The p38 Pathway: From Biology to Cancer Therapy

 , and
, and
Abstract
:1. Introduction
2. p38 MAPK Diversity
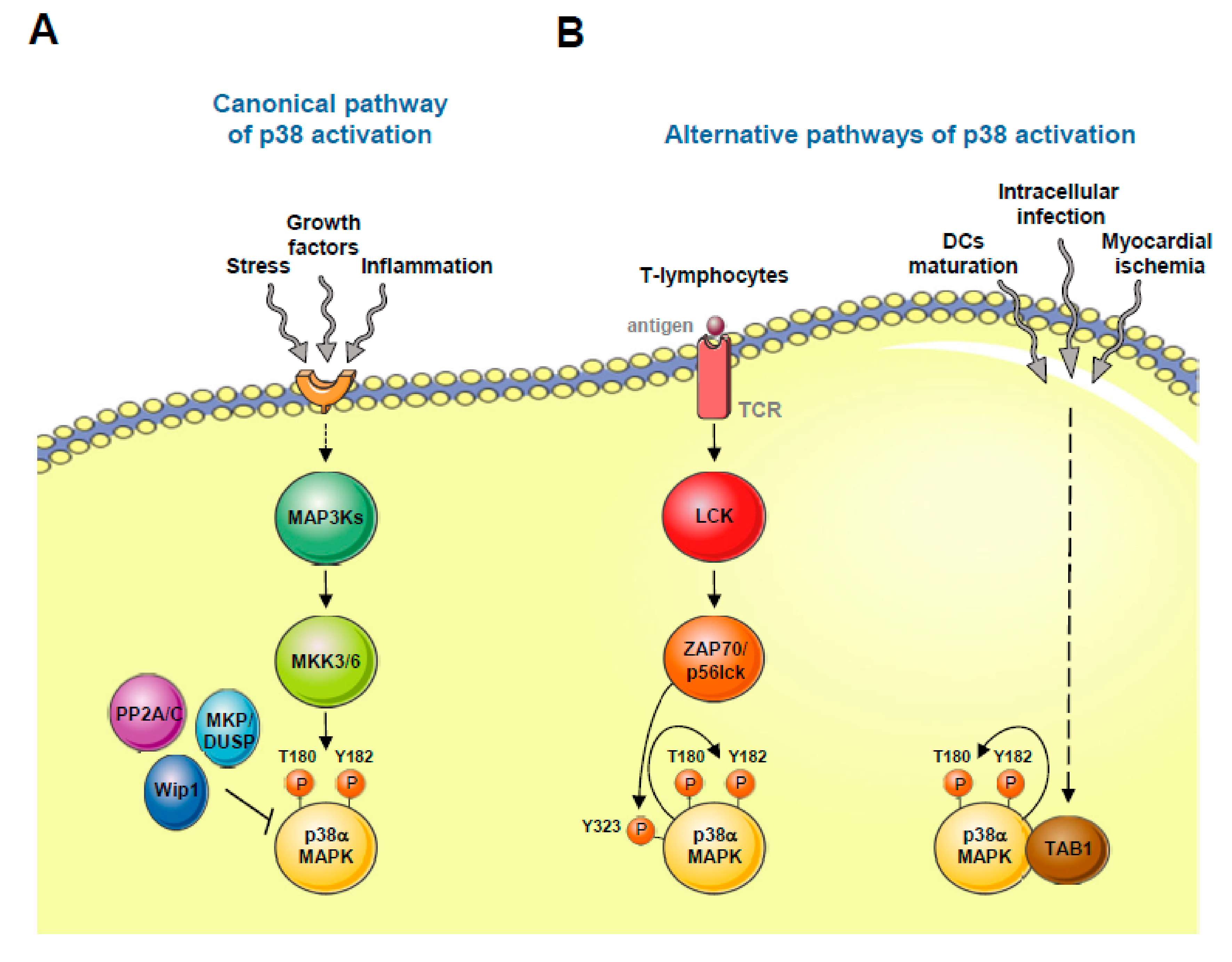
3. p38 MAPK Activation
4. Substrate Recognition and p38 MAPK Signaling Downregulation
5. p38 MAPK Activators and Physiological and Cellular Functions
6. Regulation of Cell Cycle by p38 MAPK
7. p38 MAPK as a Tumor Suppressor
8. p38 MAPK as a Tumor Promoter
9. Targeting p38 MAPK for Cancer Therapy
10. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-Activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuenda, A.; Rousseau, S. p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuenda, A.; Sanz-Ezquerro, J.J. p38gamma and p38delta: From Spectators to Key Physiological Players. Trends Biochem. Sci. 2017, 42, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Del Barco Barrantes, I.; Coya, J.M.; Maina, F.; Arthur, J.S.; Nebreda, A.R. Genetic analysis of specific and redundant roles for p38alpha and p38beta MAPKs during mouse development. Proc. Natl. Acad. Sci. USA 2011, 108, 12764–12769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-Year update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [Green Version]
- Sumara, G.; Formentini, I.; Collins, S.; Sumara, I.; Windak, R.; Bodenmiller, B.; Ramracheya, R.; Caille, D.; Jiang, H.; Platt, K.A.; et al. Regulation of PKD by the MAPK p38delta in insulin secretion and glucose homeostasis. Cell 2009, 136, 235–248. [Google Scholar] [CrossRef]
- Cuenda, A.; Nebreda, A.R. p38delta and PKD1: Kinase switches for insulin secretion. Cell 2009, 136, 209–210. [Google Scholar] [CrossRef]
- Schindler, E.M.; Hindes, A.; Gribben, E.L.; Burns, C.J.; Yin, Y.; Lin, M.H.; Owen, R.J.; Longmore, G.D.; Kissling, G.E.; Arthur, J.S.; et al. p38delta Mitogen-Activated protein kinase is essential for skin tumor development in mice. Cancer Res. 2009, 69, 4648–4655. [Google Scholar] [CrossRef] [Green Version]
- Efimova, T.; Broome, A.M.; Eckert, R.L. Protein kinase Cdelta regulates keratinocyte death and survival by regulating activity and subcellular localization of a p38delta-extracellular signal-Regulated kinase 1/2 complex. Mol. Cell Biol. 2004, 24, 8167–8183. [Google Scholar] [CrossRef] [Green Version]
- Adams, R.H.; Porras, A.; Alonso, G.; Jones, M.; Vintersten, K.; Panelli, S.; Valladares, A.; Perez, L.; Klein, R.; Nebreda, A.R. Essential role of p38alpha MAP kinase in placental but not embryonic cardiovascular development. Mol. Cell 2000, 6, 109–116. [Google Scholar] [CrossRef]
- Mudgett, J.S.; Ding, J.; Guh-Siesel, L.; Chartrain, N.A.; Yang, L.; Gopal, S.; Shen, M.M. Essential role for p38alpha mitogen-Activated protein kinase in placental angiogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 10454–10459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brancho, D.; Tanaka, N.; Jaeschke, A.; Ventura, J.J.; Kelkar, N.; Tanaka, Y.; Kyuuma, M.; Takeshita, T.; Flavell, R.A.; Davis, R.J. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003, 17, 1969–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doza, Y.N.; Cuenda, A.; Thomas, G.M.; Cohen, P.; Nebreda, A.R. Activation of the MAP kinase homologue RK requires the phosphorylation of Thr-180 and Tyr-182 and both residues are phosphorylated in chemically stressed KB cells. FEBS Lett. 1995, 364, 223–228. [Google Scholar] [PubMed] [Green Version]
- Derijard, B.; Raingeaud, J.; Barrett, T.; Wu, I.H.; Han, J.; Ulevitch, R.J.; Davis, R.J. Independent human MAP-Kinase signal transduction pathways defined by MEK and MKK isoforms. Science 1995, 267, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Enslen, H.; Raingeaud, J.; Davis, R.J. Selective activation of p38 mitogen-Activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J. Biol. Chem. 1998, 273, 1741–1748. [Google Scholar] [CrossRef] [Green Version]
