Cold Atmospheric Plasma and Silymarin Nanoemulsion Activate Autophagy in Human Melanoma Cells

 , ,
, , 
Abstract
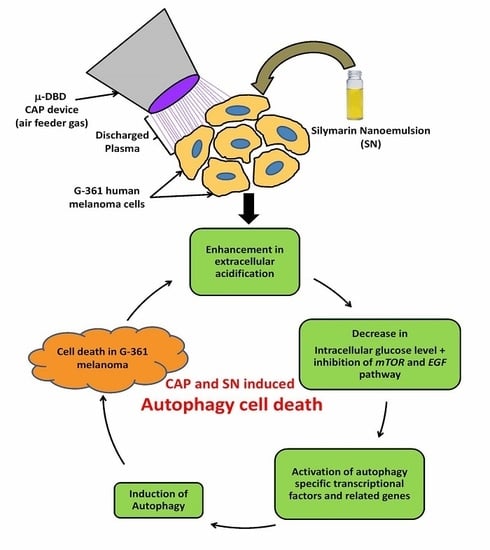
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Electrical and Optical Characteristics of the μ-DBD Plasma Instrument
2.2. Effect of CAP on Physical Parameters (Extracellular pH and Temperature)
2.3. Intracellular ATP and Glucose Estimation
2.4. PI3K Lead mTOR and EGF Signaling Arrest
2.5. Increase in Autophagic-Related Gene Expressions and Related Transcriptional Factors
2.6. Autophagy Induction in Human G-361 Melanoma Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Cell Culture
4.3. Characterization of the μ-Dielectric Barrier Discharge (μ-DBD) Plasma System Device
4.4. Estimation of Extracellular pH and Temperature and Number of Melanoma Cells
4.5. Glucose Uptake
4.6. qPCR Gene Analysis (Autophagy Related and Transcriptional Factor)
4.7. Evaluation of Autophagy by Immunocytochemistry (ICC), Flow Cytometry, and ELISA
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CAP | Cold Atmospheric Plasma |
| SN | Silymarin Nanoemulsion |
| RONS | Reactive Oxygen and Nitrogen Species |
| PI3K | Phosphatidyl Inositol 3 Kinase |
| mTOR | Mammalian Target of Rapamycin |
| MEK | Mitogen-Activated Protein Kinase |
| ATP | Adenosine Triphosphate |
| ZKSCAN3 | Zinc Finger with KRAB And SCAN Domains 3 |
| TFEB | Transcription Factor EB |
| FOXO1 | Forkhead Box Protein O1 |
| CRTC2 | cAMP-Response Element Binding Protein Regulated Transcription Coactivator 2 |
| CREBBP | cAMP-Response Element Binding Protein |
| BECN-1 | Beclin 1 |
| AMBRA-1 | Autophagy and Beclin 1 Regulator 1 |
| MAP1LC3A | Microtubule Associated Protein 1 Light Chain 3 Alpha |
| SQSTM | Sequestosome-1 |
| EGFR | Epidermal Growth Factor Receptor |
References
- Yamaguchi, Y.; Hearing, V.J. Melanocytes and their Diseases. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The Role of BRAF V600 Mutation in Melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Ndoye, A.; Weeraratna, A.T. Autophagy—An Emerging Target for Melanoma Therapy. F1000Res 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Lugowska, I.; Teterycz, P.; Rutkowski, P. Immunotherapy of Melanoma. Contemp. Oncol. (Pozn) 2018, 22, 61–67. [Google Scholar] [CrossRef]
- Chakraborty, R.; Wieland, C.N.; Comfere, N.I. Molecular Targeted Therapies in Metastatic Melanoma. Pharmgenomics Pers. Med. 2013, 6, 49–56. [Google Scholar]
- Gay-Mimbrera, J.; Garcia, M.C.; Isla-Tejera, B.; Rodero-Serrano, A.; Garcia-Nieto, A.V.; Ruano, J. Clinical and Biological Principles of Cold Atmospheric Plasma Application in Skin Cancer. Adv. Ther. 2016, 33, 894–909. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Guo, L.; Chen, Q.; Zhang, K.; Wang, T.; An, G.; Zhang, X.; Li, H.; Ding, G. Effects and Mechanisms of Cold Atmospheric Plasma on Skin Wound Healing of Rats. Contrib. Plasma Phy. 2019, 59, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, R.; Krishnamraju, P.V.; Shankar, T.; Gowd, S. Nonthermal Plasma in Dentistry: An Update. J. Int. Soc. Prev. Community Dent. 2017, 7, 71–75. [Google Scholar]
