Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis—A Novel Form of Non-Apoptotic Cell Death?

 , ,
, , 
Abstract
:1. Introduction
2. General Mechanisms of Liver Cell Death
3. Apoptosis
4. Necrosis and Necroptosis
5. Autophagy
6. Pyroptosis
7. Iron Metabolism
8. Ferroptosis
8.1. The Role of Lipids in Ferroptosis
8.2. Keap1-Nrf2 System
8.3. Interaction between Iron and Oxidative Stress
9. Proposed Biomarkers of Ferroptotic Cell Death
10. Ferroptosis and Liver Disease
10.1. Ferroptosis in Metabolic Liver Diseases: Hereditary Hemochromatosis | Non-Alcoholic Fatty Liver Disease (NAFLD)
10.1.1. Hemochromatosis (HH)
10.1.2. Non-Alcoholic Fatty Liver Disease (NAFLD)
10.2. Ferroptosis in Alcoholic Liver Disease (ALD)
10.3. Ferroptosis and Viral Hepatitis
10.4. Ferroptosis and Drug-Induced Liver Injury (DILI)
10.5. Ferroptosis and Hepatocellular Carcinoma (HCC)
11. Experimental Models of Iron Overload and Liver Damage
12. Clinical Implications for the Study of Ferroptosis
13. Pharmacological Modulation of Ferroptosis
14. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Acsl4 | Acyl-CoA Synthetase Long Chain Family Member 4 |
| APAF1 | Apoptotic Protease-Activating Factor-I |
| APAP | Acetaminophen |
| AREs | Antioxidant Response Elements |
| ASH | Alcoholic Steatohepatitis |
| ATGs | autophagosomes |
| ATP | Adenosine Triphosphate |
| Bcl-2 | B-cell Lymphoma 2 |
| BH3 | Bcl-2 Homologous 3 |
| BMP | Bone Morphogenetic Protein |
| Cbr3 | Carbonyl Reductase 3 |
| CNS | Central Nervous System |
| COX | Cyclooxygenase |
| Cul3 | Cullin 3 |
| DAMP | Damage-Associated Molecular Pattern |
| DATP | Deoxyadenosine Triphosphate |
| D-cytb | Duodenal Cytochrome b |
| DFO | Deferoxamine |
| DILI | Drug-Induced Liver Injury |
| DIOS | Dysmetabolic Iron Overload Syndrome |
| DMT1 | Divalent Metal-Ion Transporter 1 |
| DNA | Deoxyribonucleic Acid |
| DNICs | Dinitrosyl-Dithiolato-Fe Complexes |
| DR | Death Receptor |
| DsRNA | Double Stranded RNA |
| EpREs | Electrophile Response Elements |
| ER | Endoplasmic Reticulum |
| FADD | Fas-Associated Death-Domain |
| FAS | Fas Cell Surface Death Receptor |
| Fe3+ | Ferric Iron |
| Fe2+ | Ferrous Iron |
| FINs | Ferroptosis-Inducing Compounds |
| FLIP | FLICE-Inhibitory Protein |
| Fpn1 | Ferroportin |
| GCL | Y-Glutamyl-Cysteine Ligase |
| GGC | Y-Glutamyl-Cysteine |
| GPX | Glutathione Peroxidase |
| Gpx4 | Glutathione Peroxidase 4 |
| GSS | Glutathione synthetase |
| GSDM | Gasdermin |
| GSH | Glutathione |
| GST | Glutathione S-Transferase |
| H2O2 | Hydrogen Peroxide |
| HCC | Hepatocellular Carcinoma |
| HCV | Hepatitis C Virus Infection |
| HH | Hereditary Hemochromatosis |
| HFE | Homeostatic Iron Regulator |
| HNE | 4-Hydroxynonenal |
| INF | Interferon |
| IRP1 | Iron Regulatory Protein 1 |
| Ipr2-/- | Regulatory Protein 2 |
| 5-LOX | 5-Lipoxygenase |
| KCs | Kupffer Cells |
| Keap-1 | Kelch-Like Erythroid Cell-Derived Protein 1 |
| LOX | Lipoxygenases |
| LPO | lipid peroxidation |
| MDA | Malondialdehyde |
| MLKL | Mixed Lineage kinase Domain-Like |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NAFLD | Non-Alcoholic Fatty Liver Disease |
| NASH | Non-Alcoholic Steatohepatitis |
| NAPQI | N-Acetyl-P-Benzoquinone Imine |
| NF-κB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| NLR | Nucleotide-Binding Domain–Like Receptors |
| NO | Nitrogen Monoxide |
| Nrf2 | NF-E2-Related Factor 2 |
| NTBI | non-transferrin-bound iron |
| PAMPs | Pathogen-Associated Molecular Patterns |
| PIR | Pirin |
| Pro-IL | Pro-Inflammatory Cytokine Interleukin |
| PRRs | Pattern Recognition Receptors |
| Ptgs2 | Prostaglandin-Endoperoxide Synthase 2 |
| PUFA-PL | Polyunsaturated-Fatty-Acid-Containing Phospholipids |
| PYHIN | Pyrin and HIN Domain |
| Rb | Retinoblastoma |
| RIPK | Receptor-Interacting Serine/Threonine-Protein Kinase |
| RHIM | RIP Homotypic Interaction Motif |
| ROS | Reactive Oxygen Species |
| RNS | Reactive Nitrogen Species |
| RSL3 | Ras-Selective Lethal 3 |
| SiRNA | Small Interfering RNA |
| SLC3A2 | Glutamate/Cystine Antiporter Solute Carrier Family 3 Member 2 |
| SLC7A11 | Glutamate/Cystine Antiporter Solute Carrier Family 7 Member 11 |
| sMAF | Heterodimer with Small MAF |
| TfR1 | Transferrin Receptor 1 |
| TLR | Toll Like Receptors |
| TNF | Tumor Necrosis Factor |
| TNFR1 | TNF Receptor Superfamily Member 1A |
| TRADD | TNF-R Adopter Protein via Death Domain |
| TRAIL | TNF-Related Apoptosis-Inducing Ligand, |
| TRIM | Tripartite Motif |
References
- Green, R.M.; Flamm, S. AGA technical review on the evaluation of liver chemistry tests. Gastroenterology 2002, 123, 1367–1384. [Google Scholar] [CrossRef] [Green Version]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef]
- Moriyama, M.; Matsumura, H.; Aoki, H.; Shimizu, T.; Yamagami, H.; Shioda, A.; Kaneko, M.; Goto, I.; Tanaka, N.; Arakawa, Y. Decreased risk of hepatocellular carcinoma in patients with chronic hepatitis C whose serum alanine aminotransferase levels became less than twice the upper limit of normal following interferon therapy. Liver Int. 2005, 25, 85–90. [Google Scholar] [CrossRef]
- Miyake, Y.; Iwasaki, Y.; Terada, R.; Okamaoto, R.; Ikeda, H.; Makino, Y.; Kobashi, H.; Takaguchi, K.; Sakaguchi, K.; Shiratori, Y. Persistent elevation of serum alanine aminotransferase levels leads to poor survival and hepatocellular carcinoma development in type 1 autoimmune hepatitis. Aliment. Pharmacol. Ther. 2006, 24, 1197–1205. [Google Scholar] [CrossRef]
