On the Trail of Tetu1: Genome-Wide Discovery of CACTA Transposable Elements in Sunflower Genome

 , ,
, ,
Abstract
:1. Introduction
2. Results
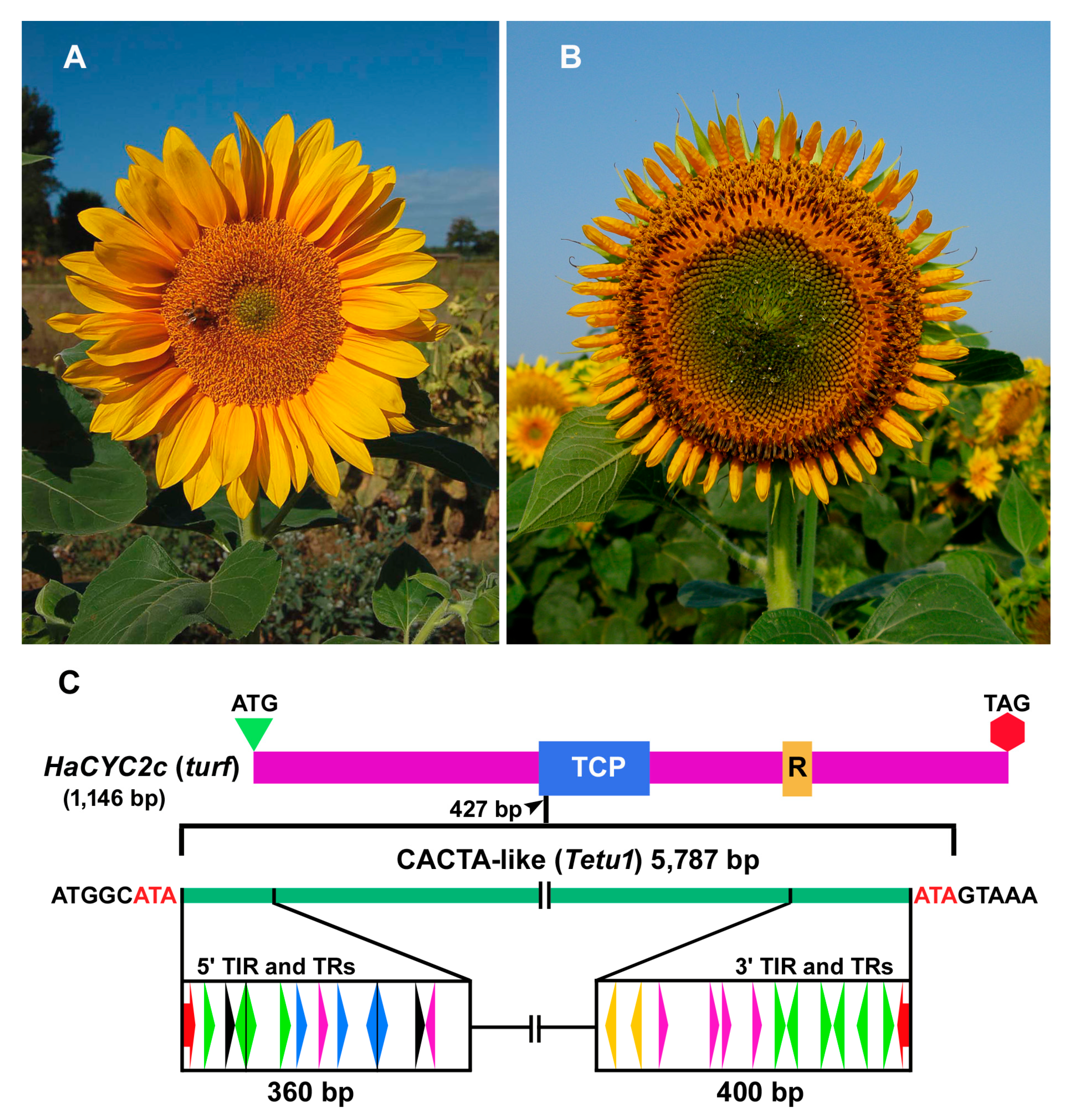
2.1. Identification of Putative CACTA Transposon Sequences in the Sunflower Genome
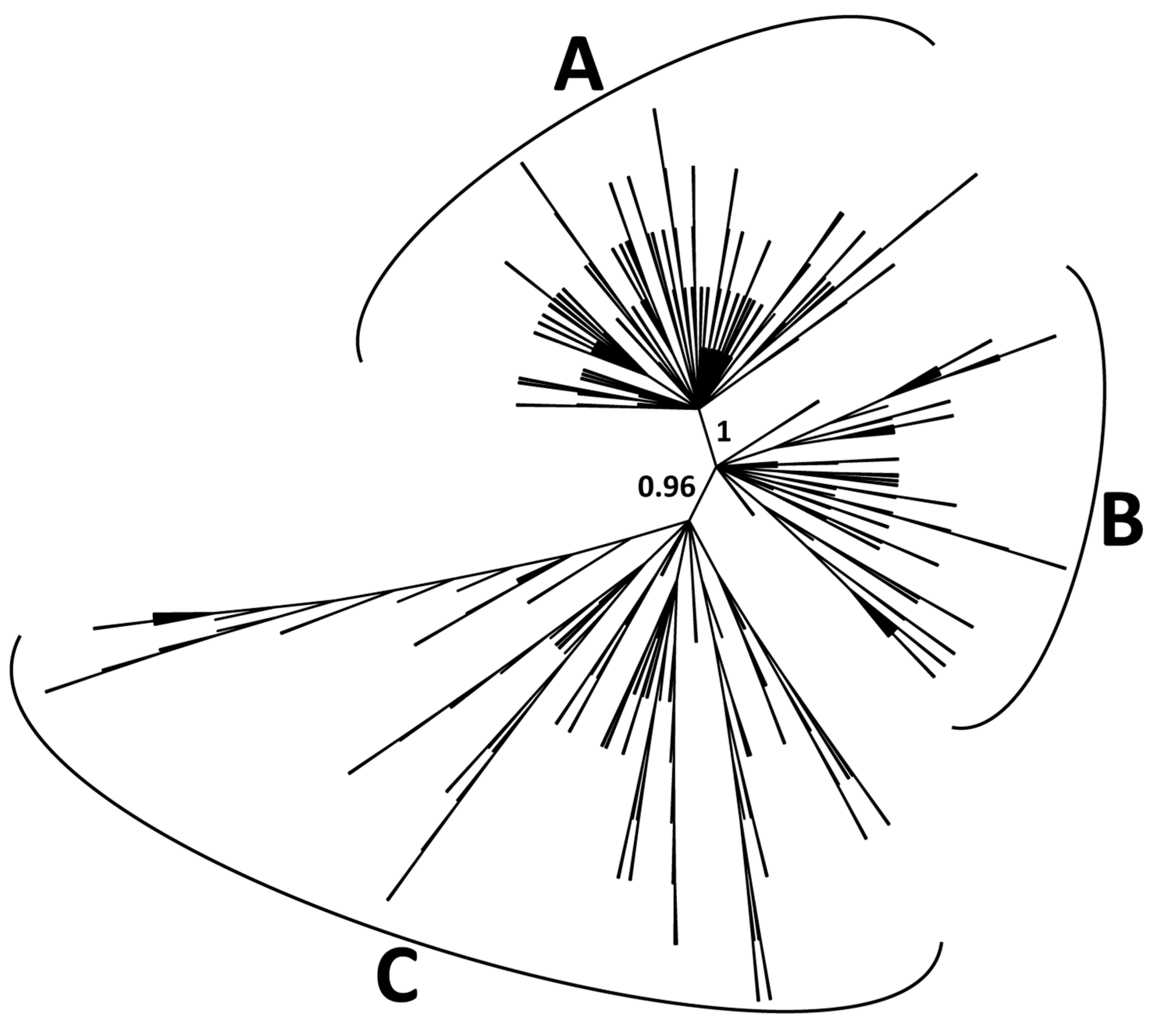
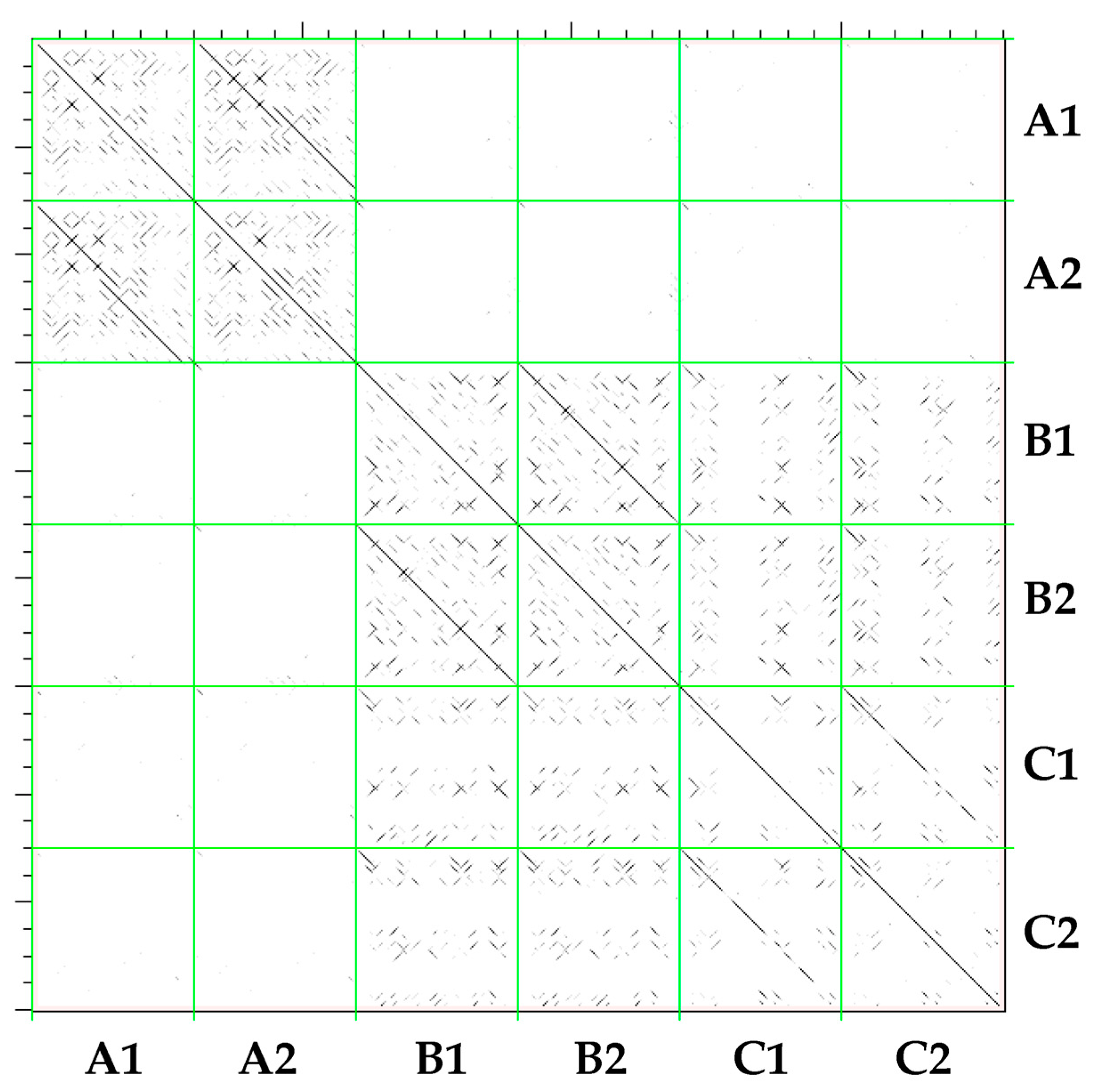
2.2. Classification of CACTA Transposons Based on their TR Sequences and Abundance Estimation
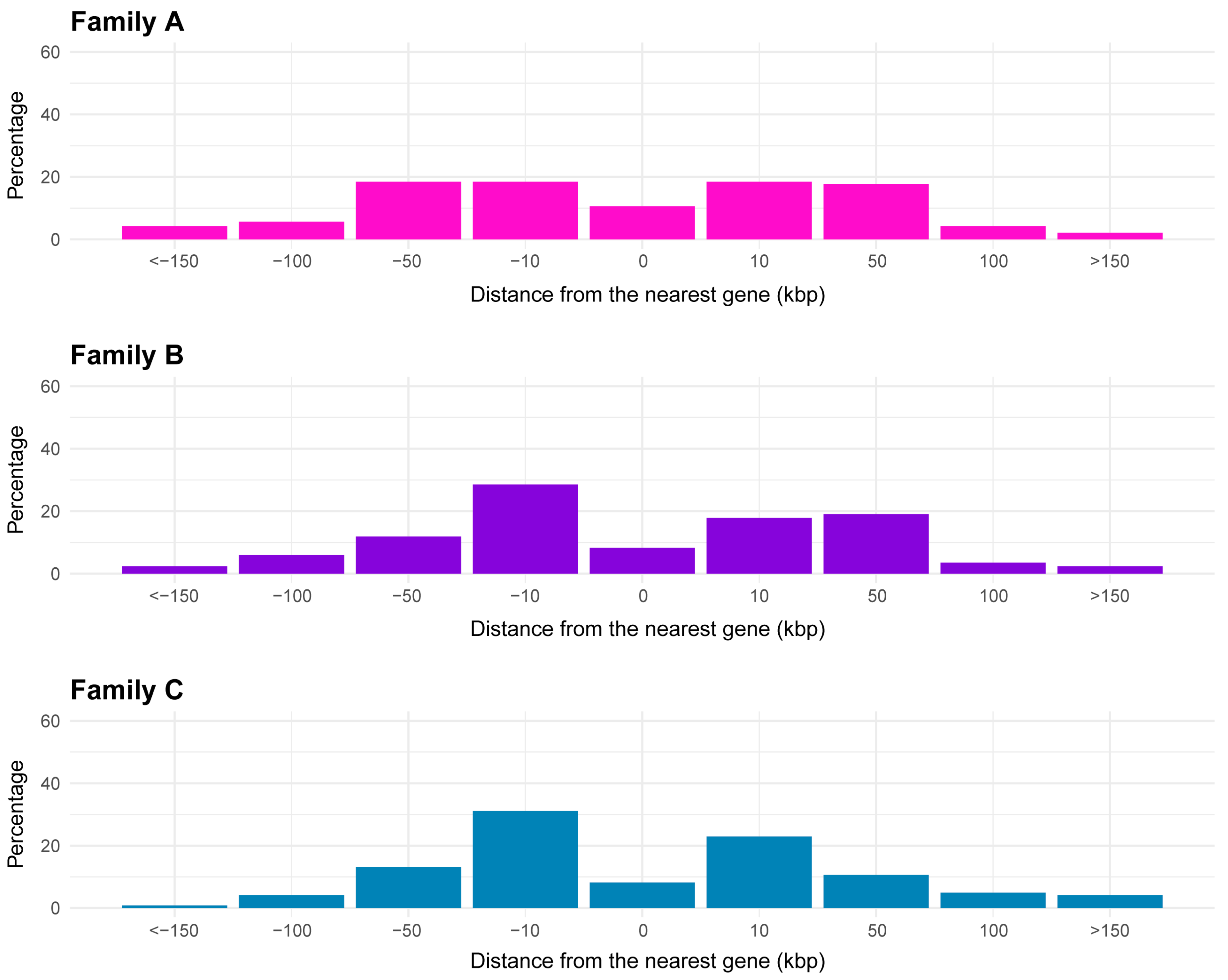
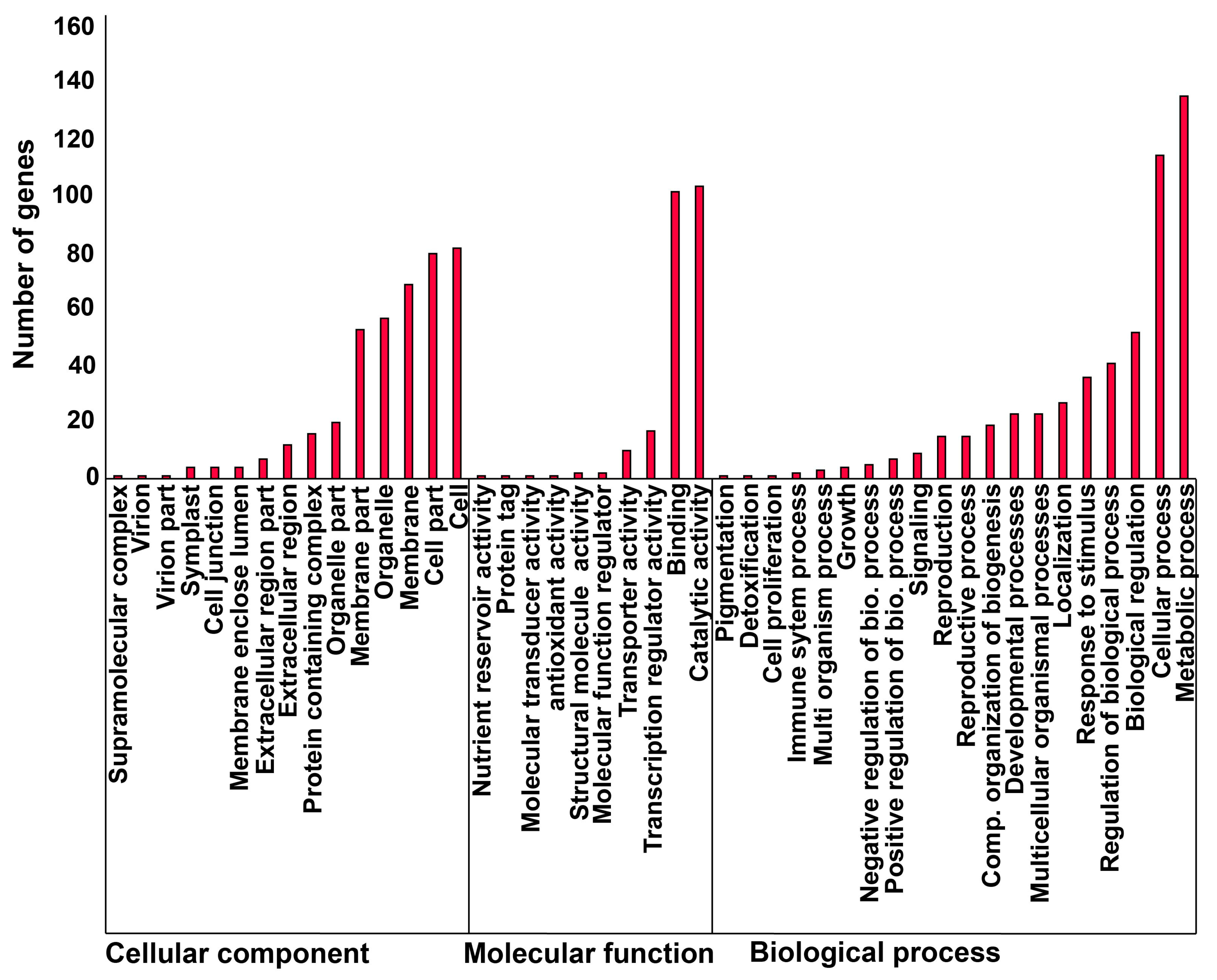
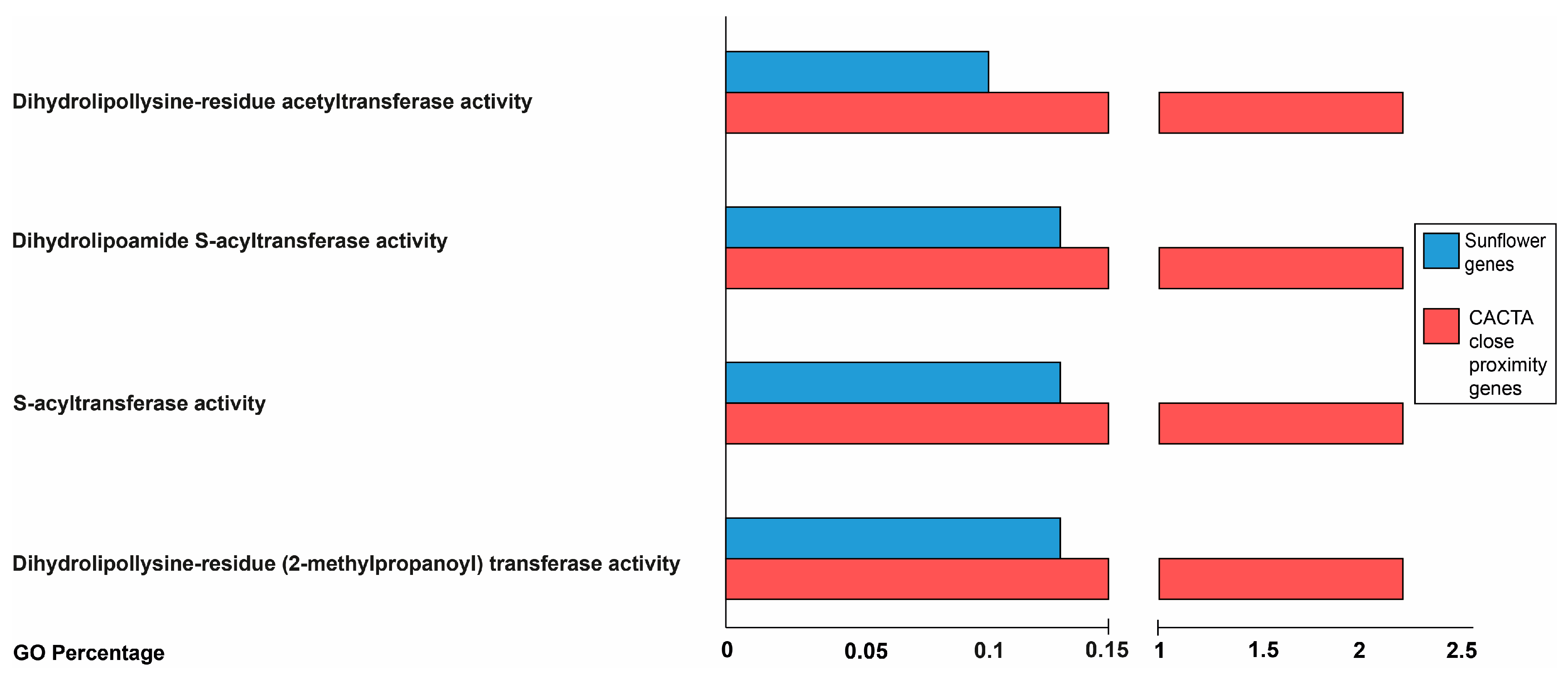
2.3. Proximity of CACTA Transposons to Genes and Functional Analysis
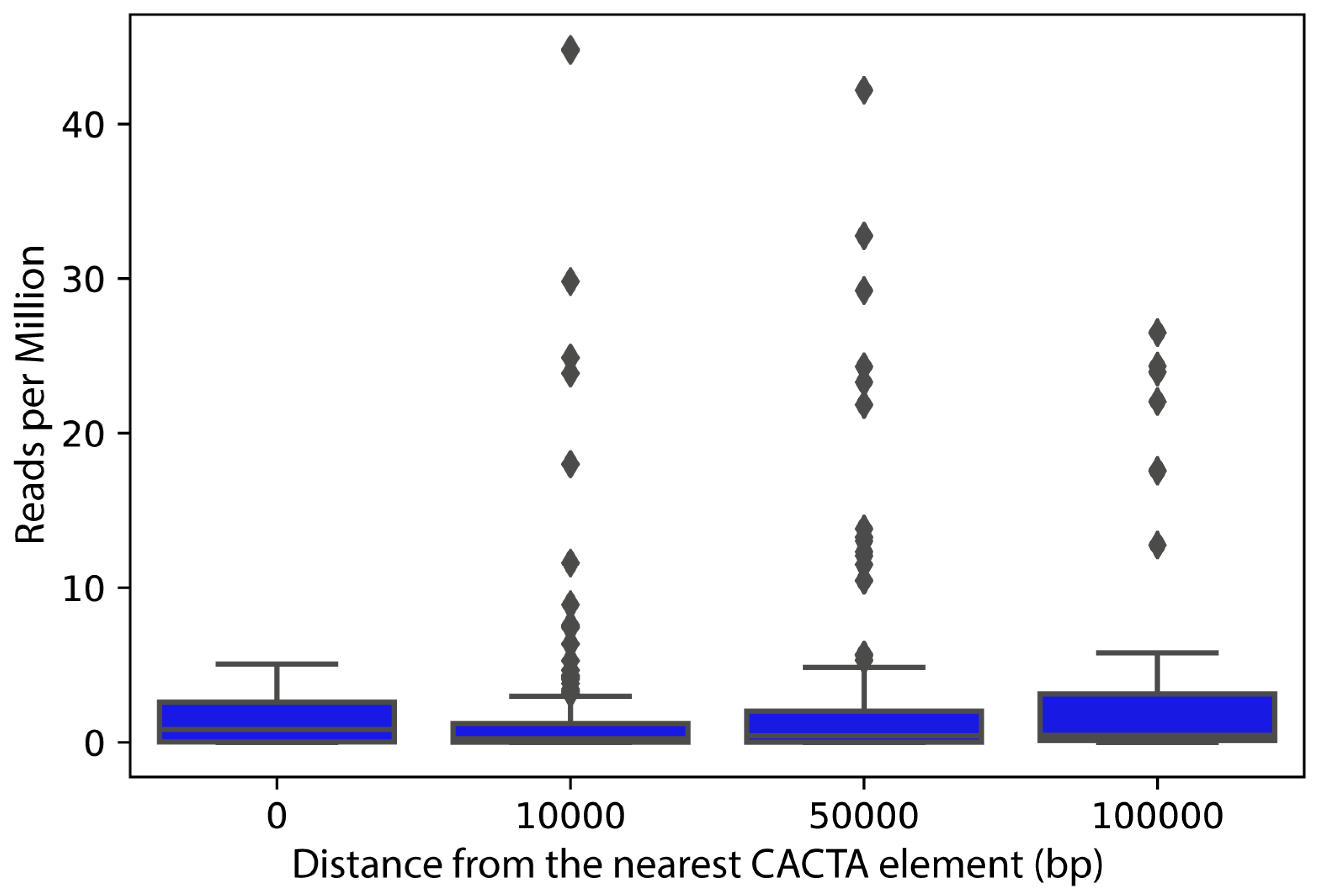
2.4. Expression Analysis of CACTA Transposons and Genes Closest to CACTA Transposons in the Sunflower Genome
3. Discussion
4. Material and Methods
4.1. Sequence Collection
4.2. Abundance Estimation and DNA Mapping Procedure
4.3. Evolutionary Analysis using the Maximum Likelihood Method
4.4. Analysis of Proximity of CACTA Elements to Genes
4.5. TPase Domain Identification and CACTA Transposon Expression Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Chry | Chrysanthemoides |
| CYC | CYCLOIDEA gene |
| GO | Gene ontology |
| LTR | Long terminal repeats |
| REs | Retrotransposons |
