MicroRNA (miRNA): A New Dimension in the Pathogenesis of Antiphospholipid Syndrome (APS)



Abstract
:1. Introduction
2. Search Strategy and Inclusion Criteria
3. Origin and Function of miRNAs
4. The Role of miRNA in Immune Response
5. Innate Immune System
6. Adaptive Immune System
6.1. T-Cells
6.2. B-Cells
7. MicroRNA in Autoimmunity
8. Antiphospholipid Syndrome
9. APS: Genetic Predisposition and Family Studies
10. MicroRNA and Antiphospholipid Syndrome
11. Future Direction
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| miRNAs | MicroRNAs |
| mRNAs | Messenger RNAs |
| SLE | Systemic lupus erythematosus |
| RA | Rheumatoid arthritis |
| SS | Systemic sclerosis |
| MS | Multiple sclerosis |
| APS | Antiphospholipid syndrome |
| Pri-miRNA | Primary miRNA |
| dsRNA | Double-stranded RNA |
| DGCR8 | DiGeorge syndrome critical region gene 8 |
| Pre-miRNA | Precursor miRNA |
| TRBP | Trans-activator RNA binding protein |
| RISC | RNA-induced silencing complex |
| Ago | Argonaute |
| 3′-UTR | 3′-untranslated region |
| NK | Natural killer |
| PAMPs | Pathogen-associated molecular patterns |
| DAMPs | Danger-associated molecular patterns |
| TLRs | Toll-like receptors |
| NF-κB | Nuclear factor kappa-B |
| MAPK | Mitogen-activated protein kinase |
| MHC | Major histocompatibility complex |
| TCR | T-cell receptor |
| SOCS | Suppressor of cytokine signaling |
| Regulatory T | Treg |
| IFN-γ | Interferon gamma |
| BAFF | B-cell activating factor |
| aPLs | Antiphospholipid antibodies |
| LA | Lupus anticoagulants |
| aCL | Anticardiolipin |
| β2GPI | β2-glycoprotein I |
| TF | Tissue factor |
| mTOR | Mechanistic target of rapamycin |
| HLA | Human leukocyte antigen |
| TGF | Transforming growth factor |
References
- Lu, T.X.; Rothenberg, M.E. MicroRNA. J. Allergy Clin. Immunol. 2018, 141, 1202–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, H.; Suzuki, H.I. Systems and Synthetic microRNA Biology: From Biogenesis to Disease Pathogenesis. Int. J. Mol. Sci. 2019, 21, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef]
- O’connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010, 10, 111–122. [Google Scholar] [CrossRef]
- Wang, Q.; Lin, W.; Tang, X.; Li, S.; Guo, L.; Lin, Y.; Kwok, H.F. The roles of microRNAs in regulating the expression of PD-1/PD-L1 immune checkpoint. Int. J. Mol. Sci. 2017, 18, 2540. [Google Scholar] [CrossRef] [Green Version]
- Ramassone, A.; Pagotto, S.; Veronese, A.; Visone, R. Epigenetics and microRNAs in cancer. Int. J. Mol. Sci. 2018, 19, 459. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Zhang, W.; Jia, J.; Lu, Q.; Gershwin, M.E. Exosomal microRNA in autoimmunity. Cell Mol. Immunol. 2019, 16, 932–934. [Google Scholar] [CrossRef]
- Venkatesha, S.H.; Dudics, S.; Song, Y.; Mahurkar, A.; Moudgil, K.D. The miRNA expression profile of experimental autoimmune encephalomyelitis reveals novel potential disease biomarkers. Int. J. Mol. Sci. 2018, 19, 3990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-Q.; Papp, G.; Póliska, S.; Szabó, K.; Tarr, T.; Bálint, B.L.; Szodoray, P.; Zeher, M. MicroRNA expression profiles identify disease-specific alterations in systemic lupus erythematosus and primary Sjögren’s syndrome. PLoS ONE 2017, 12, e0174585. [Google Scholar] [CrossRef] [PubMed]
- Evangelatos, G.; Fragoulis, G.E.; Koulouri, V.; Lambrou, G.I. MicroRNAs in rheumatoid arthritis: From pathogenesis to clinical impact. Autoimmun. Rev. 2019, 18, 102391. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Zuo, X.X.; Li, Y.S.; Gao, S.M.; Dai, X.D.; Zhu, H.L.; Luo, H. Integration of microRNA and mRNA expression profiles in the skin of systemic sclerosis patients. Sci. Rep. 2017, 7, 42899. [Google Scholar] [CrossRef] [Green Version]