- Eguchi, A.; Wree, A.; Feldstein, A.E. Biomarkers of liver cell death. J. Hepatol. 2014, 60, 1063–1074. [Google Scholar] [CrossRef] [Green Version]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783.e4. [Google Scholar] [CrossRef] [Green Version]
- Wang, K. Molecular mechanisms of hepatic apoptosis regulated by nuclear factors. Cell. Signal. 2015, 27, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhi, H.; Guicciardi, M.E.; Gores, G.J. Hepatocyte death: A clear and present danger. Physiol. Rev. 2010, 90, 1165–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.-S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Mehta, K.J.; Je Farnaud, S.; Sharp, P.A. Iron and liver fibrosis: Mechanistic and clinical aspects. World J. Gastroenterol. 2019, 25, 521–538. [Google Scholar] [CrossRef]
- Pietrangelo, A. Iron in NASH, chronic liver diseases and HCC: How much iron is too much? J. Hepatol. 2009, 50, 249–251. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Tan, H.-Y.; Wang, N.; Zhang, Z.-J.; Lao, L.; Wong, C.-W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.-D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol. 2016, 16, 7–21. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Strasser, A.; McDunn, J.E.; Swanson, P.E. Cell Death. N. Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Rautou, P.E.; Cazals-Hatem, D.; Moreau, R.; Francoz, C.; Feldmann, G.; Lebrec, D.; Ogier-Denis, É.; Bedossa, P.; Valla, D.; Durand, F. Acute Liver Cell Damage in Patients With Anorexia Nervosa: A Possible Role of Starvation-Induced Hepatocyte Autophagy. Gastroenterology 2008, 135, 840–848. [Google Scholar] [CrossRef]
- Vande Walle, L.; Lamkanfi, M. Pyroptosis. Curr. Biol. 2016, 26, R568–R572. [Google Scholar] [CrossRef] [Green Version]
- Wallach, D.; Kang, T.-B.; Dillon, C.P.; Green, D.R. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science 2016, 352, aaf2154. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef]
- Cadwell, K. Crosstalk between autophagy and inflammatory signalling pathways: Balancing defence and homeostasis. Nat. Rev. Immunol. 2016, 16, 661. [Google Scholar] [CrossRef]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guicciardi, M.E.; Gores, G.J. Apoptosis: A mechanism of acute and chronic liver injury. Gut 2005, 54, 1024–1033. [Google Scholar] [CrossRef] [Green Version]
- Wang, K. Molecular mechanisms of hepatic apoptosis. Cell Death Dis. 2014, 5, e996. [Google Scholar] [CrossRef]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Canbay, A.; Feldstein, A.E.; Higuchi, H.; Werneburg, N.; Grambihler, A.; Bronk, S.F.; Gores, G.J. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology 2003, 38, 1188–1198. [Google Scholar] [CrossRef]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef] [Green Version]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of Bcl-xL-Bak peptide complex: Recognition between regulators of apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Bouchier-Hayes, L.; Green, D.R. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, Q.; Shi, Y. Apoptosome: A platform for the activation of initiator caspases. Cell Death Differ. 2007, 14, 56–65. [Google Scholar] [CrossRef]
- Ow, Y.-L.P.; Green, D.R.; Hao, Z.; Mak, T.W. Cytochrome c: Functions beyond respiration. Nat. Rev. Mol. Cell Biol. 2008, 9, 532–542. [Google Scholar] [CrossRef]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413. [Google Scholar] [CrossRef]
- Wilson, N.S.; Dixit, V.; Ashkenazi, A. Death receptor signal transducers: Nodes of coordination in immune signaling networks. Nat. Immunol. 2009, 10, 348–355. [Google Scholar] [CrossRef]
- Scott, F.L.; Stec, B.; Pop, C.; Dobaczewska, M.K.; Lee, J.J.; Monosov, E.; Robinson, H.; Salvesen, G.S.; Schwarzenbacher, R.; Riedl, S.J. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 2009, 457, 1019–1022. [Google Scholar] [CrossRef] [Green Version]
- Bordon, Y. Cell death and immunity: Gasdermins: The hole picture emerges. Nat. Rev. Immunol. 2016, 16, 401. [Google Scholar] [CrossRef]
- Thome, M.; Schneider, P.; Hofmann, K.; Fickenscher, H.; Meinl, E.; Neipel, F.; Mattmann, C.; Burns, K.; Bodmer, J.L.; Schröter, M.; et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997, 386, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Chan, F.K.-M.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Vanden Berghe, T.; D’Herde, K.; Vandenabeele, P. Apoptosis and necrosis: Detection, discrimination and phagocytosis. Methods 2008, 44, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Tomaipitinca, L.; Mandatori, S.; Mancinelli, R.; Giulitti, F.; Petrungaro, S.; Moresi, V.; Facchiano, A.; Ziparo, E.; Gaudio, E.; Giampietri, C. The Role of Autophagy in Liver Epithelial Cells and Its Impact on Systemic Homeostasis. Nutrients 2019, 11, 827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. Permeases recycle amino acids resulting from autophagy. Autophagy 2007, 3, 149–150. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptopic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D. The role of NCOA4-mediated ferritinophagy in health and disease. Pharmaceuticals 2018, 11, 114. [Google Scholar] [CrossRef] [Green Version]
