.; Maddaloni, E.; Buzzetti, R. Combination immunotherapies for type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2015, 11, 289–297. [Google Scholar] [CrossRef]
- Skyler, J.S. Immune therapy for treating type 1 diabetes: Challenging existing paradigms. J. Clin. Investig. 2015, 125, 94–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hypponen, E.; Laara, E.; Reunanen, A.; Jarvelin, M.R.; Virtanen, S.M. Intake of vitamin D and risk of type 1 diabetes: A birth-cohort study. Lancet 2001, 358, 1500–1503. [Google Scholar] [CrossRef]
- Tapia, G.; Mårild, K.; Dahl, S.R.; Lund-Blix, N.A.; Viken, M.K.; Lie, B.A.; Njølstad, P.R.; Joner, G.; Skrivarhaug, T.; Cohen, A.S.; et al. Maternal and Newborn Vitamin D-Binding Protein, Vitamin D Levels, Vitamin D Receptor Genotype, and Childhood Type 1 Diabetes. Diabetes Care 2019, 42, 553–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zipitis, C.S.; Akobeng, A.K. Vitamin D supplementation in early childhood and risk of type 1 diabetes: A systematic review and meta-analysis. Arch. Dis. Child. 2008, 93, 512–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgartl, H.J.; Standl, E.; Schmidt-Gayk, H.; Kolb, H.J.; Janka, H.U.; Ziegler, A.G. Changes of vitamin D3 serum concentrations at the onset of immune-mediated type 1 (insulin-dependent) diabetes mellitus. Diabetes Res. 1991, 16, 145–148. [Google Scholar] [PubMed]
- Munger, K.L.; Levin, L.I.; Massa, J.; Horst, R.; Orban, T.; Ascherio, A. Preclinical serum 25-hydroxyvitamin D levels and risk of type 1 diabetes in a cohort of US military personnel. Am. J. Epidemiol. 2013, 177, 411–419. [Google Scholar] [CrossRef]
- Mathieu, C.; Gysemans, C.; Giulietti, A.; Bouillon, R. Vitamin D and diabetes. Diabetologia 2005, 48, 1247–1257. [Google Scholar] [CrossRef] [Green Version]
- Teegarden, D.; Donkin, S.S. Vitamin D: Emerging new roles in insulin sensitivity. Nutr. Res. Rev. 2009, 22, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.; Kaupper, T.; Adler, K.; Foersch, J.; Bonifacio, E.; Ziegler, A.G. No Effect of the 1α,25-Dihydroxyvitamin D3 on β-Cell Residual Function and Insulin Requirement in Adults With New-Onset Type 1 Diabetes. Diabetes Care 2010, 33, 1443–1448. [Google Scholar] [CrossRef] [Green Version]
- Bizzarri, C.; Pitocco, D.; Napoli, N.; Di Stasio, E.; Maggi, D.; Manfrini, S.; Suraci, C.; Cavallo, M.G.; Cappa, M.; Ghirlanda, G.; et al. No Protective Effect of Calcitriol on β-Cell Function in Recent-Onset Type 1 Diabetes: The IMDIAB XIII trial. Diabetes Care 2010, 33, 1962–1963. [Google Scholar] [CrossRef] [Green Version]
- Piemonti, L.; Monti, P.; Sironi, M.; Fraticelli, P.; Leone, B.E.; Dal Cin, E.; Allavena, P.; Di Carlo, V. Vitamin D3 affects differentiation, maturation, and function of human monocyte-derived dendritic cells. J. Immunol. 2000, 164, 4443–4451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penna, G.; Adorini, L. 1α,25-Dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J. Immunol. 2000, 164, 2405–2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, M.D.; Lutz, W.; Phan, V.A.; Bachman, L.A.; McKean, D.J.; Kumar, R. Dendritic cell modulation by 1α,25 dihydroxyvitamin D3 and its analogs: A vitamin D receptor-dependent pathway that promotes a persistent state of immaturity in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 6800–6805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penna, G.; Amuchastegui, S.; Giarratana, N.; Daniel, K.C.; Vulcano, M.; Sozzani, S.; Adorini, L. 1,25-Dihydroxyvitamin D3 selectively modulates tolerogenic properties in myeloid but not plasmacytoid dendritic cells. J. Immunol. 