Generation of a Model to Predict Differentiation and Migration of Lymphocyte Subsets under Homeostatic and CNS Autoinflammatory Conditions

 , ,
, , 
Abstract
:1. Introduction
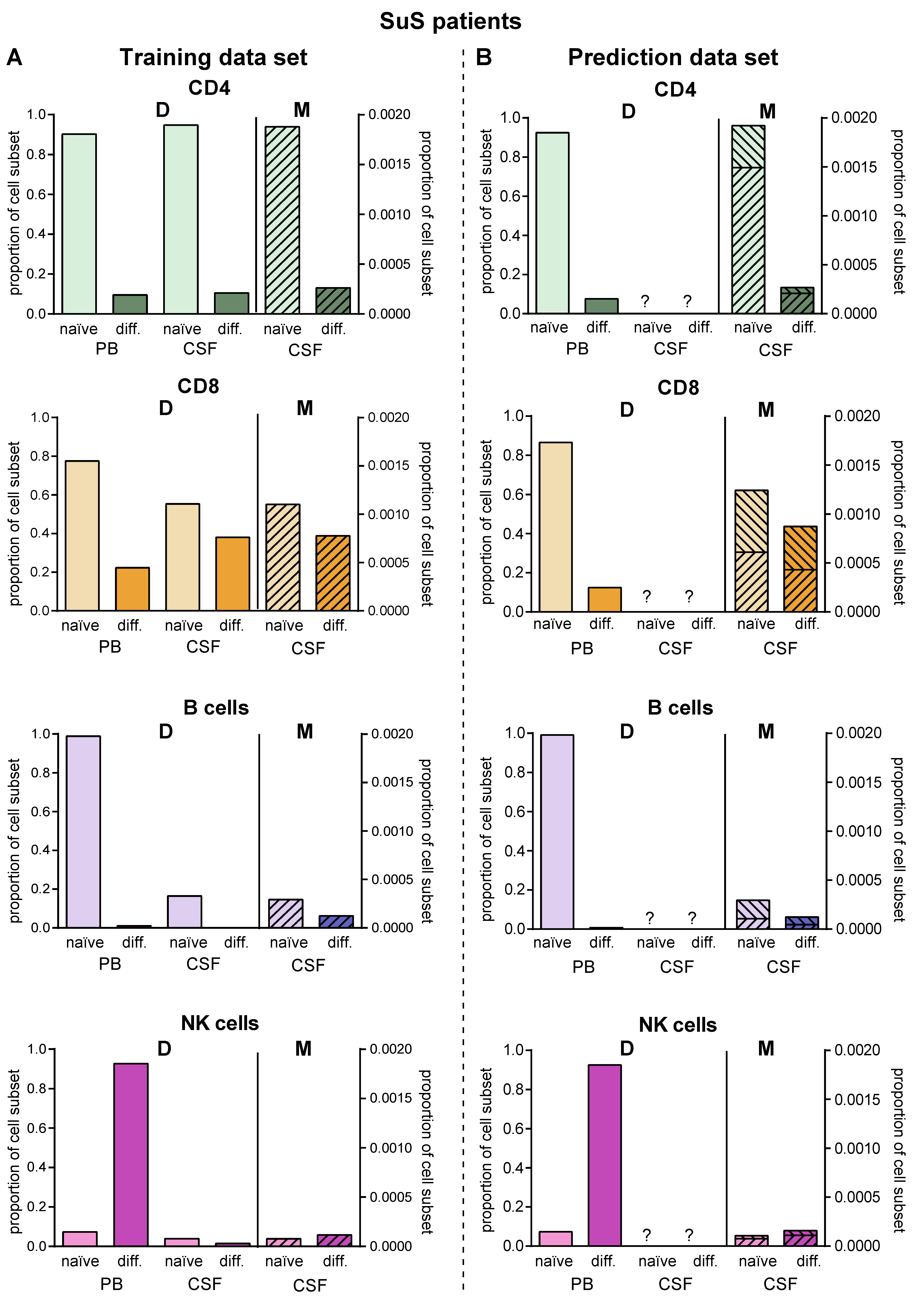
2. Results
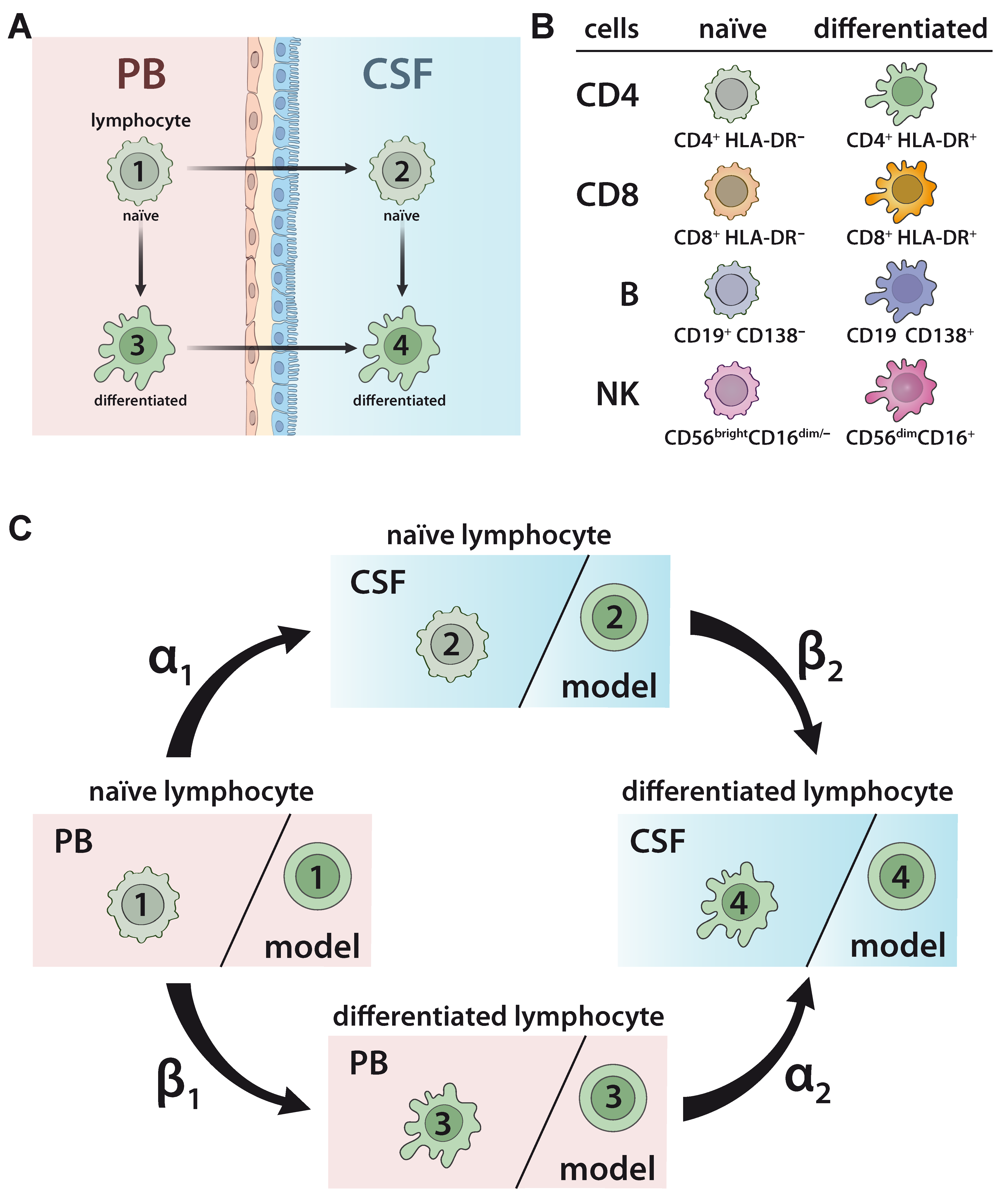
2.1. Developing a Model to Predict the Relationship between Naïve and Differentiated Lymphocyte Subsets in the PB and CSF
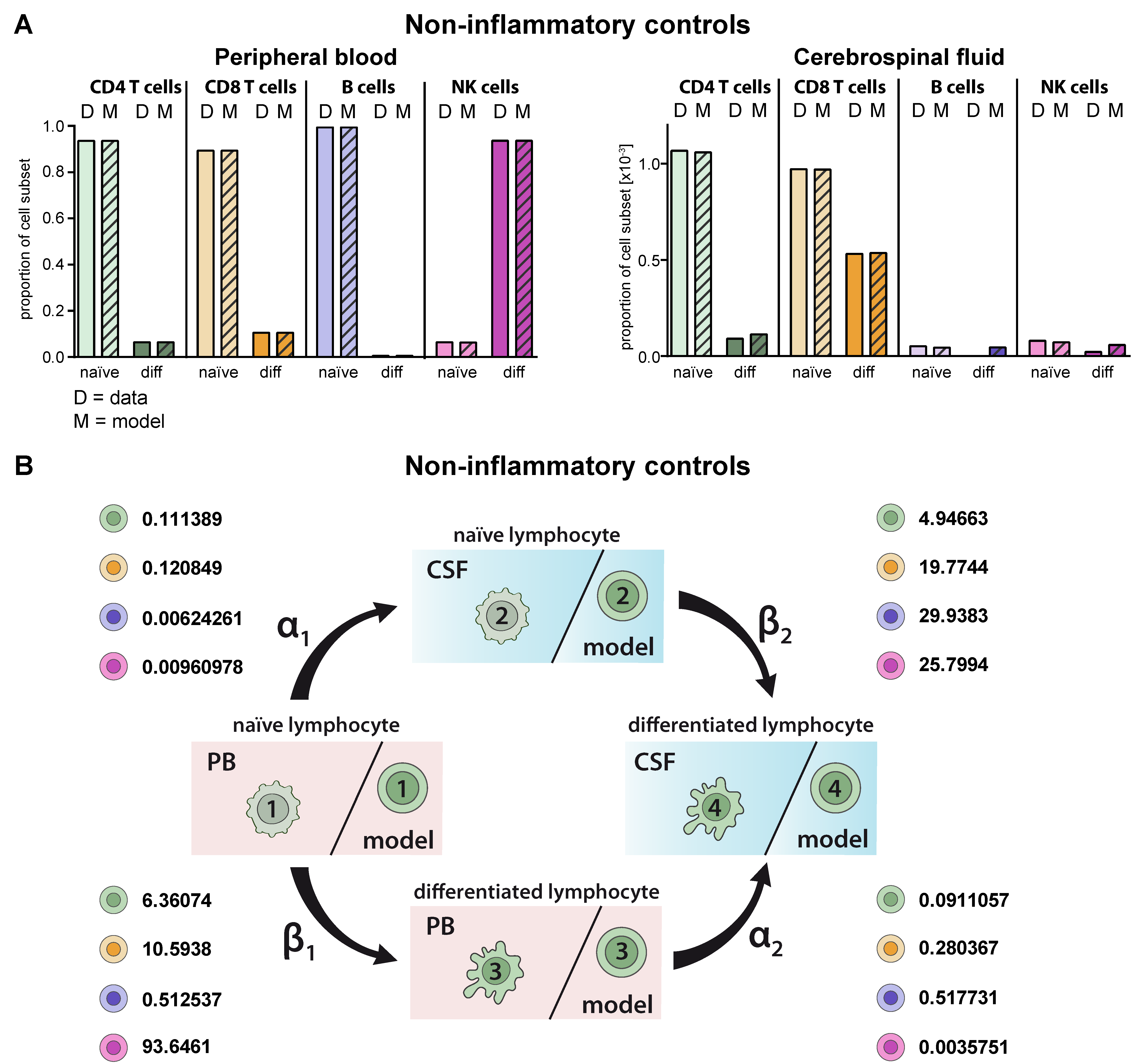
2.2. Model Verification on the Distribution of Lymphocyte Subsets in the PB and CSF under Homeostatic Conditions
2.3. Model Predicting RRMS-Related Changes in Circulating and Intrathecal Lymphocyte Subsets
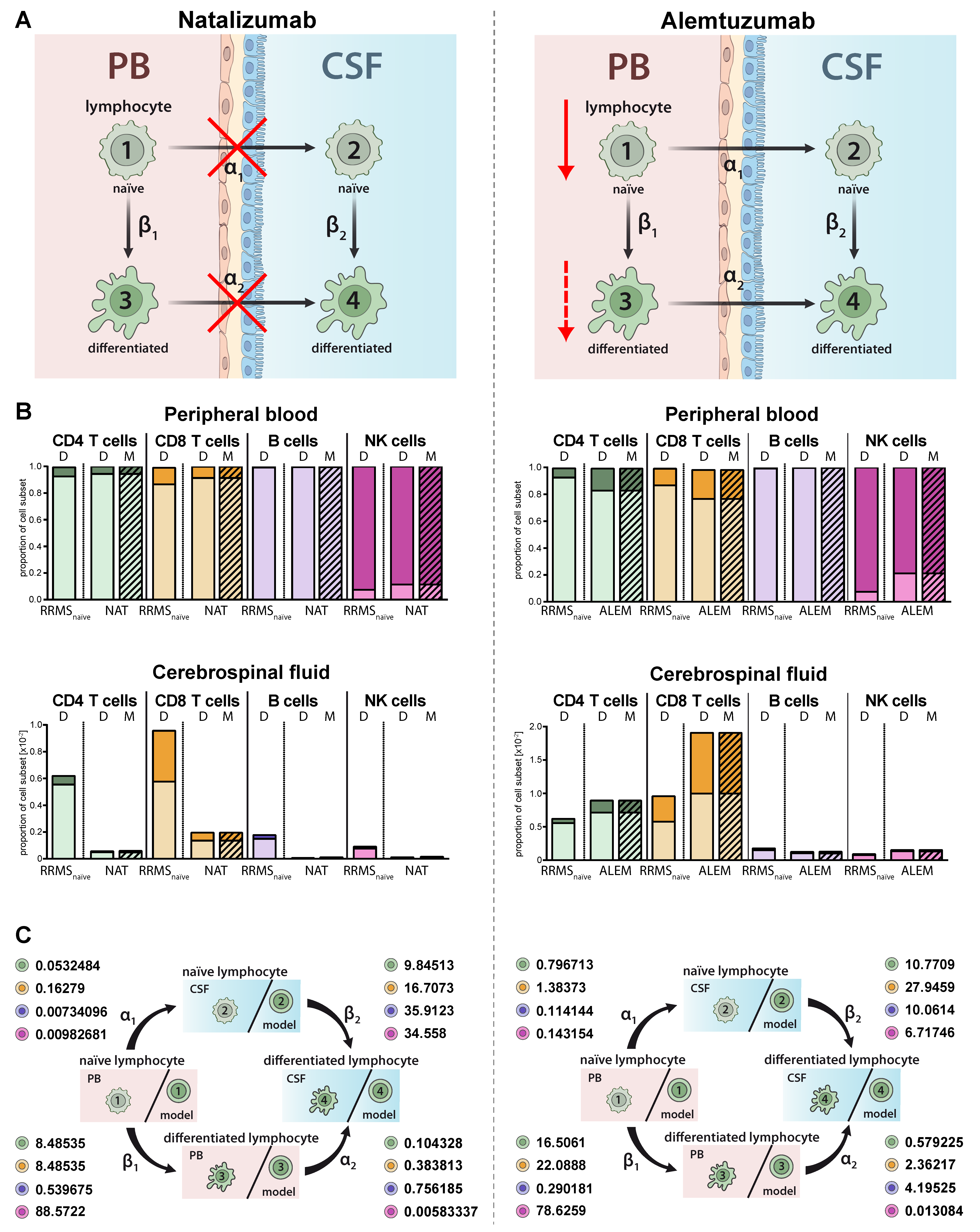
2.4. Model Predictions for Treatment-Related Changes in the Peripheral and Intrathecal Immune Profile
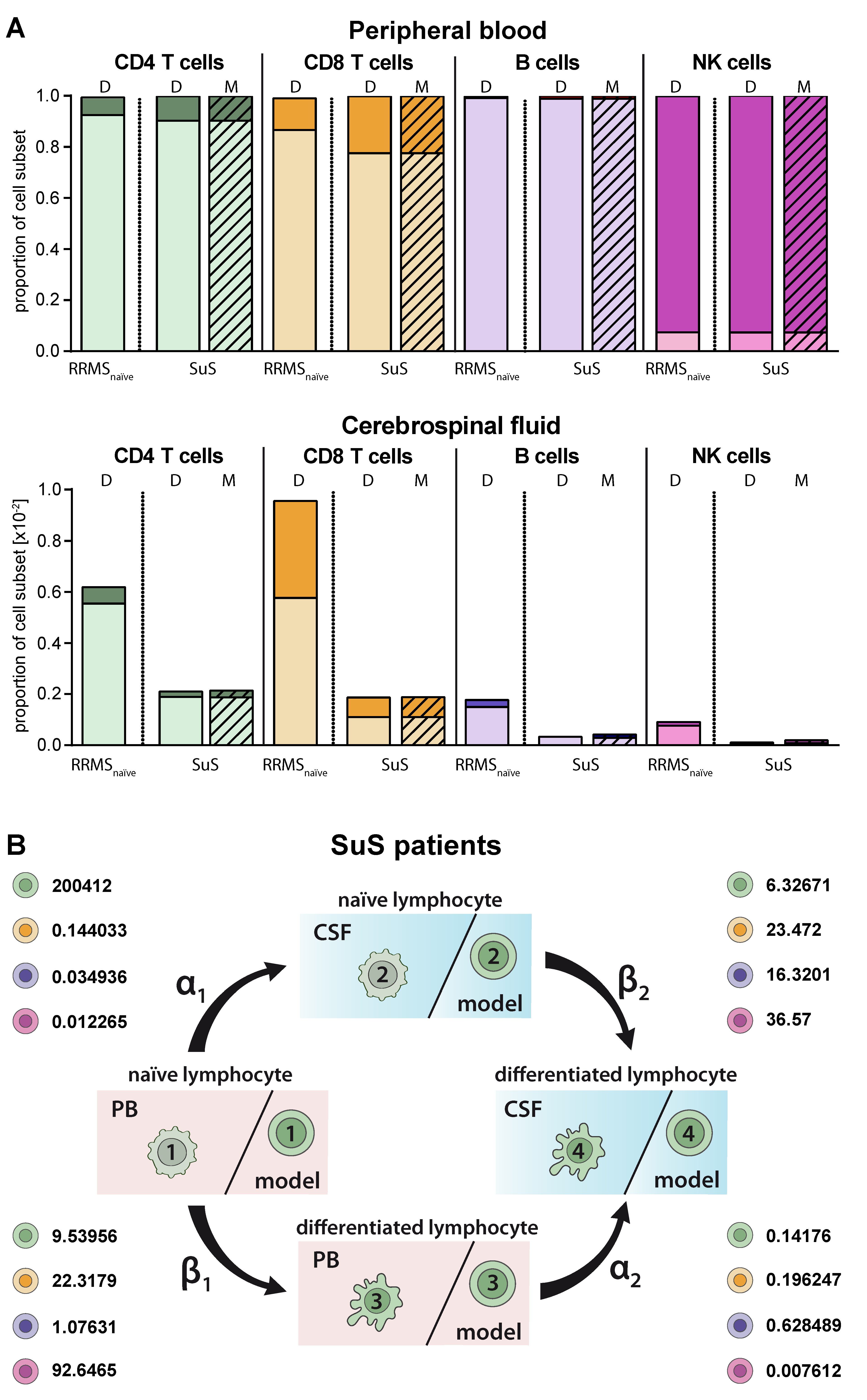
2.5. Model Predicting Distribution of Lymphocytes in the CSF Compartment in the Absence of CSF Data
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Material from Controls and Patients
4.3. Flow Cytometry
4.4. Gating Strategy
4.5. Mathematical Modelling
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Kleine, T.O.; Benes, L. Immune surveillance of the human central nervous system (CNS): Different migration pathways of immune cells through the blood-brain barrier and blood-cerebrospinal fluid barrier in healthy persons. Cytometry A 2006, 69, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Kleine, T.O. Cellular immune surveillance of central nervous system bypasses blood-brain barrier and blood-cerebrospinal-fluid barrier: Revealed with the New Marburg cerebrospinal-fluid model in healthy humans. Cytometry A 2015, 87, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Herich, S.; Schneider-Hohendorf, T.; Rohlmann, A.; Khaleghi Ghadiri, M.; Schulte-Mecklenbeck, A.; Zondler, L.; Janoschka, C.; Ostkamp, P.; Richter, J.; Breuer, J.; et al. Human CCR5high effector memory cells perform CNS parenchymal immune surveillance via GZMK-mediated transendothelial diapedesis. Brain 2019, 142, 3411–3427. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider-Hohendorf, T.; Mohan, H.; Bien, C.G.; Breuer, J.; Becker, A.; Görlich, D.; Kuhlmann, T.; Widman, G.; Herich, S.; Elpers, C.; et al. CD8(+) T-cell pathogenicity in Rasmussen encephalitis elucidated by large-scale T-cell receptor sequencing. Nat. Commun. 2016, 7, 11153. [Google Scholar] [CrossRef]
- Gross, C.C.; Meyer, C.; Bhatia, U.; Yshii, L.; Kleffner, I.; Bauer, J.; Tröscher, A.R.; Schulte-Mecklenbeck, A.; Herich, S.; Schneider-Hohendorf, T.; et al. CD8+ T cell-mediated endotheliopathy is a targetable mechanism of neuro-inflammation in Susac syndrome. Nat. Commun. 2019, 10, 5779. [Google Scholar] [CrossRef] [Green Version]