- Alonso, G.; Ambrosino, C.; Jones, M.; Nebreda, A.R. Differential activation of p38 mitogen-Activated protein kinase isoforms depending on signal strength. J. Biol. Chem. 2000, 275, 40641–40648. [Google Scholar] [CrossRef] [Green Version]
- Remy, G.; Risco, A.M.; Inesta-Vaquera, F.A.; Gonzalez-Teran, B.; Sabio, G.; Davis, R.J.; Cuenda, A. Differential activation of p38MAPK isoforms by MKK6 and MKK3. Cell Signal. 2010, 22, 660–667. [Google Scholar] [CrossRef]
- Cuevas, B.D.; Abell, A.N.; Johnson, G.L. Role of mitogen-Activated protein kinase kinase kinases in signal integration. Oncogene 2007, 26, 3159–3171. [Google Scholar] [CrossRef] [Green Version]
- Salvador, J.M.; Mittelstadt, P.R.; Guszczynski, T.; Copeland, T.D.; Yamaguchi, H.; Appella, E.; Fornace, A.J., Jr.; Ashwell, J.D. Alternative p38 activation pathway mediated by T cell receptor-Proximal tyrosine kinases. Nat. Immunol. 2005, 6, 390–395. [Google Scholar] [CrossRef]
- Jirmanova, L.; Sarma, D.N.; Jankovic, D.; Mittelstadt, P.R.; Ashwell, J.D. Genetic disruption of p38alpha Tyr323 phosphorylation prevents T-cell receptor-mediated p38alpha activation and impairs interferon-Gamma production. Blood 2009, 113, 2229–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, B.; Gram, H.; Di, P.F.; Huang, B.; New, L.; Ulevitch, R.J.; Luo, Y.; Han, J. MAPKK-Independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science 2002, 295, 1291–1294. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zheng, M.; Chen, J.; Xie, C.; Kolatkar, A.R.; Zarubin, T.; Ye, Z.; Akella, R.; Lin, S.; Goldsmith, E.J.; et al. Determinants that control the specific interactions between TAB1 and p38alpha. Mol. Cell Biol. 2006, 26, 3824–3834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, P.C.; Campbell, D.G.; Nebreda, A.R.; Cohen, P. Feedback control of the protein kinase TAK1 by SAPK2a/p38alpha. EMBO J. 2003, 22, 5793–5805. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.F.; Martin, E.D.; Chaikuad, A.; Bassi, R.; Clark, J.; Martino, L.; Verma, S.; Sicard, P.; Tata, R.; Atkinson, R.A.; et al. Mechanism and consequence of the autoactivation of p38alpha mitogen-Activated protein kinase promoted by TAB1. Nat. Struct. Mol. Biol. 2013, 20, 1182–1190. [Google Scholar] [CrossRef] [Green Version]
- Tanno, M.; Bassi, R.; Gorog, D.A.; Saurin, A.T.; Jiang, J.; Heads, R.J.; Martin, J.L.; Davis, R.J.; Flavell, R.A.; Marber, M.S. Diverse mechanisms of myocardial p38 mitogen-Activated protein kinase activation: Evidence for MKK-Independent activation by a TAB1-Associated mechanism contributing to injury during myocardial ischemia. Circ. Res. 2003, 93, 254–261. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Miller, E.J.; Ninomiya-Tsuji, J.; Russell, R.R., III; Young, L.H. AMP-Activated protein kinase activates p38 mitogen-Activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ. Res. 2005, 97, 872–879. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, W.; Faure, M.; Yoshimura, T. Activation of discoidin domain receptor 1 facilitates the maturation of human monocyte-Derived dendritic cells through the TNF receptor associated factor 6/TGF-beta-Activated protein kinase 1 binding protein 1 beta/p38 alpha mitogen-activated protein kinase signaling cascade. J. Immunol. 2003, 171, 3520–3532. [Google Scholar]
- Kim, L.; Del Rio, L.; Butcher, B.A.; Mogensen, T.H.; Paludan, S.R.; Flavell, R.A.; Denkers, E.Y. p38 MAPK autophosphorylation drives macrophage IL-12 production during intracellular infection. J. Immunol. 2005, 174, 4178–4184. [Google Scholar] [CrossRef] [Green Version]
- Im, J.S.; Lee, J.K. ATR-Dependent activation of p38 MAP kinase is responsible for apoptotic cell death in cells depleted of Cdc7. J. Biol. Chem. 2008, 283, 25171–25177. [Google Scholar] [CrossRef] [Green Version]