- Öngel, C.; Keleş, M.; Acar, E.; Birer, Ö. Atmospheric Pressure Plasma Jet Treatment of Human Hair Fibers. J. Bio. Tribo. Corros. 2015, 1, 1–10. [Google Scholar] [CrossRef]
- Yan, D.; Sherman, J.H.; Keidar, M. Cold Atmospheric Plasma, a Novel Promising Anti-Cancer Treatment Modality. Oncotarget 2017, 8, 15977–15995. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, M.; Kaushik, N.; Ghimire, B.; Adhikari, B.; Baboota, S.; Al-Khedhairy, A.A.; Wahab, R.; Lee, S.J.; Kaushik, N.K.; Choi, E.H. Cold Atmospheric Plasma and Silymarin Nanoemulsion Synergistically Inhibits Human Melanoma Tumorigenesis Via Targeting HGF/C-MET Downstream Pathway. Cell. Commun. Signal. 2019, 17, 52–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.; Bekeschus, S.; Jablonowski, H.; Barton, A.; Weltmann, K.D.; Wende, K. Role of Ambient Gas Composition on Cold Physical Plasma-Elicited Cell Signaling in Keratinocytes. Biophys. J. 2017, 112, 2397–2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klochkov, S.G.; Neganova, M.E.; Nikolenko, V.N.; Chen, K.; Somasundaram, S.G.; Kirkland, C.E.; Aliev, G. Implications of Nanotechnology for the Treatment of Cancer: Recent Advances. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, N.K.; Kaushik, N.; Yoo, K.C.; Uddin, N.; Kim, J.S.; Lee, S.J.; Choi, E.H. Low Doses of PEG-Coated Gold Nanoparticles Sensitize Solid Tumors to Cold Plasma by Blocking the PI3K/AKT-Driven Signaling Axis to Suppress Cellular Transformation by Inhibiting Growth and EMT. Biomaterials 2016, 87, 118–130. [Google Scholar] [CrossRef]
- Vargas-Mendoza, N.; Madrigal-Santillan, E.; Morales-Gonzalez, A.; Esquivel-Soto, J.; Esquivel-Chirino, C.; Garcia-Luna Y Gonzalez-Rubio, M.; Gayosso-de-Lucio, J.A.; Morales-Gonzalez, J.A. Hepatoprotective Effect of Silymarin. World J. Hepatol. 2014, 6, 144–149. [Google Scholar] [CrossRef]
- Ramasamy, K.; Agarwal, R. Multitargeted Therapy of Cancer by Silymarin. Cancer Lett. 2008, 269, 352–362. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, N.K.; Kaushik, N.; Linh, N.N.; Ghimire, B.; Pengkit, A.; Sornsakdanuphap, J.; Lee, S.J.; Choi, E.H. Plasma and Nanomaterials: Fabrication and Biomedical Applications. Nanomaterials (Basel) 2019, 9, 98. [Google Scholar] [CrossRef] [Green Version]
- Bogaerts, A.; Yusupov, M.; Razzokov, J.; Van der Paal, J. Plasma for Cancer Treatment: How can RONS Penetrate through the Cell Membrane? Answers from Computer Modeling. Front. Chem. Sci. Eng. 2019, 13, 253–263. [Google Scholar] [CrossRef]
- Pai, K.; Timmons, C.; Roehm, K.D.; Ngo, A.; Narayanan, S.S.; Ramachandran, A.; Jacob, J.D.; Ma, L.M.; Madihally, S.V. Investigation of the Roles of Plasma Species Generated by Surface Dielectric Barrier Discharge. Sci. Rep. 2018, 8, 16674. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Rajjoub, K.; Sherman, J.; Canady, J.; Recek, N.; Yan, D.; Bian, K.; Murad, F.; Keidar, M. Cold Plasma Accelerates the Uptake of Gold Nanoparticles into Glioblastoma Cells. Plasma Processes Polym. 2015, 12, 1364–1369. [Google Scholar] [CrossRef]