- Cepok, S.; Jacobsen, M.; Schock, S.; Omer, B.; Jaekel, S.; Böddeker, I.; Oertel, W.H.; Sommer, N.; Hemmer, B. Patterns of cerebrospinal fluid pathology correlate with disease progression in multiple sclerosis. Brain 2001, 124, 2169–2176. [Google Scholar] [CrossRef] [Green Version]
- Cepok, S.; Rosche, B.; Grummel, V.; Vogel, F.; Zhou, D.; Sayn, J.; Sommer, N.; Hartung, H.-P.; Hemmer, B. Short-lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain 2005, 128, 1667–1676. [Google Scholar] [CrossRef] [Green Version]
- Obermeier, B.; Lovato, L.; Mentele, R.; Brück, W.; Forne, I.; Imhof, A.; Lottspeich, F.; Turk, K.W.; Willis, S.N.; Wekerle, H.; et al. Related B cell clones that populate the CSF and CNS of patients with multiple sclerosis produce CSF immunoglobulin. J. Neuroimmunol. 2011, 233, 245–248. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Lin, Y.C.; Wu, T.; Salgado, A.D.; Mexhitaj, I.; Wuest, S.C.; Romm, E.; Ohayon, J.; Goldbach-Mansky, R.; Vanderver, A.; et al. Comprehensive immunophenotyping of cerebrospinal fluid cells in patients with neuroimmunological diseases. J. Immunol. 2014, 192, 2551–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider-Hohendorf, T.; Rossaint, J.; Mohan, H.; Böning, D.; Breuer, J.; Kuhlmann, T.; Gross, C.C.; Flanagan, K.; Sorokin, L.; Vestweber, D.; et al. VLA-4 blockade promotes differential routes into human CNS involving PSGL-1 rolling of T cells and MCAM-adhesion of TH17 cells. J. Exp. Med. 2014, 211, 1833–1846. [Google Scholar] [CrossRef] [PubMed]