| TCP | TEOSINTE BRANCHED1/CYCLOIDEA/PROLIFERATING NUCLEAR ANTIGEN CELL FACTOR |
| TEs | Transposable elements |
| Tetu1 | Transposable element of turf1 |
| TIRs | Terminal inverted repeats |
| TRs | Sub-terminal repeats |
| TPase | Transposase domain |
| TSD | Target-site duplication motif |
| turf | Tubular ray flower |
References
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenaillon, M.I.; Hollister, J.D.; Gaut, B.S. A triptych of the evolution of plant transposable elements. Trends Plant Sci. 2010, 15, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Novák, P.; Hoštáková, N.; Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef]
- Butelli, E.; Licciardello, C.; Zhang, Y.; Liu, J.; Mackay, S.; Bailey, P.; Reforgiato-Recupero, G.; Martin, C. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 2012, 24, 1242–1255. [Google Scholar] [CrossRef] [Green Version]
- Falchi, R.; Vendramin, E.; Zanon, L.; Scalabrin, S.; Verde, I.; Vizzotto, G.; Morgante, M. Three distinct mutational mechanisms acting on a single gene underpin the origin of yellow flesh in peach. Plant J. 2013, 76, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef]
- Catoni, M.; Jonesman, T.; Cerruti, E.; Paszkowski, J. Mobilization of Pack-CACTA transposons in Arabidopsis suggests the mechanism of gene shuffling. Nucleic Acids Res. 2019, 47, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Ueki, N.; Nishii, I. Idaten is a new cold-inducible transposon of Volvox carteri that can be used for tagging developmentally important genes. Genetics 2008, 180, 1343–1353. [Google Scholar] [CrossRef] [Green Version]
- Pereira, A.; Cuypers, H.; Gierl, A.; Schwarz-Sommer, Z.; Saedler, H. Molecular analysis of the En/Spm transposable element system of Zea mays. EMBO J. 1986, 5, 835–841. [Google Scholar] [CrossRef]
- Snowden, K.C.; Napoli, C.A. Psl: a novel Spm-like transposable element from Petunia hybrida. Plant J. 1998, 14, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S.; Brendel, V.; Zhang, J.; Axtell, J.D.; Peterson, T. Molecular characterization of a mutable pigmentation phenotype and isolation of the first active transposable element from Sorghum bicolor. Proc. Natl. Acad. Sci. USA 1999, 96, 15330–15335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, A.; Yonebayashi, S.; Watanabe, K.; Toyama, T.; Shimada, H.; Kakutani, T. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 2001, 411, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Novick, P.A.; Smith, J.D.; Floumanhaft, M.; Ray, D.A.; Boissinot, S. The evolution and diversity of DNA transposons in the genome of the lizard Anolis