- Fleming, R.E.; Ponka, P. Iron overload in human disease. N. Engl. J. Med. 2012, 366, 348–359. [Google Scholar] [CrossRef] [Green Version]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meynard, D.; Babitt, J.L.; Lin, H.Y. The liver: Conductor of systemic iron balance. Blood 2014, 123, 168–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, N.; Zhang, A.-S.; Enns, C.A. Iron regulation by hepcidin. J. Clin. Invest. 2013, 123, 2337–2343. [Google Scholar] [CrossRef]
- Wang, C.Y.; Babitt, J.L. Liver iron sensing and body iron homeostasis. Blood 2019, 133, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganz, T.; Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Dalleau, S.; Baradat, M.; Guéraud, F.; Huc, L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013, 20, 1615–1630. [Google Scholar] [CrossRef] [Green Version]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Shimada, K.; Hayano, M.; Pagano, N.C.; Stockwell, B.R. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem. Biol. 2016, 23, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Barrientos, T.; Andrews, N.C. Iron and copper in mitochondrial diseases. Cell Metab. 2013, 17, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beard, J.L. Iron Biology in Immune Function, Muscle Metabolism and Neuronal Functioning. J. Nutr. 2001, 131, 568S–580S. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta - Gen. Subj. 2013, 1830, 3289–3303. [Google Scholar]
- Wang, H.; An, P.; Xie, E.; Wu, Q.; Fang, X.; Gao, H.; Zhang, Z.; Li, Y.; Wang, X.; Zhang, J.; et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017, 66, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.; Welsch, M.; Skouta, R.; Lee, E.; Hayano, M.; Thomas, A.G.; Gleason, C.; Tatonetti, N.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.Y.S.; Lee, D.H.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J.Y.S. Ferroptosis-induced endoplasmic reticulum stress: Cross-talk between ferroptosis and apoptosis. Mol. Cancer Res. 2018, 16, 1073–1076. [Google Scholar] [CrossRef] [Green Version]
- Amri, E.Z.; Ailhaud, G.; Grimaldi, P.A. Fatty acids as signal transducing molecules: Involvement in the differentiation of preadipose to adipose cells. J. Lipid Res. 1994, 35, 930–937. [Google Scholar]
- De Carvalho, C.C.C.R.; Caramujo, M.J. The various roles of fatty acids. Molecules 2018, 23, 2583. [Google Scholar] [CrossRef] [Green Version]
- Sellmayer, A.; Danesch, U.; Weber, P.C. Effects of different polunsaturated fatty acids on growth-related early gene expression and cell growth. Lipids 1996, 31, S37. [Google Scholar] [CrossRef]
- Brash, A.R. Arachidonic acid as a bioactive molecule. J. Clin. Invest. 2001, 107, 1339–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.G.; Chen, Y.Q.; Honn, K.V. Arachidonate lipoxygenases as essential regulators of cell survival and apoptosis. Proc. Natl. Acad. Sci. USA. 1996, 93, 5241–5246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magtanong, L.; Ko, P.J.; Dixon, S.J. Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ. 2016, 23, 1099–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, A.J.; Fullerton, J.N.; Massey, K.A.; Auld, G.; Sewell, G.; James, S.; Newson, J.; Karra, E.; Winstanley, A.; Alazawi, W.; et al. Immunosuppression in acutely decompensated cirrhosis is mediated by prostaglandin E2. Nat. Med. 2014, 20, 518. [Google Scholar]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Chanas, S.A.; Henderson, C.J.; McLellan, L.I.; Wolf, C.R.; Cavin, C.; Hayes, J.D. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001, 61, 3299–3307. [Google Scholar]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef]
- Leone, A.; Roca, M.S.; Ciardiello, C.; Terranova-Barberio, M.; Vitagliano, C.; Ciliberto, G.; Mancini, R.; Di Gennaro, E.; Bruzzese, F.; Budillon, A. Vorinostat synergizes with EGFR inhibitors in NSCLC cells by increasing ROS via up-regulation of the major mitochondrial porin VDAC1 and modulation of the c-Myc-NRF2-KEAP1 pathway. Free Radic. Biol. Med. 2015, 89, 287–299. [Google Scholar] [CrossRef]
- Kang, M.-I.; Kobayashi, A.; Wakabayashi, N.; Kim, S.-G.; Yamamoto, M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2046–2051. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Long, M.J.; Lin, H.-Y.; Parvez, S.; Zhao, Y.; Poganik, J.R.; Huang, P.; Aye, Y. β-TrCP1 Is a Vacillatory Regulator of Wnt Signaling. Cell Chem. Biol. 2017, 24, 944–957.e7. [Google Scholar] [CrossRef] [Green Version]
- Takaya, K.; Suzuki, T.; Motohashi, H.; Onodera, K.; Satomi, S.; Kensler, T.W.; Yamamoto, M. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2012, 53, 817–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol. Cell. Biol. 2016, 36, 271–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, K.; Yamamoto, M. The KEAP1NRF2 system in cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Chiu, D.T.-Y.; Wei, Y.-H. Special issue on “Oxidative stress and mitochondrial alterations in aging and disease”. Free Radic. Res. 2014, 48, 967–969. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-H.; Kim, J.-H.; Kim, E.-H.; Na, H.-K.; Cha, Y.-N.; Chung, J.H.; Surh, Y.-J. 15-Deoxy-Delta12,14-prostaglandin J2 upregulates the expression of heme oxygenase-1 and subsequently matrix metalloproteinase-1 in human breast cancer cells: Possible roles of iron and ROS. Carcinogenesis 2009, 30, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Keleku-Lukwete, N.; Suzuki, M.; Yamamoto, M. An Overview of the Advantages of KEAP1-NRF2 System Activation During Inflammatory Disease Treatment. Antioxid. Redox Signal. 2018, 29, 1746–1755. [Google Scholar] [CrossRef]