- Purvis, J.E.; Lahav, G. Encoding and decoding cellular information through signaling dynamics. Cell 2013, 152, 945–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regot, S.; Hughey, J.J.; Bajar, B.T.; Carrasco, S.; Covert, M.W. High-Sensitivity measurements of multiple kinase activities in live single cells. Cell 2014, 157, 1724–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, P.L.; Wu, Z.; Zhang, P.; Wood, L.D.; Bhakta, K.S.; Han, J.; Feramisco, J.R.; Karin, M.; Wang, J.Y. Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes Dev. 2000, 14, 574–584. [Google Scholar] [PubMed]
- Haq, R.; Brenton, J.D.; Takahashi, M.; Finan, D.; Finkielsztein, A.; Damaraju, S.; Rottapel, R.; Zanke, B. Constitutive p38HOG mitogen-Activated protein kinase activation induces permanent cell cycle arrest and senescence. Cancer Res. 2002, 62, 5076–5082. [Google Scholar] [PubMed]
- Trempolec, N.; Dave-Coll, N.; Nebreda, A.R. SnapShot: p38 MAPK substrates. Cell 2013, 152, 924. [Google Scholar] [CrossRef] [Green Version]
- Tanoue, T.; Adachi, M.; Moriguchi, T.; Nishida, E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell Biol. 2000, 2, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Tanoue, T.; Maeda, R.; Adachi, M.; Nishida, E. Identification of a docking groove on ERK and p38 MAP kinases that regulates the specificity of docking interactions. EMBO J. 2001, 20, 466–479. [Google Scholar] [CrossRef]
- Biondi, R.M.; Nebreda, A.R. Signalling specificity of Ser/Thr protein kinases through docking-Site-Mediated interactions. Biochem. J. 2003, 372, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Lee, J.D.; Bibbs, L.; Ulevitch, R.J. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994, 265, 808–811. [Google Scholar] [CrossRef]
- Ferrigno, P.; Silver, P.A. Regulated nuclear localization of stress-Responsive factors: How the nuclear trafficking of protein kinases and transcription factors contributes to cell survival. Oncogene 1999, 18, 6129–6134. [Google Scholar] [CrossRef] [Green Version]
- Ferrigno, P.; Posas, F.; Koepp, D.; Saito, H.; Silver, P.A. Regulated nucleo/cytoplasmic exchange of HOG1 MAPK requires the importin beta homologs NMD5 and XPO1. EMBO J. 1998, 17, 5606–5614. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Ming, X.; Deng, P.; Jiang, Y. Mechanisms regulating the nuclear translocation of p38 MAP kinase. J. Cell Biochem. 2010, 110, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Zehorai, E.; Seger, R. Beta-Like Importins Mediate the Nuclear Translocation of MAPKs. Cell Physiol. Biochem. 2019, 52, 802–821. [Google Scholar] [PubMed] [Green Version]
- Ben-Levy, R.; Hooper, S.; Wilson, R.; Paterson, H.F.; Marshall, C.J. Nuclear export of the stress-Activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr. Biol. 1998, 8, 1049–1057. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Y.; Mei, Z.Q.; Wu, J.W.; Wang, Z.X. Enzymatic activity and substrate specificity of mitogen-Activated protein kinase p38alpha in different phosphorylation states. J. Biol. Chem. 2008, 283, 26591–26601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takekawa, M.; Adachi, M.; Nakahata, A.; Nakayama, I.; Itoh, F.; Tsukuda, H.; Taya, Y.; Imai, K. p53-Inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 2000, 19, 6517–6526. [Google Scholar] [CrossRef]
- Macurek, L.; Lindqvist, A.; Voets, O.; Kool, J.; Vos, H.R.; Medema, R.H. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene 2010, 29, 2281–2291. [Google Scholar] [CrossRef] [Green Version]
- Ferreiro, I.; Joaquin, M.; Islam, A.; Gomez-Lopez, G.; Barragan, M.; Lombardia, L.; Dominguez, O.; Pisano, D.G.; Lopez-Bigas, N.; Nebreda, A.R.; et al. Whole genome analysis of p38 SAPK-Mediated gene expression upon stress. BMC. Genom. 2010, 11, 144. [Google Scholar] [CrossRef] [Green Version]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar] [CrossRef] [Green Version]
- Bonney, E.A. Mapping out p38MAPK. Am. J. Reprod. Immunol. 2017, 77. [Google Scholar] [CrossRef] [Green Version]