- Stüve, O.; Marra, C.M.; Bar-Or, A.; Niino, M.; Cravens, P.D.; Cepok, S.; Frohman, E.M.; Phillips, J.T.; Arendt, G.; Jerome, K.R.; et al. Altered CD4+/CD8+ T-cell ratios in cerebrospinal fluid of natalizumab-treated patients with multiple sclerosis. Arch. Neurol. 2006, 63, 1383–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kivisäkk, P.; Healy, B.C.; Viglietta, V.; Quintana, F.J.; Hootstein, M.A.; Weiner, H.L.; Khoury, S.J. Natalizumab treatment is associated with peripheral sequestration of proinflammatory T cells. Neurology 2009, 72, 1922–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krumbholz, M.; Meinl, I.; Kümpfel, T.; Hohlfeld, R.; Meinl, E. Natalizumab disproportionately increases circulating pre-B and B cells in multiple sclerosis. Neurology 2008, 71, 1350–1354. [Google Scholar] [CrossRef]
- Lohmann, L.; Janoschka, C.; Schulte-Mecklenbeck, A.; Klinsing, S.; Kirstein, L.; Hanning, U.; Wirth, T.; Schneider-Hohendorf, T.; Schwab, N.; Gross, C.C.; et al. Immune Cell Profiling During Switching from Natalizumab to Fingolimod Reveals Differential Effects on Systemic Immune-Regulatory Networks and on Trafficking of Non-T Cell Populations into the Cerebrospinal Fluid-Results from the ToFingo Successor Study. Front. Immunol. 2018, 9, 1560. [Google Scholar] [CrossRef]