carolinensis. Genome Biol. Evol. 2011, 3, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Fedoroff, N.V. Molecular genetics and epigenetics of CACTA elements. Methods Mol. Biol. 2013, 1057, 177–192. [Google Scholar]
- Peterson, P.A. A mutable pale green locus in maize. Genetics 1953, 38, 682–683. [Google Scholar]
- McClintock, B. Mutations in maize and chromosomal aberrations in Neurospora. Carnegie Inst. Wash. Year Book 1954, 53, 254–260. [Google Scholar]
- Wicker, T.; Guyot, R.; Yahiaoui, N.; Keller, B. CACTA transposons in Triticeae. A diverse family of high-copy repetitive elements. Plant Physiol. 2003, 132, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Fambrini, M.; Salvini, M.; Pugliesi, C. A transposon-mediate inactivation of a CYCLOIDEA-like gene originates polysymmetric and androgynous ray flowers in Helianthus annuus. Genetica 2011, 139, 1521–1529. [Google Scholar] [CrossRef]
- Fambrini, M.; Basile, A.; Salvini, M.; Pugliesi, C. Excisions of a defective transposable CACTA element (Tetu1) generate new alleles of a CYCLOIDEA-like gene of Helianthus annuus. Gene 2014, 549, 198–207. [Google Scholar] [CrossRef]
- Fambrini, M.; Pugliesi, C. CYCLOIDEA 2 clade genes: key players in the control of floral symmetry, inflorescence architecture, and reproductive organ development. Plant Mol. Biol. Rep. 2017, 35, 20–36. [Google Scholar] [CrossRef]