- Gupta, J.; Nebreda, A.R. Roles of p38alpha mitogen-Activated protein kinase in mouse models of inflammatory diseases and cancer. FEBS J. 2015, 282, 1841–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Sudo, T.; Senftleben, U.; Dadak, A.M.; Johnson, R.; Karin, M. Requirement for p38alpha in erythropoietin expression: A role for stress kinases in erythropoiesis. Cell 2000, 102, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Engel, F.B.; Schebesta, M.; Duong, M.T.; Lu, G.; Ren, S.; Madwed, J.B.; Jiang, H.; Wang, Y.; Keating, M.T. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005, 19, 1175–1187. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.S.; Le, G.X.; Demidov, O.N.; Marshall, N.T.; Wang, S.T.; Krishnamurthy, J.; Sharpless, N.E.; Dunn, N.R.; Bulavin, D.V. p38 MAPK controls expression of multiple cell cycle inhibitors and islet proliferation with advancing age. Dev. Cell 2009, 17, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Ventura, J.J.; Tenbaum, S.; Perdiguero, E.; Huth, M.; Guerra, C.; Barbacid, M.; Pasparakis, M.; Nebreda, A.R. p38alpha MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat. Genet. 2007, 39, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; He, G.; Matsuzawa, A.; Yu, G.Y.; Maeda, S.; Hardiman, G.; Karin, M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 2008, 14, 156–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, L.; Bakiri, L.; Mairhorfer, A.; Schweifer, N.; Haslinger, C.; Kenner, L.; Komnenovic, V.; Scheuch, H.; Beug, H.; Wagner, E.F. p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat. Genet. 2007, 39, 741–749. [Google Scholar] [CrossRef]
- Wu, Z.; Woodring, P.J.; Bhakta, K.S.; Tamura, K.; Wen, F.; Feramisco, J.R.; Karin, M.; Wang, J.Y.; Puri, P.L. p38 and extracellular signal-Regulated kinases regulate the myogenic program at multiple steps. Mol. Cell Biol. 2000, 20, 3951–3964. [Google Scholar] [CrossRef] [Green Version]
- Lluis, F.; Perdiguero, E.; Nebreda, A.R.; Munoz-Canoves, P. Regulation of skeletal muscle gene expression by p38 MAP kinases. Trends Cell Biol. 2006, 16, 36–44. [Google Scholar] [CrossRef]
- Simone, C.; Forcales, S.V.; Hill, D.A.; Imbalzano, A.N.; Latella, L.; Puri, P.L. p38 pathway targets SWI-SNF chromatin-Remodeling complex to muscle-Specific loci. Nat. Genet. 2004, 36, 738–743. [Google Scholar] [CrossRef] [Green Version]
- Perdiguero, E.; Ruiz-Bonilla, V.; Serrano, A.L.; Munoz-Canoves, P. Genetic deficiency of p38alpha reveals its critical role in myoblast cell cycle exit: The p38alpha-JNK connection. Cell Cycle 2007, 6, 1298–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, M.A.; Le, G.F.; Scime, A.; Kuang, S.; von Maltzahn, J.; Seale, V.; Cuenda, A.; Ranish, J.A.; Rudnicki, M.A. p38-{gamma}-Dependent gene silencing restricts entry into the myogenic differentiation program. J. Cell Biol. 2009, 187, 991–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnum, K.J.; O′Connell, M.J. Cell cycle regulation by checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [PubMed] [Green Version]
- Thoms, H.C.; Dunlop, M.G.; Stark, L.A. p38-Mediated inactivation of cyclin D1/cyclin-Dependent kinase 4 stimulates nucleolar translocation of RelA and apoptosis in colorectal cancer cells. Cancer Res. 2007, 67, 1660–1669. [Google Scholar] [CrossRef] [Green Version]
- Bollaert, E.; de Rocca, S.A.; Demoulin, J.B. The HMG box transcription factor HBP1: A cell cycle inhibitor at the crossroads of cancer signaling pathways. Cell Mol. Life Sci. 2019, 76, 1529–1539. [Google Scholar] [CrossRef]
- Kishi, H.; Nakagawa, K.; Matsumoto, M.; Suga, M.; Ando, M.; Taya, Y.; Yamaizumi, M. Osmotic shock induces G1 arrest through p53 phosphorylation at Ser33 by activated p38MAPK without phosphorylation at Ser15 and Ser20. J. Biol. Chem. 2001, 276, 39115–39122. [Google Scholar] [CrossRef] [Green Version]
- Stramucci, L.; Pranteda, A.; Bossi, G. Insights of Crosstalk between p53 Protein and the MKK3/MKK6/p38 MAPK Signaling Pathway in Cancer. Cancers (Basel) 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.; Benchimol, S. Transcriptional repression mediated by the p53 tumour suppressor. Cell Death Differ. 2003, 10, 404–408. [Google Scholar] [CrossRef]