- Kawatani, Y.; Suzuki, T.; Shimizu, R.; Kelly, V.P.; Yamamoto, M. Nrf2 and selenoproteins are essential for maintaining oxidative homeostasis in erythrocytes and protecting against hemolytic anemia. Blood 2011, 117, 986–996. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Rehmani, I.; Esaki, S.; Fu, R.; Chen, L.; de Serrano, V.; Liu, A. Pirin is an iron-dependent redox regulator of NF-κB. Proc. Natl. Acad. Sci. USA 2013, 110, 9722–9727. [Google Scholar] [CrossRef] [Green Version]
- Meneghini, R. Iron homeostasis, oxidative stress, and DNA damage. Free Radic. Biol. Med. 1997, 23, 783–792. [Google Scholar] [CrossRef]
- Nakajima, S.; Kitamura, M. Bidirectional regulation of NF-κB by reactive oxygen species: A role of unfolded protein response. Free Radic. Biol. Med. 2013, 65, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Suryo Rahmanto, Y.; Kalinowski, D.S.; Lane, D.J.R.; Lok, H.C.; Richardson, V.; Richardson, D.R. Nitrogen monoxide (NO) storage and transport by dinitrosyl-dithiol-iron complexes: Long-lived NO that is trafficked by interacting proteins. J. Biol. Chem. 2012, 287, 6960–6968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, D.R.; Lok, H.C. The nitric oxide-iron interplay in mammalian cells: Transport and storage of dinitrosyl iron complexes. Biochim. Biophys. Acta 2008, 1780, 638–651. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Demple, B. Direct nitric oxide signal transduction via nitrosylation of iron-sulfur centers in the SoxR transcription activator. Proc. Natl. Acad. Sci. USA 2000, 97, 5146–5150. [Google Scholar] [CrossRef] [Green Version]
- Osler, W. Clinical Remarks on Hypertrophic Cirrhosis of the Liver with Bronzing of the Skin: Haemochromatosis. Br. Med. J. 1899, 2, 1595–1596. [Google Scholar] [CrossRef]
- Anderson, E.R.; Shah, Y.M. Iron homeostasis in the liver. Compr. Physiol. 2013, 3, 315–330. [Google Scholar]
- Niederau, C.; Fischer, R.; Pürschel, A.; Stremmel, W.; Häussinger, D.; Strohmeyer, G. Long-term survival in patients with hereditary hemochromatosis. Gastroenterology 1996, 110, 1107–1119. [Google Scholar] [CrossRef]
- Powell, L.W.; Seckington, R.C.; Deugnier, Y. Haemochromatosis. Lancet 2016, 388, 706–716. [Google Scholar] [CrossRef]
- Fracanzani, A.L.; Fargion, S.; Romano, R.; Conte, D.; Piperno, A.; D’Alba, R.; Mandelli, C.; Fraquelli, M.; Pacchetti, S.; Braga, M. Portal hypertension and iron depletion in patients with genetic hemochromatosis. Hepatology 1995, 22, 1127–1131. [Google Scholar] [CrossRef]
- Cooksey, R.C.; Jouihan, H.A.; Ajioka, R.S.; Hazel, M.W.; Jones, D.L.; Kushner, J.P.; McClain, D.A. Oxidative stress, beta-cell apoptosis, and decreased insulin secretory capacity in mouse models of hemochromatosis. Endocrinology 2004, 145, 5305–5312. [Google Scholar] [CrossRef]
- Fernández-Real, J.M.; Peñarroja, G.; Castro, A.; García-Bragado, F.; Hernández-Aguado, I.; Ricart, W. Blood letting in high-ferritin type 2 diabetes: Effects on insulin sensitivity and beta-cell function. Diabetes 2002, 51, 1000–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, L.; Moscatiello, S.; Vanni, E.; Fracanzani, A.L.; Bugianesi, E.; Fargion, S.; Marchesini, G. Venesection for non-alcoholic fatty liver disease unresponsive to lifestyle counselling--a propensity score-adjusted observational study. QJM 2011, 104, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Bozzini, C.; Girelli, D.; Olivieri, O.; Martinelli, N.; Bassi, A.; De Matteis, G.; Tenuti, I.; Lotto, V.; Friso, S.; Pizzolo, F.; et al. Prevalence of body iron excess in the metabolic syndrome. Diabetes Care 2005, 28, 2061–2063. [Google Scholar] [CrossRef] [Green Version]
- Simcox, J.A.; McClain, D.A. Iron and diabetes risk. Cell Metab. 2013, 17, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deugnier, Y.; Bardou-Jacquet, É.; Lainé, F. Dysmetabolic iron overload syndrome (DIOS). Presse Med. 2017, 46, e306–e311. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Real, J.M.; Ricart-Engel, W.; Arroyo, E.; Balançá, R.; Casamitjana-Abella, R.; Cabrero, D.; Fernández-Castañer, M.; Soler, J. Serum ferritin as a component of the insulin resistance syndrome. Diabetes Care 1998, 21, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Turlin, B.; Mendler, M.H.; Moirand, R.; Guyader, D.; Guillygomarc’h, A.; Deugnier, Y. Histologic features of the liver in insulin resistance-associated iron overload. A study of 139 patients. Am. J. Clin. Pathol. 2001, 116, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Marmur, J.; Beshara, S.; Eggertsen, G.; Onelöv, L.; Albiin, N.; Danielsson, O.; Hultcrantz, R.; Stål, P. Hepcidin levels correlate to liver iron content, but not steatohepatitis, in non-alcoholic fatty liver disease. BMC Gastroenterol. 2018, 18, 78. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Belt, P.; Wilson, L.A.; Yeh, M.M.; Neuschwander-Tetri, B.A.; Chalasani, N.; Sanyal, A.J.; Nelson, J.E. NASH Clinical Research Network Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Valenti, L.; Fracanzani, A.L.; Bugianesi, E.; Dongiovanni, P.; Galmozzi, E.; Vanni, E.; Canavesi, E.; Lattuada, E.; Roviaro, G.; Marchesini, G.; et al. HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 2010, 138, 905–912. [Google Scholar] [CrossRef] [Green Version]