- Chapman, M.A.; Tang, S.; Draeger, D.; Nambeesan, S.; Shaffer, H.; Barb, J.G.; Knapp, S.J.; Burke, J.M. Genetic analysis of floral symmetry in Van Gogh’s sunflowers reveals independent recruitment of CYCLOIDEA genes in the Asteraceae. PLoS Genet. 2012, 8, e1002628. [Google Scholar] [CrossRef] [PubMed]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Mascagni, F.; Barghini, E.; Giordani, T.; Rieseberg, L.H.; Cavallini, A.; Natali, L. Repetitive DNA and plant domestication: variation in copy number and proximity to genes of LTR-retrotransposons among wild and cultivated sunflower (Helianthus annuus) genotypes. Genome Biol. Evol. 2015, 7, 3368–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natali, L.; Cossu, R.M.; Barghini, E.; Giordani, T.; Buti, M.; Mascagni, F.; Morgante, M.; Gill, N.; Kane, N.C.; Rieseberg, L.; et al. The repetitive component of the sunflower genome as revealed by different procedures for assembling next generation sequencing reads. BMC Genomics 2013, 14, 686. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.-J.; Hong, S.-W.; Son, J.-H.; Lee, J.K.; Cha, Y.-S.; Eun, M.-Y.; Kim, N.-S. CACTA and MITE transposon distributions on a genetic map of rice using F15 RILs derived from Milyang 23 and Gihobyeo hybrids. Mol. Cells 2006, 21, 360–366. [Google Scholar]