- Wong, D.J.; Ribas, A. Targeted Therapy for Melanoma. Cancer Treat. Res. 2016, 167, 251–262. [Google Scholar]
- Grossman, D.; Altieri, D. Drug Resistance in Melanoma: Mechanisms, Apoptosis, and New Potential Therapeutic Targets. Cancer Metastasis Rev. 2001, 20, 3–11. [Google Scholar] [CrossRef]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef]
- Mak, T.W.; Okada, H. Pathways of Apoptotic and Non-Apoptotic Death in Tumour Cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [Green Version]
- Lorin, S.; Hamai, A.; Mehrpour, M.; Codogno, P. Autophagy Regulation and its Role in Cancer. Semin. Cancer Biol. 2013, 23, 361–379. [Google Scholar] [CrossRef]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and Function of Autophagy during Cell Survival and Cell Death. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [Green Version]
- Yen, W.L.; Klionsky, D.J. How to Live Long and Prosper: Autophagy, Mitochondria, and Aging. Physiology (Bethesda) 2008, 23, 248–262. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of Autophagy in Cancer. Nat. Rev. Cancer. 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Ghimire, B.; Szili, E.J.; Lamichhane, P.; Short, R.D.; Lim, J.S.; Attri, P.; Masur, K.; Weltmann, K.; Hong, S.; Choi, E.H. The Role of UV Photolysis and Molecular Transport in the Generation of Reactive Species in a Tissue Model with a Cold Atmospheric Pressure Plasma Jet. App. Phys. Lett. 2019, 114, 93701. [Google Scholar] [CrossRef] [Green Version]
- Ghimire, B.; Sornsakdanuphap, J.; Hong, Y.J.; Uhm, H.S.; Weltmann, K.; Choi, E.H. The Effect of the Gap Distance between an Atmospheric-Pressure Plasma Jet Nozzle and Liquid Surface on OH and N2 Species Concentrations. Phys. Plasmas 2017, 24, 73502. [Google Scholar] [CrossRef]
- Ghimire, B.; Lamichhane, P.; Lim, J.S.; Min, B.; Paneru, R.; Weltmann, K.; Choi, E.H. An Atmospheric Pressure Plasma Jet Operated by Injecting Natural Air. Appl. Phys. Lett. 2018, 113, 194101. [Google Scholar] [CrossRef]
- Papadopoli, D.; Boulay, K.; Kazak, L.; Pollak, M.; Mallette, F.; Topisirovic, I.; Hulea, L. mTOR as a Central Regulator of Lifespan and Aging. F1000Resesrch 2019, 8. [Google Scholar] [CrossRef]
- Füllgrabe, J.; Ghislat, G.; Cho, D.; Rubinsztein, D.C. Transcriptional Regulation of Mammalian Autophagy at a Glance. J. Cell Sci. 2016, 129, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; White, E. Autophagy in Tumorigenesis and Energy Metabolism: Friend by Day, Foe by Night. Curr. Opin. Genet. Dev. 2011, 21, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Guo, X.; Xie, X.; Zhao, X.; Li, D.; Deng, W.; Song, Y.; Shen, F.; Wu, M.; Wei, L. Autophagy in Hypoxia Protects Cancer Cells Against Apoptosis Induced by Nutrient Deprivation through a Beclin1-Dependent Way in Hepatocellular Carcinoma. J. Cell. Biochem. 2011, 112, 3406–3420. [Google Scholar] [CrossRef]
- Meijer, A.J. Amino Acid Regulation of Autophagosome Formation. Methods Mol. Biol. 2008, 445, 89–109. [Google Scholar]
- Meijer, A.J.; Codogno, P. Autophagy: Regulation and Role in Disease. Crit. Rev. Clin. Lab. Sci. 2009, 46, 210–240. [Google Scholar] [CrossRef] [Green Version]