- Carlsson, A.E.; Sept, D. Mathematical modeling of cell migration. Methods Cell Biol. 2008, 84, 911–937. [Google Scholar] [CrossRef]
- Ganusov, V.V.; Auerbach, J. Mathematical modeling reveals kinetics of lymphocyte recirculation in the whole organism. PLoS Comput. Biol. 2014, 10, e1003586. [Google Scholar] [CrossRef] [Green Version]
- Ruck, T.; Bittner, S.; Meuth, S.G.; Herty, M. Insights from mathematical modelling for T cell migration into the central nervous system. Math. Med. Biol. 2017, 34, 39–58. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Gross, C.C.; Schulte-Mecklenbeck, A.; Rünzi, A.; Kuhlmann, T.; Posevitz-Fejfár, A.; Schwab, N.; Schneider-Hohendorf, T.; Herich, S.; Held, K.; Konjević, M.; et al. Impaired NK-mediated regulation of T-cell activity in multiple sclerosis is reconstituted by IL-2 receptor modulation. Proc. Natl. Acad. Sci. USA 2016, 113, E2973–E2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.H.; Khan, O.A.; Sheremata, W.A.; Blumhardt, L.D.; Rice, G.P.A.; Libonati, M.A.; Willmer-Hulme, A.J.; Dalton, C.M.; Miszkiel, K.A.; O’Connor, P.W. A Controlled Trial of Natalizumab for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2003, 348, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coles, A.J.; Compston, D.A.S.; Selmaj, K.W.; Lake, S.L.; Moran, S.; Margolin, D.H.; Norris, K.; Tandon, P.K. Alemtuzumab