- Lafarga, V.; Cuadrado, A.; de Silanes, I.L.; Bengoechea, R.; Fernandez-Capetillo, O.; Nebreda, A.R. p38 Mitogen-Activated protein kinase- and HuR-Dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol. Cell Biol. 2009, 29, 4341–4351. [Google Scholar] [CrossRef] [Green Version]
- Swat, A.; Dolado, I.; Rojas, J.M.; Nebreda, A.R. Cell density-Dependent inhibition of epidermal growth factor receptor signaling by p38alpha mitogen-activated protein kinase via Sprouty2 downregulation. Mol. Cell Biol. 2009, 29, 3332–3343. [Google Scholar] [CrossRef] [Green Version]
- Joaquin, M.; Gubern, A.; Gonzalez-Nunez, D.; Josue, R.E.; Ferreiro, I.; de Nadal, E.; Nebreda, A.R.; Posas, F. The p57 CDKi integrates stress signals into cell-Cycle progression to promote cell survival upon stress. EMBO J. 2012, 31, 2952–2964. [Google Scholar] [CrossRef] [PubMed]
- Gubern, A.; Joaquin, M.; Marques, M.; Maseres, P.; Garcia-Garcia, J.; Amat, R.; Gonzalez-Nunez, D.; Oliva, B.; Real, F.X.; de Nadal, E.; et al. The N-Terminal Phosphorylation of RB by p38 Bypasses Its Inactivation by CDKs and Prevents Proliferation in Cancer Cells. Mol. Cell 2016, 64, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joaquin, M.; de Nadal, E.; Posas, F. An RB insensitive to CDK regulation. Mol. Cell. Oncol. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raman, M.; Earnest, S.; Zhang, K.; Zhao, Y.; Cobb, M.H. TAO kinases mediate activation of p38 in response to DNA damage. EMBO J. 2007, 26, 2005–2014. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Tong, T.; Fan, W.; Fan, F.; Antinore, M.J.; Zhu, X.; Mazzacurati, L.; Li, X.; Petrik, K.L.; Rajasekaran, B.; et al. GADD45-Induced cell cycle G2-M arrest associates with altered subcellular distribution of cyclin B1 and is independent of p38 kinase activity. Oncogene 2002, 21, 8696–8704. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. p53-Deficient cells rely on ATM- and ATR-Mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [Google Scholar] [CrossRef] [Green Version]
- Donzelli, M.; Draetta, G.F. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003, 4, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer. 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Faust, D.; Schmitt, C.; Oesch, F.; Oesch-Bartlomowicz, B.; Schreck, I.; Weiss, C.; Dietrich, C. Differential p38-Dependent signalling in response to cellular stress and mitogenic stimulation in fibroblasts. Cell Commun. Signal. 2012, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Hitomi, M.; Han, J.; Stacey, D.W. The p38 pathway provides negative feedback for Ras proliferative signaling. J. Biol. Chem. 2000, 275, 38973–38980. [Google Scholar] [CrossRef] [Green Version]
- Dolado, I.; Swat, A.; Ajenjo, N.; De, V.G.; Cuadrado, A.; Nebreda, A.R. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell 2007, 11, 191–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacNeil, A.J.; Jiao, S.C.; McEachern, L.A.; Yang, Y.J.; Dennis, A.; Yu, H.; Xu, Z.; Marshall, J.S.; Lin, T.J. MAPK kinase 3 is a tumor suppressor with reduced copy number in breast cancer. Cancer Res. 2014, 74, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu-Lee, L.Y.; Yu, G.; Lee, Y.C.; Lin, S.C.; Pan, J.; Pan, T.; Yu, K.J.; Liu, B.; Creighton, C.J.; Rodriguez-Canales, J.; et al. Osteoblast-Secreted Factors Mediate Dormancy of Metastatic Prostate Cancer in the Bone via Activation of the TGFbetaRIII-p38MAPK-pS249/T252RB Pathway. Cancer Res. 2018, 78, 2911–2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulavin, D.V.; Phillips, C.; Nannenga, B.; Timofeev, O.; Donehower, L.A.; Anderson, C.W.; Appella, E.; Fornace, A.J., Jr. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-Mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat. Genet. 2004, 36, 343–350. [Google Scholar] [CrossRef]