- Balgi, A.D.; Diering, G.H.; Donohue, E.; Lam, K.K.Y.; Fonseca, B.D.; Zimmerman, C.; Numata, M.; Roberge, M. Regulation of mTORC1 Signaling by pH. PLoS ONE 2011, 6, e21549. [Google Scholar] [CrossRef]
- Orvedahl, A.; Levine, B. Eating the Enemy within: Autophagy in Infectious Diseases. Cell Death Differentiation 2009, 16, 57–69. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and Function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Thorburn, A. Autophagy and its Effects: Making Sense of Double-Edged Swords. PLoS Biol. 2014, 12, e1001967. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.S.; Vats, S.; Chia, A.Y.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual Role of Autophagy in Hallmarks of Cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- ] Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Autophagy in Cancer Metastasis. Oncogene 2017, 36, 1619–1630. [Google Scholar] [CrossRef]
- Dower, C.M.; Wills, C.A.; Frisch, S.M.; Wang, H.G. Mechanisms and Context Underlying the Role of Autophagy in Cancer Metastasis. Autophagy 2018, 14, 1110–1128. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, R.M.; Henquin, J.C. The Role of Metabolism, Cytoplasmic Ca2+, and pH-Regulating Exchangers in Glucose-Induced Rise of Cytoplasmic pH in Normal Mouse Pancreatic Islets. J. Biol. Chem. 1995, 270, 7915–7921. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J. Energy Metabolism of Cancer: Glycolysis Versus Oxidative Phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [Green Version]
- Zou, C.; Wang, Y.; Shen, Z. 2-NBDG as a Fluorescent Indicator for Direct Glucose Uptake Measurement. J. Biochem. Biophys. Methods 2005, 64, 207–215. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do Cancers have High Aerobic Glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Gillies, R.J.; Raghunand, N.; Karczmar, G.S.; Bhujwalla, Z.M. MRI of the Tumor Microenvironment. J. Magnetic Res. Imag. 2002, 16, 430–450. [Google Scholar] [CrossRef]
- Rofstad, E.K.; Mathiesen, B.; Kindem, K.; Galappathi, K. Acidic Extracellular pH Promotes Experimental Metastasis of Human Melanoma Cells in Athymic Nude Mice. Cancer Res. 2006, 66, 6699–6707. [Google Scholar] [CrossRef] [Green Version]
- Nezich, C.L.; Wang, C.; Fogel, A.I.; Youle, R.J. MiT/TFE Transcription Factors are Activated during Mitophagy Downstream of Parkin and Atg5. J. Cell Biol. 2015, 210, 435–450. [Google Scholar] [CrossRef] [Green Version]
- Di Malta, C.; Cinque, L.; Settembre, C. Transcriptional Regulation of Autophagy: Mechanisms and Diseases. Front. Cell. Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef]
- Li, S.; Song, Y.; Quach, C.; Guo, H.; Jang, G.B.; Maazi, H.; Zhao, S.; Sands, N.A.; Liu, Q.; In, G.K.; et al. Transcriptional Regulation of Autophagy-Lysosomal Function in BRAF-Driven Melanoma Progression and Chemoresistance. Nat. Commun. 2019, 10, 1693–1698. [Google Scholar] [CrossRef] [Green Version]
- McCain, J. The MAPK (ERK) Pathway: Investigational Combinations for the Treatment of BRAF-Mutated Metastatic Melanoma. P T A Peer-Rev. J. Formul. Manag. 2013, 38, 96–108. [Google Scholar]