vs. Interferon Beta-1a in Early Multiple Sclerosis. N. Engl. J. Med. 2008, 359, 1786–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, C.C.; Ahmetspahic, D.; Ruck, T.; Schulte-Mecklenbeck, A.; Schwarte, K.; Jörgens, S.; Scheu, S.; Windhagen, S.; Graefe, B.; Melzer, N.; et al. Alemtuzumab treatment alters circulating innate immune cells in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e289. [Google Scholar] [CrossRef] [Green Version]
- Alvermann, S.; Hennig, C.; Stüve, O.; Wiendl, H.; Stangel, M. Immunophenotyping of cerebrospinal fluid cells in multiple sclerosis: In search of biomarkers. JAMA Neurol. 2014, 71, 905–912. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Kleffner, I.; Dörr, J.; Ringelstein, M.; Gross, C.C.; Böckenfeld, Y.; Schwindt, W.; Sundermann, B.; Lohmann, H.; Wersching, H.; Promesberger, J.; et al. Diagnostic criteria for Susac syndrome. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1287–1295. [Google Scholar] [CrossRef]
- Cercignani, C.; Illner, R.; Pulvirenti, M. The Mathematical Theory of Dilute Gases; Springer: New York, NY, USA, 1994. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | N | Females (%) | Age (Median/IQR) |
|---|---|---|---|
| Controls | 75 | 62.7% | 33.9/21.4 |
| RRMS naïve | 221 | 74.0% | 31.9/17.2 |
| RRMS NAT | 41 | 73.2% | 35.1/17.4 |