- Yu, W.; Imoto, I.; Inoue, J.; Onda, M.; Emi, M.; Inazawa, J. A novel amplification target, DUSP26, promotes anaplastic thyroid cancer cell growth by inhibiting p38 MAPK activity. Oncogene 2007, 26, 1178–1187. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yang, Y.; Peng, Y.; Austin, R.J.; Van Eyndhoven, W.G.; Nguyen, K.C.; Gabriele, T.; McCurrach, M.E.; Marks, J.R.; Hoey, T.; et al. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat. Genet. 2002, 31, 133–134. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Inflammatory cytokines in cancer: Tumour necrosis factor and interleukin 6 take the stage. Ann. Rheum. Dis. 2011, 70 (Suppl 1), i104–i108. [Google Scholar] [CrossRef]
- Del Reino, P.; Alsina-Beauchamp, D.; Escos, A.; Cerezo-Guisado, M.I.; Risco, A.; Aparicio, N.; Zur, R.; Fernandez-Estevez, M.; Collantes, E.; Montans, J.; et al. Pro-Oncogenic role of alternative p38 mitogen-Activated protein kinases p38gamma and p38delta, linking inflammation and cancer in colitis-Associated colon cancer. Cancer Res. 2014, 74, 6150–6160. [Google Scholar] [CrossRef] [Green Version]
- Yin, N.; Qi, X.; Tsai, S.; Lu, Y.; Basir, Z.; Oshima, K.; Thomas, J.P.; Myers, C.R.; Stoner, G.; Chen, G. p38gamma MAPK is required for inflammation-Associated colon tumorigenesis. Oncogene 2016, 35, 1039–1048. [Google Scholar] [CrossRef]
- Igea, A.; Nebreda, A.R. The Stress Kinase p38alpha as a Target for Cancer Therapy. Cancer Res. 2015, 75, 3997–4002. [Google Scholar] [CrossRef] [Green Version]
- Gawrzak, S.; Rinaldi, L.; Gregorio, S.; Arenas, E.J.; Salvador, F.; Urosevic, J.; Figueras-Puig, C.; Rojo, F.; Del Barco Barrantes, I.; Cejalvo, J.M.; et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER(+) breast cancer. Nat. Cell Biol. 2018, 20, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Canovas, B.; Igea, A.; Sartori, A.A.; Gomis, R.R.; Paull, T.T.; Isoda, M.; Perez-Montoyo, H.; Serra, V.; Gonzalez-Suarez, E.; Stracker, T.H.; et al. Targeting p38alpha Increases DNA Damage, Chromosome Instability, and the Anti-Tumoral Response to Taxanes in Breast Cancer Cells. Cancer Cell 2018, 33, 1094–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, M.; Canals, D.; Adada, M.; Coant, N.; Salama, M.F.; Helke, K.L.; Arthur, J.S.; Shroyer, K.R.; Kitatani, K.; Obeid, L.M.; et al. P38 delta MAPK promotes breast cancer progression and lung metastasis by enhancing cell proliferation and cell detachment. Oncogene 2017, 36, 6649–6657. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, A.K.; Basu, S.; Hu, J.; Yie, T.A.; Tchou-Wong, K.M.; Rom, W.N.; Lee, T.C. Selective p38 activation in human non-small cell lung cancer. Am. J. Respir. Cell Mol. Biol. 2002, 26, 558–564. [Google Scholar] [CrossRef]
- Leelahavanichkul, K.; Amornphimoltham, P.; Molinolo, A.A.; Basile, J.R.; Koontongkaew, S.; Gutkind, J.S. A role for p38 MAPK in head and neck cancer cell growth and tumor-Induced angiogenesis and lymphangiogenesis. Mol. Oncol. 2014, 8, 105–118. [Google Scholar] [CrossRef]
- Tang, J.; Qi, X.; Mercola, D.; Han, J.; Chen, G. Essential role of p38gamma in K-Ras transformation independent of phosphorylation. J. Biol. Chem. 2005, 280, 23910–23917. [Google Scholar] [CrossRef] [Green Version]
- Tomas-Loba, A.; Manieri, E.; Gonzalez-Teran, B.; Mora, A.; Leiva-Vega, L.; Santamans, A.M.; Romero-Becerra, R.; Rodriguez, E.; Pintor-Chocano, A.; Feixas, F.; et al. p38gamma is essential for cell cycle progression and liver tumorigenesis. Nature 2019, 568, 557–560. [Google Scholar] [CrossRef] [Green Version]
- Pramanik, R.; Qi, X.; Borowicz, S.; Choubey, D.; Schultz, R.M.; Han, J.; Chen, G. p38 isoforms have opposite effects on AP-1-Dependent transcription through regulation of c-Jun. The determinant roles of the isoforms in the p38 MAPK signal specificity. J. Biol. Chem. 2003, 278, 4831–4839. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, A.L.; Hayward, N.K. Molecular pathways: Mitogen-Activated protein kinase pathway mutations and drug resistance. Clin. Cancer Res. 2013, 19, 2301–2309. [Google Scholar] [CrossRef] [Green Version]