- Sabot, F.; Simon, D.; Bernard, M. Plant transposable elements, with an emphasis on grass species. Euphytica 2004, 139, 227–247. [Google Scholar] [CrossRef]
- Sergeeva, E.M.; Salina, E.A.; Adonina, I.G.; Chalhoub, B. Evolutionary analysis of the CACTA DNA-transposon Caspar across wheat species using sequence comparison and in situ hybridization. Mol. Genet. Genomics 2010, 284, 11–23. [Google Scholar] [CrossRef]
- Chapman, M.A.; Leebens-Mack, J.H.; Burke, J.M. Positive selection and expression divergence following gene duplication in the sunflower CYCLOIDEA gene family. Mol. Biol. Evol. 2008, 25, 1260–1273. [Google Scholar] [CrossRef] [Green Version]
- Fambrini, M.; Salvini, M.; Basile, A.; Pugliesi, C. Transposon-dependent induction of Vincent van Gogh’s sunflowers: Exceptions revealed. genesis 2014, 52, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Fambrini, M.; Pugliesi, C. Mobilization of the Tetu1 transposable element of Helianthus annuus: evidence for excision in different developmental stages. Biol. Plantarum 2017, 61, 55–63. [Google Scholar] [CrossRef]
- Bennetzen, J.L. Transposable element contributions to plant gene and genome evolution. Plant Mol. Biol. 2000, 42, 251–269. [Google Scholar] [CrossRef] [PubMed]