| RRMS ALEM | 28 | 60.7% | 35.8/15.9 |
| SuS | 33 | 72.7% | 34.7/13.7 |
| Transition Rates (%) | ||||||
|---|---|---|---|---|---|---|
| Cells | Controls | RRMS Naïve | RRMS NAT | RRMS ALEM | SuS | |
| CD4 | α1 | 0.111389 | 0.586281 | 0.0532484 | 0.796713 | 0.200412 |
| α2 | 0.0911057 | 0.472904 | 0.104328 | 0.579225 | 0.14176 | |
| β1 | 6.36074 | 6.97785 | 5.56686 | 16.5061 | 9.53956 | |
| β2 | 4.94663 | 5.36697 | 9.84513 | 10.7709 | 6.32671 | |
| CD8 | α1 | 0.120849 | 0.743036 | 0.16279 | 1.38373 | 0.144033 |
| α2 | 0.280367 | 1.68937 | 0.383813 | 2.36217 | 0.196247 | |
| β1 | 10.5938 | 12.6372 | 8.48535 | 22.0888 | 22.3179 | |
| β2 | 19.7744 | 22.4608 | 16.7073 | 27.9459 | 23.472 | |
| B | α1 | 0.00624261 | 0.163174 | 0.00734096 | 0.114144 | 0.034936 |
| α2 | 0.517731 | 2.05505 | 0.756185 | 4.19525 | 0.628489 | |
| β1 | 0.512537 | 0.706717 | 0.539675 | 0.290181 | 1.07631 | |
| β2 | 29.9383 | 8.32669 | 35.9123 | 10.0614 | 16.3201 | |
| NK | α1 | 0.00960978 | 0.083438 | 0.00982681 | 0.143154 | 0.012265 |
| α2 | 0.0035751 | 0.008764 | 0.00583337 | 0.013084 | 0.007612 | |
| β1 | 93.6461 | 92.5525 | 88.5722 | 78.6259 | 92.6465 | |
| β2 | 25.7994 | 8.89219 | 34.558 | 6.71746 | 36.57 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gross, C.C.; Pawlitzki, M.; Schulte-Mecklenbeck, A.; Rolfes, L.; Ruck, T.; Hundehege, P.; Wiendl, H.; Herty, M.; Meuth, S.G. Generation