- Fracanzani, A.L.; Valenti, L.; Bugianesi, E.; Vanni, E.; Grieco, A.; Miele, L.; Consonni, D.; Fatta, E.; Lombardi, R.; Marchesini, G.; et al. Risk of nonalcoholic steatohepatitis and fibrosis in patients with nonalcoholic fatty liver disease and low visceral adiposity. J. Hepatol. 2011, 54, 1244–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fargion, S.; Mattioli, M.; Fracanzani, A.L.; Sampietro, M.; Tavazzi, D.; Fociani, P.; Taioli, E.; Valenti, L.; Fiorelli, G. Hyperferritinemia, iron overload, and multiple metabolic alterations identify patients at risk for nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2001, 96, 2448–2455. [Google Scholar] [CrossRef] [PubMed]
- Manousou, P.; Kalambokis, G.; Grillo, F.; Watkins, J.; Xirouchakis, E.; Pleguezuelo, M.; Leandro, G.; Arvaniti, V.; Germani, G.; Patch, D.; et al. Serum ferritin is a discriminant marker for both fibrosis and inflammation in histologically proven non-alcoholic fatty liver disease patients. Liver Int. 2011, 31, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Hoki, T.; Miyanishi, K.; Tanaka, S.; Takada, K.; Kawano, Y.; Sakurada, A.; Sato, M.; Kubo, T.; Sato, T.; Sato, Y.; et al. Increased duodenal iron absorption through up-regulation of divalent metal transporter 1 from enhancement of iron regulatory protein 1 activity in patients with nonalcoholic steatohepatitis. Hepatology 2015, 62, 751–761. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Fracanzani, A.L.; Fargion, S.; Valenti, L. Iron in fatty liver and in the metabolic syndrome: A promising therapeutic target. J. Hepatol. 2011, 55, 920–932. [Google Scholar] [CrossRef]
- Beaton, M.D.; Chakrabarti, S.; Levstik, M.; Speechley, M.; Marotta, P.; Adams, P. Phase II clinical trial of phlebotomy for non-Alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2013. [Google Scholar] [CrossRef]
- Adams, L.A.; Crawford, D.H.; Stuart, K.; House, M.J.; St. Pierre, T.G.; Webb, M.; Ching, H.L.I.; Kava, J.; Bynevelt, M.; Macquillan, G.C.; et al. The impact of phlebotomy in nonalcoholic fatty liver disease: A prospective, randomized, controlled trial. Hepatology 2015. [Google Scholar] [CrossRef]
- Harrison-Findik, D.D.; Schafer, D.; Klein, E.; Timchenko, N.A.; Kulaksiz, H.; Clemens, D.; Fein, E.; Andriopoulos, B.; Pantopoulos, K.; Gollan, J. Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J. Biol. Chem. 2006, 281, 22974–22982. [Google Scholar] [CrossRef] [Green Version]
- Harrison-Findik, D.D.; Klein, E.; Crist, C.; Evans, J.; Timchenko, N.; Gollan, J. Iron-mediated regulation of liver hepcidin expression in rats and mice is abolished by alcohol. Hepatology 2007, 46, 1979–1985. [Google Scholar] [CrossRef]
- Eng, S.C.; Taylor, S.L.; Reyes, V.; Raaka, S.; Berger, J.; Kowdley, K. V Hepatic iron overload in alcoholic end-stage liver disease is associated with iron deposition in other organs in the absence of HFE-1 hemochromatosis. Liver Int. 2005, 25, 513–517. [Google Scholar] [CrossRef]
- Costa-Matos, L.; Batista, P.; Monteiro, N.; Simões, M.; Egas, C.; Pereira, J.; Pinho, H.; Santos, N.; Ribeiro, J.; Cipriano, M.A.; et al. Liver hepcidin mRNA expression is inappropriately low in alcoholic patients compared with healthy controls. Eur. J. Gastroenterol. Hepatol. 2012, 24, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Stål, P.; Hultcrantz, R. Iron increases ethanol toxicity in rat liver. J. Hepatol. 1993, 17, 108–115. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Horne, W.; Kamimura, S.; Niemelä, O.; Parkkila, S.; Ylä-Herttuala, S.; Brittenham, G.M. Experimental liver cirrhosis induced by alcohol and iron. J. Clin. Invest. 1995, 96, 620–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, L.M.; Dixon, J.L.; Purdie, D.M.; Powell, L.W.; Crawford, D.H.G. Excess alcohol greatly increases the prevalence of cirrhosis in hereditary hemochromatosis. Gastroenterology 2002, 122, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Iron-induced oxidant stress in alcoholic liver fibrogenesis. Alcohol 2003, 30, 121–129. [Google Scholar] [CrossRef]
- Raynard, B.; Balian, A.; Fallik, D.; Capron, F.; Bedossa, P.; Chaput, J.-C.; Naveau, S. Risk factors of fibrosis in alcohol-induced liver disease. Hepatology 2002, 35, 635–638. [Google Scholar] [CrossRef]
- Ganne-Carrié, N.; Christidis, C.; Chastang, C.; Ziol, M.; Chapel, F.; Imbert-Bismut, F.; Trinchet, J.C.; Guettier, C.; Beaugrand, M. Liver iron is predictive of death in alcoholic cirrhosis: A multivariate study of 229 consecutive patients with alcoholic and/or hepatitis C virus cirrhosis: A prospective follow up study. Gut 2000, 46, 277–282. [Google Scholar] [CrossRef]
- Ma, L.; Zou, T.; Yuan, Y.; Lv, J.; Dong, X.; Yang, G.; Zhu, Y.; Luo, J.; Zhang, Z.; Yang, J. Duodenal ferroportin is up-regulated in patients with chronic hepatitis C. PLoS One 2014, 9, e110658. [Google Scholar] [CrossRef]