- Langdon, T.; Jenkins, G.; Hasterok, R.; Jones, R.N.; King, I.P. A high-copy-number CACTA family transposon in temperate grasses and cereals. Genetics 2003, 163, 1097–1108. [Google Scholar] [PubMed]
- Bonas, U.; Sommer, H.; Harrison, B.J.; Saedler, H. The transposable element Tam1 of Antirrhinum majus is 17 kb long. Mol. Gen. Genet. 1984, 194, 138–143. [Google Scholar] [CrossRef]
- Roccaro, M.; Li, Y.; Sommer, H.; Saedler, H. ROSINA (RSI) is part of a CACTA transposable element, TamRSI, and links flower development to transposon activity. Mol. Genet. Genomics 2007, 278, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabot, F.; Guyot, R.; Wicker, T.; Chantret, N.; Laubin, B.; Chalhoub, B.; Leroy, P.; Sourdille, P.; Bernard, M. Updating of transposable element annotations from large wheat genomic sequences reveals diverse activities and gene associations. Mol. Genet. Genomics 2005, 274, 119–130. [Google Scholar] [CrossRef]
- Studer, A.; Zhao, Q.; Ross-Ibarra, J.; Doebly, J. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 2011, 43, 1160–1164. [Google Scholar] [CrossRef] [Green Version]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013–2015. Available online: http://repeatmasker.org/ (accessed on 1 December 2019).