of a Model to Predict Differentiation and Migration of Lymphocyte Subsets under Homeostatic and CNS Autoinflammatory Conditions. Int. J. Mol. Sci. 2020, 21, 2046. https://doi.org/10.3390/ijms21062046
Gross CC, Pawlitzki M, Schulte-Mecklenbeck A, Rolfes L, Ruck T, Hundehege P, Wiendl H, Herty M, Meuth SG. Generation of a Model to Predict Differentiation and Migration of Lymphocyte Subsets under Homeostatic and CNS Autoinflammatory Conditions. International Journal of Molecular Sciences. 2020; 21(6):2046. https://doi.org/10.3390/ijms21062046
Chicago/Turabian StyleGross, Catharina C., Marc Pawlitzki, Andreas Schulte-Mecklenbeck, Leoni Rolfes, Tobias Ruck, Petra Hundehege, Heinz Wiendl, Michael Herty, and Sven G. Meuth. 2020. "Generation of a Model to Predict Differentiation and Migration of Lymphocyte Subsets under Homeostatic and CNS Autoinflammatory Conditions" International Journal of Molecular Sciences 21, no. 6: 2046. https://doi.org/10.3390/ijms21062046
APA StyleGross, C. C., Pawlitzki, M., Schulte-Mecklenbeck, A., Rolfes, L., Ruck, T., Hundehege, P., Wiendl, H., Herty, M., & Meuth, S. G. (2020). Generation of a Model to Predict Differentiation and Migration of Lymphocyte Subsets under Homeostatic and CNS Autoinflammatory Conditions. International Journal of Molecular Sciences, 21(6), 2046. https://doi.org/10.3390/ijms21062046