- Fujita, N.; Horiike, S.; Sugimoto, R.; Tanaka, H.; Iwasa, M.; Kobayashi, Y.; Hasegawa, K.; Ma, N.; Kawanishi, S.; Adachi, Y.; et al. Hepatic oxidative DNA damage correlates with iron overload in chronic hepatitis C patients. Free Radic. Biol. Med. 2007, 42, 353–362. [Google Scholar] [CrossRef]
- Lambrecht, R.W.; Sterling, R.K.; Naishadham, D.; Stoddard, A.M.; Rogers, T.; Morishima, C.; Morgan, T.R.; Bonkovsky, H.L. HALT-C Trial Group Iron levels in hepatocytes and portal tract cells predict progression and outcomes of patients with advanced chronic hepatitis C. Gastroenterology 2011, 140, 1490–1500. [Google Scholar] [CrossRef] [Green Version]
- Guyader, D.; Thirouard, A.-S.; Erdtmann, L.; Rakba, N.; Jacquelinet, S.; Danielou, H.; Perrin, M.; Jouanolle, A.-M.; Brissot, P.; Deugnier, Y. Liver iron is a surrogate marker of severe fibrosis in chronic hepatitis C. J. Hepatol. 2007, 46, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.N.; Uprichard, S.L. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl. Acad. Sci. USA 2013, 110, 10777–10782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurl, I.; Zoller, H.; Obrist, P.; Datz, C.; Bachmann, F.; Elliott, R.M.; Weiss, G. Iron regulates hepatitis C virus translation via stimulation of expression of translation initiation factor 3. J. Infect. Dis. 2004, 190, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Urban, S. Entry of hepatitis B and hepatitis D virus into hepatocytes: Basic insights and clinical implications. J. Hepatol. 2016, 64, S32–S40. [Google Scholar] [CrossRef]
- Ryan, J.D.; Altamura, S.; Devitt, E.; Mullins, S.; Lawless, M.W.; Muckenthaler, M.U.; Crowe, J. Pegylated interferon-α induced hypoferremia is associated with the immediate response to treatment in hepatitis C. Hepatology 2012, 56, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, F.; Ventura, P.; Vegetti, A.; Guido, M.; Abbati, G.; Corradini, E.; Fattovich, G.; Ferrari, C.; Tagliazucchi, M.; Carbonieri, A.; et al. Serum ferritin as a predictor of treatment outcome in patients with chronic hepatitis C. Am. J. Gastroenterol. 2009, 104, 605–616. [Google Scholar]
- Lange, C.M.; Kutalik, Z.; Morikawa, K.; Bibert, S.; Cerny, A.; Dollenmaier, G.; Dufour, J.F.; Gerlach, T.J.; Heim, M.H.; Malinverni, R.; et al. Serum ferritin levels are associated with a distinct phenotype of chronic hepatitis C poorly responding to pegylated interferon-alpha and ribavirin therapy. Hepatology 2012, 55, 1038–1047. [Google Scholar] [CrossRef]
- Kullak-Ublick, G.A.; Andrade, R.J.; Merz, M.; End, P.; Benesic, A.; Gerbes, A.L.; Aithal, G.P. Drug-induced liver injury: Recent advances in diagnosis and risk assessment. Gut 2017, 66, 1154–1164. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.M. Acetaminophen (APAP) hepatotoxicity-Isn’t it time for APAP to go away? J. Hepatol. 2017, 67, 1324–1331. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Ramachandran, A.; Jaeschke, H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol. 2016, 10, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Lőrincz, T.; Jemnitz, K.; Kardon, T.; Mandl, J.; Szarka, A. Ferroptosis is Involved in Acetaminophen Induced Cell Death. Pathol. Oncol. Res. 2015, 21, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Heidari, R.; Esmailie, N.; Azarpira, N.; Najibi, A.; Niknahad, H. Effect of Thiol-reducing Agents and Antioxidants on Sulfasalazine-induced Hepatic Injury in Normotermic Recirculating Isolated Perfused Rat Liver. Toxicol. Res. 2016, 32, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramappa, V.; Aithal, G.P. Hepatotoxicity Related to Anti-tuberculosis Drugs: Mechanisms and Management. J. Clin. Exp. Hepatol. 2013, 3, 37–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Swelm, R.P.L.; Laarakkers, C.M.M.; Blous, L.; Peters, J.G.P.; Blaney Davidson, E.N.; van der Kraan, P.M.; Swinkels, D.W.; Masereeuw, R.; Russel, F.G.M. Acute acetaminophen intoxication leads to hepatic iron loading by decreased hepcidin synthesis. Toxicol. Sci. 2012, 129, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahon, P.; Sutton, A.; Rufat, P.; Ziol, M.; Thabut, G.; Schischmanoff, P.O.; Vidaud, D.; Charnaux, N.; Couvert, P.; Ganne-Carrie, N.; et al. Liver Iron, HFE Gene Mutations, and Hepatocellular Carcinoma Occurrence in Patients With Cirrhosis. Gastroenterology 2008, 134, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Hellerbrand, C.; Pöppl, A.; Hartmann, A.; Schölmerich, J.; Lock, G. HFE C282Y heterozygosity in hepatocellular carcinoma: Evidence for an increased prevalence. Clin. Gastroenterol. Hepatol. 2003, 1, 279–284. [Google Scholar] [CrossRef]
- Kowdley, K.V. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology 2004. [Google Scholar] [CrossRef]
- Lu, B.; Chen, X.B.; Ying, M.D.; He, Q.J.; Cao, J.; Yang, B. The Role of Ferroptosis in Cancer Development and Treatment Response. Front. Pharmacol. 2018, 8, 992. [Google Scholar] [CrossRef]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.-C.; Mazière, J.-C.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014, 34, 6417–6422. [Google Scholar]