- Caporali, S.; Alvino, E.; Lacal, P.M.; Levati, L.; Giurato, G.; Memoli, D.; Caprini, E.; Antonini Cappellini, G.C.; D’Atri, S. Targeting the PI3K/AKT/mTOR Pathway Overcomes the Stimulating Effect of Dabrafenib on the Invasive Behavior of Melanoma Cells with Acquired Resistance to the BRAF Inhibitor. Int. J. Oncol. 2016, 49, 1164–1174. [Google Scholar] [CrossRef]
- Kapuy, O.; Vinod, P.K.; Banhegyi, G. mTOR Inhibition Increases Cell Viability Via Autophagy Induction during Endoplasmic Reticulum Stress - an Experimental and Modeling Study. FEBS Open Bio. 2014, 4, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Steven, A.; Seliger, B. Control of CREB Expression in Tumors: From Molecular Mechanisms and Signal Transduction Pathways to Therapeutic Target. Oncotarget 2016, 7, 35454–35465. [Google Scholar] [CrossRef] [Green Version]
- Henriksson, E.; Sall, J.; Gormand, A.; Wasserstrom, S.; Morrice, N.A.; Fritzen, A.M.; Foretz, M.; Campbell, D.G.; Sakamoto, K.; Ekelund, M.; et al. SIK2 Regulates CRTCs, HDAC4 and Glucose Uptake in Adipocytes. J. Cell. Sci. 2015, 128, 472–486. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.L. Ambra1 in Autophagy and Apoptosis: Implications for Cell Survival and Chemotherapy Resistance. Oncol. Lett. 2016, 12, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Menon, M.B.; Dhamija, S. Beclin 1 Phosphorylation - at the Center of Autophagy Regulation. Front. Cell. Dev. Biol. 2018, 6, 137. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.Y.; Ellis, R.A.; Lovat, P.E. Prognostic Impact of Autophagy Biomarkers for Cutaneous Melanoma. Front. Oncol. 2016, 6, 236. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Monitoring Autophagic Degradation of p62/SQSTM1. Method. Enzymol. 2009, 452, 181–197. [Google Scholar]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-Positive Structures are Prominent in Autophagy-Deficient Cells. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adhikari, M.; Adhikari, B.; Ghimire, B.; Baboota, S.; Choi, E.H. Cold Atmospheric Plasma and Silymarin Nanoemulsion Activate Autophagy in Human Melanoma Cells. Int. J. Mol. Sci. 2020, 21, 1939. https://doi.org/10.3390/ijms21061939
Adhikari M, Adhikari B, Ghimire B, Baboota S, Choi EH. Cold Atmospheric Plasma and Silymarin Nanoemulsion Activate Autophagy in Human Melanoma Cells. International Journal of Molecular Sciences. 2020; 21(6):1939. https://doi.org/10.3390/ijms21061939
Chicago/Turabian StyleAdhikari, Manish, Bhawana Adhikari, Bhagirath Ghimire, Sanjula Baboota, and Eun Ha Choi. 2020. "Cold Atmospheric Plasma and Silymarin Nanoemulsion Activate Autophagy in Human Melanoma Cells" International Journal of Molecular Sciences 21, no. 6: 1939. https://doi.org/10.3390/ijms21061939
APA StyleAdhikari, M., Adhikari, B., Ghimire, B., Baboota, S., & Choi, E. H. (2020). Cold Atmospheric Plasma and Silymarin Nanoemulsion Activate Autophagy in Human Melanoma Cells. International Journal of Molecular Sciences, 21(6), 1939. https://doi.org/10.3390/ijms21061939