2007, 178, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonstra, A.; Barrat, F.J.; Crain, C.; Heath, V.L.; Savelkoul, H.F.; O’Garra, A. 1α,25-Dihydroxyvitamin D3 has a direct effect on naive CD4+ T cells to enhance the development of Th2 cells. J. Immunol. 2001, 167, 4974–4980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, M.A.; Roep, B.O.; Posgai, A.; Wheeler, D.C.S.; Peakman, M. The challenge of modulating β-cell autoimmunity in type 1 diabetes. Lancet Diabetes Endocrinol. 2019, 7, 52–64. [Google Scholar] [CrossRef]
- Tuomilehto, J. The emerging global epidemic of type 1 diabetes. Curr. Diabetes Rep. 2013, 13, 795–804. [Google Scholar] [CrossRef]
- Diaz-Valencia, P.A.; Bougnères, P.; Valleron, A.J. Global epidemiology of type 1 diabetes in young adults and adults: A systematic review. BMC Public Health 2015, 15, 255. [Google Scholar] [CrossRef]
- Gomez-Lopera, N.; Pineda-Trujillo, N.; Diaz-Valencia, P.A. Correlating the global increase in type 1 diabetes incidence across age groups with national economic prosperity: A systematic review. World J. Diabetes 2019, 10, 560–580. [Google Scholar] [CrossRef]
- Pociot, F.; Lernmark, Å. Genetic risk factors for type 1 diabetes. Lancet 2016, 387, 2331–2339. [Google Scholar] [CrossRef]
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, Å.; et al. Staging presymptomatic type 1 diabetes: A scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rewers, M.; Ludvigsson, J. Environmental risk factors for type 1 diabetes. Lancet 2016, 387, 2340–2348. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Commonly Discussed “Old”Autoantigens | More Recently Described Antigens/Neoepitopes |
|---|---|
| Insulin | Tetraspanin-7, Glima38 |
| Proinsulin | Islet Amyloid Polypeptide (IAPP) |
| Insulin B-chain | Human IAPP Precursor Protein (ppIAPP) |
| Proinsulin peptides eg C19-A3 | ChgA (Chromogranin A) |
| Glutamic Acid Decarboxylase (GAD65) | IGRP Islet-Specific Glucose-6-Phosphatase Catalytic Subunit-Related Protein |
| Insulinoma-Associated Antigen; (IA-2) | Insulin-Gene Enhancer Protein Isl-1 |
| Tyrosine Phosphatase (IA-2) | Peripherin |
| Zinc Transporter 8-Antigen. | P4Hb (Prolyl 4-Hydroxylase Subunit Beta) |
| Islet Cell Antigen (ICA); a mixture | P4Hb (Prolyl 4-Hydroxylase Subunit Beta) |
| GRP78 (Glucose-Regulated Protein 78) | |
| Urocortin-3 | |
| Oxidative Post-Translational Modifications (oxPTM)-Insulin |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ludvigsson, J. Autoantigen Treatment in Type 1 Diabetes: Unsolved Questions on How to Select Autoantigen and Administration Route. Int. J. Mol. Sci. 2020, 21, 1598. https://doi.org/10.3390/ijms21051598
Ludvigsson J. Autoantigen Treatment in Type 1 Diabetes: Unsolved Questions on How to Select Autoantigen and Administration Route. International Journal of Molecular Sciences. 2020; 21(5):1598. https://doi.org/10.3390/ijms21051598
Chicago/Turabian StyleLudvigsson, Johnny. 2020. "Autoantigen Treatment in Type 1 Diabetes: Unsolved Questions on How to Select Autoantigen and Administration Route" International Journal of Molecular Sciences 21, no. 5: 1598. https://doi.org/10.3390/ijms21051598
APA StyleLudvigsson, J. (2020). Autoantigen Treatment in Type 1 Diabetes: Unsolved Questions on How to Select Autoantigen and Administration Route. International Journal of Molecular Sciences, 21(5), 1598. https://doi.org/10.3390/ijms21051598