- Gil-Araujo, B.; Toledo Lobo, M.V.; Gutierrez-Salmeron, M.; Gutierrez-Pitalua, J.; Ropero, S.; Angulo, J.C.; Chiloeches, A.; Lasa, M. Dual specificity phosphatase 1 expression inversely correlates with NF-kappaB activity and expression in prostate cancer and promotes apoptosis through a p38 MAPK dependent mechanism. Mol. Oncol. 2014, 8, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Del Barco Barrantes, I.; Igea, A.; Sakellariou, S.; Pateras, I.S.; Gorgoulis, V.G.; Nebreda, A.R. Dual function of p38alpha MAPK in colon cancer: Suppression of colitis-Associated tumor initiation but requirement for cancer cell survival. Cancer Cell 2014, 25, 484–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, M.; Kenny, H.A.; Ashcroft, B.; Mukherjee, A.; Johnson, A.; Zhang, Y.; Helou, Y.; Batlle, R.; Liu, X.; Gutierrez, N.; et al. Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis. Cell Metab 2019, 29, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [Green Version]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef]
- Kumar, B.; Koul, S.; Petersen, J.; Khandrika, L.; Hwa, J.S.; Meacham, R.B.; Wilson, S.; Koul, H.K. p38 mitogen-Activated protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer by modulation of MMP-2 and MMP-9 activity. Cancer Res. 2010, 70, 832–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez-Cuervo, C.; Merrell, M.A.; Watson, L.; Harris, K.W.; Rosenthal, E.L.; Vaananen, H.K.; Selander, K.S. Breast cancer cells with inhibition of p38alpha have decreased MMP-9 activity and exhibit decreased bone metastasis in mice. Clin. Exp. Metastasis 2004, 21, 525–533. [Google Scholar] [CrossRef]
- Arechederra, M.; Priego, N.; Vazquez-Carballo, A.; Sequera, C.; Gutierrez-Uzquiza, A.; Cerezo-Guisado, M.I.; Ortiz-Rivero, S.; Roncero, C.; Cuenda, A.; Guerrero, C.; et al. p38 MAPK down-Regulates fibulin 3 expression through methylation of gene regulatory sequences: Role in migration and invasion. J. Biol. Chem. 2015, 290, 4383–4397. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, S.; Dolado, I.; Beardmore, V.; Shpiro, N.; Marquez, R.; Nebreda, A.R.; Arthur, J.S.; Case, L.M.; Tessier-Lavigne, M.; Gaestel, M.; et al. CXCL12 and C5a trigger cell migration via a PAK1/2-p38alpha MAPK-MAPKAP-K2-HSP27 pathway. Cell Signal. 2006, 18, 1897–1905. [Google Scholar] [CrossRef]
- Limoge, M.; Safina, A.; Truskinovsky, A.M.; Aljahdali, I.; Zonneville, J.; Gruevski, A.; Arteaga, C.L.; Bakin, A.V. Tumor p38 MAPK signaling enhances breast carcinoma vascularization and growth by promoting expression and deposition of pro-Tumorigenic factors. Oncotarget 2017, 8, 61969–61981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urosevic, J.; Garcia-Albeniz, X.; Planet, E.; Real, S.; Cespedes, M.V.; Guiu, M.; Fernandez, E.; Bellmunt, A.; Gawrzak, S.; Pavlovic, M.; et al. Colon cancer cells colonize the lung from established liver metastases through p38 MAPK signalling and PTHLH. Nat. Cell Biol. 2014, 16, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Vergote, I.; Heitz, F.; Buderath, P.; Powell, M.; Sehouli, J.; Lee, C.M.; Hamilton, A.; Fiorica, J.; Moore, K.N.; Teneriello, M.; et al. A randomized, double-Blind, placebo-Controlled phase 1b/2 study of ralimetinib, a p38 MAPK inhibitor, plus gemcitabine and carboplatin versus gemcitabine and carboplatin for women with recurrent platinum-Sensitive ovarian cancer. Gynecol. Oncol. 2020, 156, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. p38 (MAPK): Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soni, S.; Anand, P.; Padwad, Y.S. MAPKAPK2: The master regulator of RNA-Binding proteins modulates transcript stability and tumor progression. J. Exp. Clin. Cancer Res. 2019, 38, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Ma, N.; Wang, J.; Song, J.; Bu, X.; Cheng, Y.; Sun, K.; Xiong, H.; Jiang, G.; Zhang, B. Increased p38-MAPK is responsible for chemotherapy resistance in human gastric cancer cells. BMC Cancer 2008, 8, 375. [Google Scholar] [CrossRef] [Green Version]