- Louandre, C.; Marcq, I.; Bouhlal, H.; Lachaier, E.; Godin, C.; Saidak, Z.; François, C.; Chatelain, D.; Debuysscher, V.; Barbare, J.-C.; et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015, 356, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Okuma, Y.; Kashima, J.; Watanabe, K.; Homma, S. Survival analysis and pathological features of advanced non-small cell lung cancer with miliary pulmonary metastases in patients harboring epidermal growth factor receptor mutations. J. Cancer Res. Clin. Oncol. 2018, 144, 1601–1611. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Craven, C.M.; Alexander, J.; Eldridge, M.; Kushner, J.P.; Bernstein, S.; Kaplan, J. Tissue distribution and clearance kinetics of non-transferrin-bound iron in the hypotransferrinemic mouse: A rodent model for hemochromatosis. Proc. Natl. Acad. Sci. USA. 1987. [Google Scholar] [CrossRef] [Green Version]
- Raja, K.B.; Simpson, R.J.; Peters, T.J. Intestinal iron absorption studies in mouse models of iron-overload. Br. J. Haematol. 1994, 86, 156–162. [Google Scholar] [CrossRef]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef] [Green Version]
- Fleming, R.E.; Feng, Q.; Britton, R.S. Knockout Mouse Models of Iron Homeostasis. Annu. Rev. Nutr. 2011. [Google Scholar] [CrossRef]
- Masaratana, P.; Laftah, A.H.; Latunde-Dada, G.O.; Vaulont, S.; Simpson, R.J.; McKie, A.T. Iron absorption in hepcidin1 knockout mice. Br. J. Nutr. 2011, 105, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Lunova, M.; Goehring, C.; Kuscuoglu, D.; Mueller, K.; Chen, Y.; Walther, P.; Deschemin, J.-C.; Vaulont, S.; Haybaeck, J.; Lackner, C.; et al. Hepcidin knockout mice fed with iron-rich diet develop chronic liver injury and liver fibrosis due to lysosomal iron overload. J. Hepatol. 2014, 61, 633–641. [Google Scholar] [CrossRef]
- Kalaitzakis, E.; Josefsson, A.; Castedal, M.; Henfridsson, P.; Bengtsson, M.; Andersson, B.; Björnsson, E. Hepatic encephalopathy is related to anemia and fat-free mass depletion in liver transplant candidates with cirrhosis. Scand. J. Gastroenterol. 2013, 48, 577–584. [Google Scholar] [CrossRef]
- Les, I.; Doval, E.; Flavià, M.; Jacas, C.; Cárdenas, G.; Esteban, R.; Guardia, J.; Córdoba, J. Quality of life in cirrhosis is related to potentially treatable factors. Eur. J. Gastroenterol. Hepatol. 2010, 22, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Qamar, A.A.; Grace, N.D.; Groszmann, R.J.; Garcia-Tsao, G.; Bosch, J.; Burroughs, A.K.; Ripoll, C.; Maurer, R.; Planas, R.; Escorsell, A.; et al. Incidence, Prevalence, and Clinical Significance of Abnormal Hematologic Indices in Compensated Cirrhosis. Clin. Gastroenterol. Hepatol. 2009, 7, 689–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirera, I.; Elizalde, J.I.; Piqué, J.M.; Feu, F.; Casadevall, M.; Goldin, E.; Terés, J.; Bosch, J.; Rodés, J. Anemia worsens hyperdynamic circulation of patients with cirrhosis and portal hypertension. Dig. Dis. Sci. 1997, 42, 1697–1702. [Google Scholar] [CrossRef] [PubMed]
- Bruni, A.; Pepper, A.R.; Pawlick, R.L.; Gala-Lopez, B.; Gamble, A.F.; Kin, T.; Seeberger, K.; Korbutt, G.S.; Bornstein, S.R.; Linkermann, A.; et al. Ferroptosis-inducing agents compromise in vitro human islet viability and function article. Cell Death Dis. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; O’Connor, O.A.; Stockwell, B.R. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol. 2019, 26, 623–633. [Google Scholar] [CrossRef]
- Sato, M.; Kusumi, R.; Hamashima, S.; Kobayashi, S.; Sasaki, S.; Komiyama, Y.; Izumikawa, T.; Conrad, M.; Bannai, S.; Sato, H. The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 drives ferroptosis through GPX4 inactivation and ros production in colorectal cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef] [Green Version]
- Vučković, A.; Bosello Travain, V.; Bordin, L.; Cozza, G.; Miotto, G.; Rossetto, M.; Toppo, S.; Venerando, R.; Zaccarin, M.; Maiorino, M.; et al. Inactivation of the glutathione peroxidase GPx4 by the ferroptosis-inducing molecule RSL3 requires the adaptor protein 14-3-3ε. FEBS Lett. 2019, 594, 611–624. [Google Scholar]
- Lo, M.; Wang, Y.-Z.Z.; Gout, P.W. The xc- cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J. Cell. Physiol. 2008, 215, 593–602. [Google Scholar] [CrossRef]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x-c cystine transporter: A new action for an old drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef] [Green Version]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, W.; Li, Y.; Xiao, Y.; Cheng, J.; Jia, J. The 5-Lipoxygenase Inhibitor Zileuton Confers Neuroprotection against Glutamate Oxidative Damage by Inhibiting Ferroptosis. Biol. Pharm. Bull. 2015, 38, 1234–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajarabille, N.; Latunde-Dada, G.O. Latunde-Dada Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic/Adrenic Phosphatidylethanolamines Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81. [Google Scholar] [CrossRef]
- De Avellar, I.G.J.; Magalhães, M.M.M.; Silva, A.B.; Souza, L.L.; Leitão, A.C.; Hermes-Lima, M. Reevaluating the role of 1,10-phenanthroline in oxidative reactions involving ferrous ions and DNA damage. Biochim. Biophys. Acta - Gen. Subj. 2004, 1675, 46–53. [Google Scholar] [CrossRef]
- Viganor, L.; Howe, O.; McCarron, P.; McCann, M.; Devereux, M. The Antibacterial Activity of Metal Complexes Containing 1,10- phenanthroline: Potential as Alternative Therapeutics in the Era of Antibiotic Resistance. Curr. Top. Med. Chem. 2016, 17, 1280–1302. [Google Scholar] [CrossRef]