- Quinlan, A.R.; Hall, I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Mascagni, F.; Cavallini, A.; Giordani, T.; Natali, L. Different histories of two highly variable LTR retrotransposons in sunflower species. Gene 2017, 634, 5–14. [Google Scholar]
- Mascagni, F.; Vangelisti, A.; Giordani, T.; Cavallini, A.; Natali, L. Specific LTR-retrotransposons show copy number variations between wild and cultivated sunflowers. Genes 2018, 9, 433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Giordani, T.; Buti, M.; Natali, L.; Pugliesi, C.; Cattonaro, F.; Morgante, M.; Cavallini, A. An analysis of sequence variability in eight genes putatively involved in drought response in sunflower (Helianthus annuus L.). Theor. Appl. Genet. 2011, 122, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Gouveia-Oliveira, R.; Sackett, P.W.; Pedersen, A.G. MaxAlign: maximizing usable data in an alignment. BMC Bioinformatics 2007, 8, 312. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.L.; Durbin, R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 1995, 167, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FAMILY | NO. OF ELEMENTS | COMPLETE ELEMENTS | TRUNCATED ELEMENTS | TPASE DOMAIN | SEQUENCE CONSERVATION |
|---|---|---|---|---|---|
| A | 141 | 2 | 139 | - | 64 |
| B | 84 | 13 | 71 | 27 | 52 |
| C | 122 | 19 | 103 | 12 | 45 |
| TOTAL | 347 | 34 | 313 | 39 |
| FAMILY | FAMILY PER BASE AVERAGE COVERAGE | AVERAGE NUMBER OF COPIES |
|---|---|---|
| A | 2.50 | 1.29 |
| B | 2.66 | 1.38 |
| C | 2.72 | 1.41 |
| UNKNOWN | 7.12 | 3.68 |
| FAMILY | CACTA ELEMENT NAME | MAPPED READS PER MILLION |
|---|---|---|
| B | CACTA 641 | 0.0127 |
| B | CACTA 506 | 0.0141 |
| B | CACTA 314 | 0.0150 |
| B | CACTA 203 | 0.0168 |
| B | CACTA 292 | 0.0282 |
| B | CACTA 610 | 0.0430 |
| C | CACTA 492 | 0.008 |
| C | CACTA 585 | 0.008 |
| C | CACTA 242 | 0.023 |
| C | CACTA 224 | 0.949 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ventimiglia, M.; Pugliesi, C.; Vangelisti, A.; Usai, G.; Giordani, T.; Natali, L.; Cavallini, A.; Mascagni, F. On the Trail of Tetu1: Genome-Wide Discovery of CACTA Transposable Elements in Sunflower Genome. Int. J. Mol. Sci. 2020, 21, 2021. https://doi.org/10.3390/ijms21062021
Ventimiglia M, Pugliesi C, Vangelisti A, Usai G, Giordani T, Natali L, Cavallini A, Mascagni F. On the Trail of Tetu1: Genome-Wide Discovery of CACTA Transposable Elements in Sunflower Genome. International Journal of Molecular Sciences. 2020; 21(6):2021. https://doi.org/10.3390/ijms21062021
Chicago/Turabian StyleVentimiglia, Maria, Claudio Pugliesi, Alberto Vangelisti, Gabriele Usai, Tommaso Giordani, Lucia Natali, Andrea Cavallini, and Flavia Mascagni. 2020. "On the Trail of Tetu1: Genome-Wide Discovery of CACTA Transposable Elements in Sunflower Genome" International Journal of Molecular Sciences 21, no. 6: 2021. https://doi.org/10.3390/ijms21062021
APA StyleVentimiglia, M., Pugliesi, C., Vangelisti, A., Usai, G., Giordani, T., Natali, L., Cavallini, A., & Mascagni, F. (2020). On the Trail of Tetu1: Genome-Wide Discovery of CACTA Transposable Elements in Sunflower Genome. International Journal of Molecular Sciences, 21(6), 2021. https://doi.org/10.3390/ijms21062021