- Pereira, L.; Igea, A.; Canovas, B.; Dolado, I.; Nebreda, A.R. Inhibition of p38 MAPK sensitizes tumour cells to cisplatin-Induced apoptosis mediated by reactive oxygen species and JNK. EMBO Mol. Med. 2013, 5, 1759–1774. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, H.C.; Hasskamp, P.; Schmedding, I.; Morandell, S.; van Vugt, M.A.; Wang, X.; Linding, R.; Ong, S.E.; Weaver, D.; Carr, S.A.; et al. DNA damage activates a spatially distinct late cytoplasmic cell-Cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol. Cell 2010, 40, 34–49. [Google Scholar] [CrossRef]
- Manke, I.A.; Nguyen, A.; Lim, D.; Stewart, M.Q.; Elia, A.E.; Yaffe, M.B. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell 2005, 17, 37–48. [Google Scholar] [CrossRef]
- Johansen, C.; Vestergaard, C.; Kragballe, K.; Kollias, G.; Gaestel, M.; Iversen, L. MK2 regulates the early stages of skin tumor promotion. Carcinogenesis 2009, 30, 2100–2108. [Google Scholar] [CrossRef] [Green Version]
- Henriques, A.; Koliaraki, V.; Kollias, G. Mesenchymal MAPKAPK2/HSP27 drives intestinal carcinogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E5546–E5555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalaoui, N.; Hanggi, K.; Brumatti, G.; Chau, D.; Nguyen, N.N.; Vasilikos, L.; Spilgies, L.M.; Heckmann, D.A.; Ma, C.; Ghisi, M.; et al. Targeting p38 or MK2 Enhances the Anti-Leukemic Activity of Smac-Mimetics. Cancer Cell 2016, 30, 499–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnam, M.; Goodrich, D.W. RB1, development, and cancer. Curr. Top. Dev. Biol. 2011, 94, 129–169. [Google Scholar] [PubMed] [Green Version]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-kappaB Activation and PD-L1 Expression. Mol. Cell 2019, 73, 22–35. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Cancer Type | Treatment | Phase of Study | Details |
|---|---|---|---|
| Advanced and/or metastatic cancer | LY3007113 | I | Evaluation of the safety and tolerability to different doses of treatment |
| Relapsed ovarian cancer after platinum-based chemotherapy | LY2228820 Carboplatin Gemcitabine | Ib/II | Evaluation of the safety of treatment based on inhibitor plus chemotherapy |
| Advanced cancer | LY2228820 | I | Evaluation of the safety and tolerability to different doses of treatment |
| Metastatic breast cancer | LY2228820 Tamoxifen | II | Evaluation of the efficacy of inhibitor plus tamoxifen |
| Adult glioblastoma | LY2228820 Temozolomide (TMZ) Radiotherapy | I/II | Determination of inhibitor dose with TMZ and radiotherapy (phase I). Estimating the six-month Progression-free survival (PFS) rate (phase II) |
| Relapsed multiple myeloma (MM) | SCIO-469 Bortezomib | II | Evaluation of the efficacy of inhibitor in patients with MM |
| Myelodisplastic syndrome (MDS) | SCIO-469 | II | Evaluation of the safety, tolerability and efficacy of inhibitor in patients with MDS |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Limón, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The p38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913. https://doi.org/10.3390/ijms21061913
Martínez-Limón A, Joaquin M, Caballero M, Posas F, de Nadal E. The p38 Pathway: From Biology to Cancer Therapy. International Journal of Molecular Sciences. 2020; 21(6):1913. https://doi.org/10.3390/ijms21061913
Chicago/Turabian StyleMartínez-Limón, Adrián, Manel Joaquin, María Caballero, Francesc Posas, and Eulàlia de Nadal. 2020. "The p38 Pathway: From Biology to Cancer Therapy" International Journal of Molecular Sciences 21, no. 6: 1913. https://doi.org/10.3390/ijms21061913
APA StyleMartínez-Limón, A., Joaquin, M., Caballero, M., Posas, F., & de Nadal, E. (2020). The p38 Pathway: From Biology to Cancer Therapy. International Journal of Molecular Sciences, 21(6), 1913. https://doi.org/10.3390/ijms21061913