- Taher, A.T.; Porter, J.B.; Kattamis, A.; Viprakasit, V.; Cappellini, M.D. Efficacy and safety of iron-chelation therapy with deferoxamine, deferiprone, and deferasirox for the treatment of iron-loaded patients with nontransfusion-dependent thalassemia syndromes. Drug Des. Devel. Ther. 2016, 10, 4073–4078. [Google Scholar] [CrossRef] [Green Version]
- Kontoghiorghes, G.J.; Kontoghiorghe, C.N. Prospects for the introduction of targeted antioxidant drugs for the prevention and treatment of diseases related to free radical pathology. Expert Opin. Investig. Drugs 2019, 28, 593–603. [Google Scholar] [CrossRef]
- Hider, R.C.; Hoffbrand, A.V. The role of deferiprone in iron chelation. N. Engl. J. Med. 2018, 379, 2140–2150. [Google Scholar] [CrossRef] [Green Version]


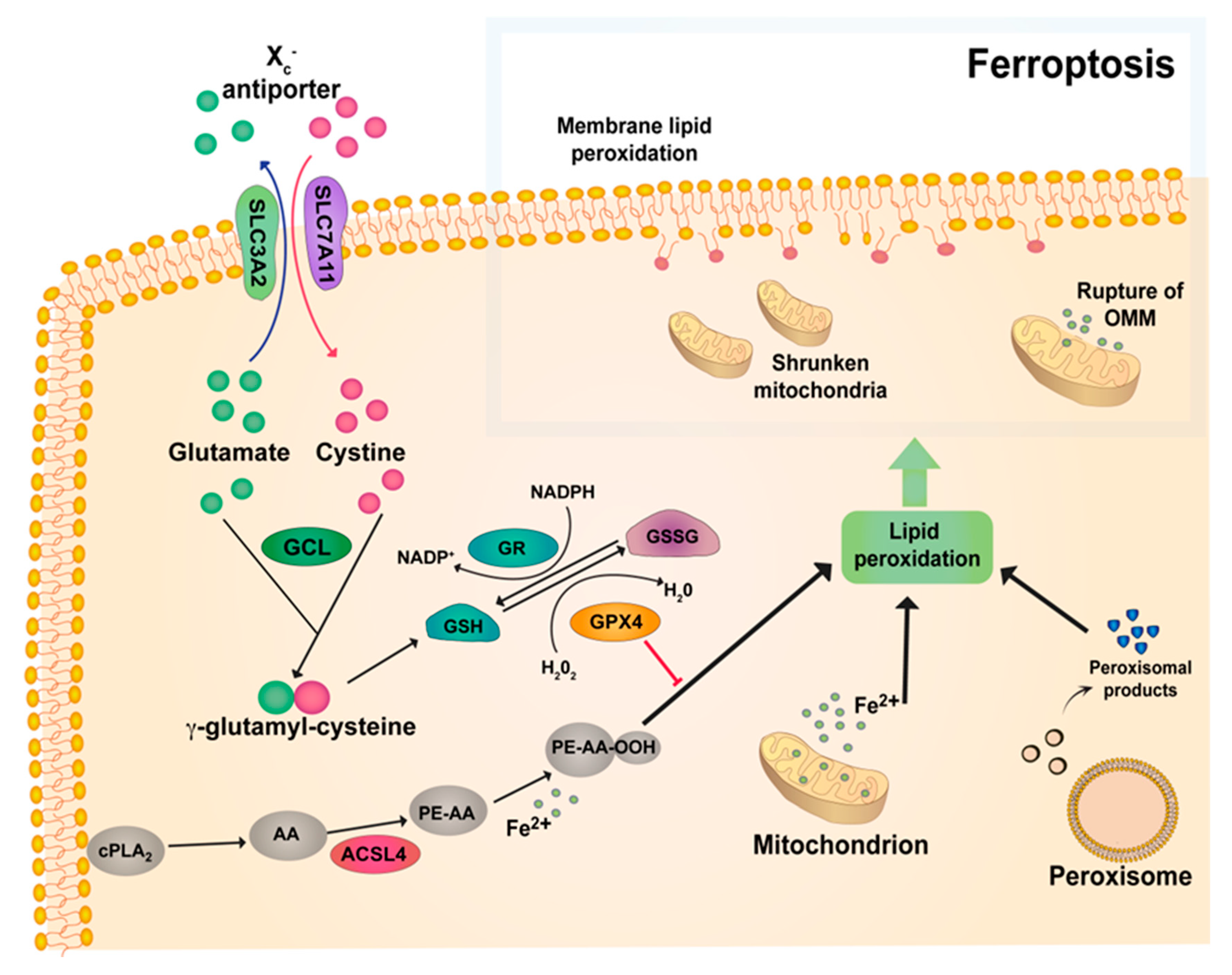{kind=link}
| Apoptosis [9,20,21] | Necrosis/Necroptosis [6,9,22,23,24] | Pyroptosis [25,26,27] | Autophagic Cell Death [28,29,30,31,32] | Ferroptosis [10,33,34] | |
|---|---|---|---|---|---|
| Morphological changes | Shrunken cells, membrane blebbing, nuclear condensation and fragmentation | Oncosis, swelling of the organelles and practically no change in the nuclei until later stages when chromatin condensation is observed | Plasma membrane rupture, pyroptotic body formation, and cell flattening | Formation of autophagosomes | Shrunken, electron-dense mitochondria and rupture of the outer mitochondrial membrane |
| Triggering stimuli | DNA damage and reactive oxygen species (ROS) overload or endoplasmic reticulum (ER) stress (intrinsic), extracellular microenvironment alterations and mediated by death receptors (DRs) (extrinsic) | Physicochemical stress in the cells, detected by TNFR1, Fas, or TLR-3/4 | Extracellular stimuli (e.g., TNF, IFNγ and TLR ligands) and different intracellular pathogens | Metabolic stressors | Glutamate, pharmacological induction (erastin, sulfasalazine, sorafenib) |
| Main components in the pathway | Caspases: initiation (caspase 2, 8, 9 and 10) and execution (caspase 3, 6 and 7) | (RIPK1), RIPK3, and mixed lineage kinase domain-like (MLKL) | Inflammasomes, caspase 1, IL-1ß, and IL-18 | ATGs proteins, acid hydrolases | Iron, GPX4, ACLS4, SLC7A11, PTGS2 |
| Compound | Molecular Target/Mechanism of Action | Common Use, Notes |
|---|---|---|
| INDUCER | ||
| Erastin [174,175,176] | Inhibits Xc- system (irreversibly) | Ferroptosis inducer in research |
| RSL3 [177,178] | Inactivates gpx4 | Ferroptosis inducer in research |
| Glutamate [179] | Competitive inhibition of the Xc- system | High concentrations inhibit the function of the antiporter, lowering the intracellular levels of GSH and therefore increasing oxidative damage. |
| Sulfasalazine [179,180] | Inhibits Xc- system | Patients with inflammatory bowel disease and arthropathies. Used in research in different types of cancer (v.gr. lymphoma, CNS tumors) |
| Sorafenib [160] | Multikinase inhibitor/inhibit Xc- system | Used mainly as a therapy in patients with advanced hepatocellular carcinoma |
| INHIBITOR | ||
| Ferrostatin-1 [181] | Interferes with ROS accumulation from lipid peroxidation | Second- and third-generation ferrostatins are more stable. |
| Liproxstatin-1 [15] | Interferes with ROS accumulation from lipid peroxidation | Relative potency stronger than Ferr-1. Inhibits FINs (RSL3, erastin). |
| Zileuton [182] | Inhibits 5-LOX (abrogates cytosolic ROS production) | Available as an oral compound. |
| DFO [34,174] | Iron chelator | Used in patients with iron overload |
| Vitamin E and analogs (v.gr. Trolox) [183,184] | Antioxidant/ROS scavenger | Some trials have tested its effect (e.g., age-related macular degeneration, dementia, metabolic diseases, NAFLD) without conclusive results |
| 1,10-phenanthroline [185,186] | Iron chelator | Used as a metal chelator and redox indicator. Mixed with different metals (Cu, Mn, Ag) has antimicrobial activity |
| deferasirox [187,188] | Iron chelator | Used in patients with iron overload |
| deferiprone [187,188,189] | Iron chelator | Used in patients with iron overload |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macías-Rodríguez, R.U.; Inzaugarat, M.E.; Ruiz-Margáin, A.; Nelson, L.J.; Trautwein, C.; Cubero, F.J. Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis—A Novel Form of Non-Apoptotic Cell Death? Int. J. Mol. Sci. 2020, 21, 1651. https://doi.org/10.3390/ijms21051651
Macías-Rodríguez RU, Inzaugarat ME, Ruiz-Margáin A, Nelson LJ, Trautwein C, Cubero FJ. Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis—A Novel Form of Non-Apoptotic Cell Death? International Journal of Molecular Sciences. 2020; 21(5):1651. https://doi.org/10.3390/ijms21051651
Chicago/Turabian StyleMacías-Rodríguez, Ricardo U., María Eugenia Inzaugarat, Astrid Ruiz-Margáin, Leonard J. Nelson, Christian Trautwein, and Francisco Javier Cubero. 2020. "Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis—A Novel Form of Non-Apoptotic Cell Death?" International Journal of Molecular Sciences 21, no. 5: 1651. https://doi.org/10.3390/ijms21051651
APA StyleMacías-Rodríguez, R. U., Inzaugarat, M. E., Ruiz-Margáin, A., Nelson, L. J., Trautwein, C., & Cubero, F. J. (2020). Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis—A Novel Form of Non-Apoptotic Cell Death? International Journal of Molecular Sciences, 21(5), 1651. https://doi.org/10.3390/ijms21051651