- Nuzziello, N.; Vilardo, L.; Pelucchi, P.; Consiglio, A.; Liuni, S.; Trojano, M.; Liguori, M. Investigating the role of MicroRNA and transcription factor co-regulatory networks in multiple sclerosis pathogenesis. Int. J. Mol. Sci. 2018, 19, 3652. [Google Scholar] [CrossRef] [Green Version]
- Perez-Sanchez, C.; Arias-de la Rosa, I.; Aguirre, M.A.; Luque-Tevar, M.; Ruiz-Limon, P.; Barbarroja, N.; Jimenez-Gomez, Y.; Abalos-Aguilera, M.C.; Collantes-Estevez, E.; Segui, P.; et al. Circulating microRNAs as biomarkers of disease and typification of the atherothrombotic status in antiphospholipid syndrome. Haematologica 2018, 103, 908–918. [Google Scholar] [CrossRef] [Green Version]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [Green Version]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Jeon, K.; Lee, J.T.; Kim, S.; Kim, V.N. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Han, J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiohama, A.; Sasaki, T.; Noda, S.; Minoshima, S.; Shimizu, N. Molecular cloning and expression analysis of a novel gene DGCR8 located in the DiGeorge syndrome chromosomal region. Biochem. Biophys. Res. Commun. 2003, 304, 184–190. [Google Scholar] [CrossRef]
- Wilson, D.I.; Burn, J.; Scambler, P.; Goodship, J. DiGeorge syndrome: Part of CATCH 22. J. Med. Genet. 1993, 30, 852–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, R.I.; Yan, K.-p.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Pauley, K.M.; Cha, S.; Chan, E.K.L. MicroRNA in autoimmunity and autoimmune diseases. J. Autoimmun. 2009, 32, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, S.; Kobayashi, M.; Yoda, M.; Sakaguchi, Y.; Katsuma, S.; Suzuki, T.; Tomari, Y. Hsc70/Hsp90 Chaperone Machinery Mediates ATP-Dependent RISC Loading of Small RNA Duplexes. Mol. Cell 2010, 39, 292–299. [Google Scholar] [CrossRef]
- Chen, J.-Q.; Papp, G.; Szodoray, P.; Zeher, M. The role of microRNAs in the pathogenesis of autoimmune diseases. Autoimmun. Rev. 2016, 15, 1171–1180. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.-J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [Green Version]
- Wee Liang, M.; Flores-Jasso, C.F.; Salomon William, E.; Zamore Phillip, D. Argonaute Divides Its RNA Guide into Domains with Distinct Functions and RNA-Binding Properties. Cell 2012, 151, 1055–1067. [Google Scholar]
- Salomon, W.E.; Jolly, S.M.; Moore, M.J.; Zamore, P.D.; Serebrov, V. Single-molecule imaging reveals that Argonaute reshapes the binding properties of its nucleic acid guides. Cell 2015, 162, 84–95. [Google Scholar] [CrossRef] [Green Version]
- Bruno, I.; Wilkinson, M.F. P-bodies react to stress and nonsense. Cell 2006, 125, 1036–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.P.; Massachi, I.; Manickavel, S.; Singh, S.; Rao, N.P.; Hasan, S.; Mc Curdy, D.K.; Sharma, S.; Wong, D.; Hahn, B.H. The role of miRNA in inflammation and autoimmunity. Autoimmun. Rev. 2013, 12, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Hukowska-Szematowicz, B.; Tokarz-Deptula, B.; Deptula, W. MicroRNA (miRNA) and the immune system. Cent. Eur. J. Immunol. 2012, 37, 387–390. [Google Scholar] [CrossRef] [Green Version]
- Curtale, G. MiRNAs at the Crossroads between Innate Immunity and Cancer: Focus on Macrophages. Cells 2018, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, R.P.; Leong, J.W.; Fehniger, T.A. MicroRNA regulation of natural killer cells. Front. Immunol. 2013, 4, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, M.T.; Hother, C.; Häger, M.; Pedersen, C.C.; Theilgaard-Mönch, K.; Borregaard, N.; Cowland, J.B. MicroRNA Profiling in Human Neutrophils during Bone Marrow Granulopoiesis and In Vivo Exudation. PLoS ONE 2013, 8, e58454. [Google Scholar] [CrossRef]
- Kumar Kingsley, S.M.; Vishnu Bhat, B. Role of MicroRNAs in the development and function of innate immune cells. Int. Rev. Immunol. 2017, 36, 154–175. [Google Scholar] [CrossRef]
- Xu, S.J.; Hu, H.T.; Li, H.L.; Chang, S. The Role of miRNAs in Immune Cell Development, Immune Cell Activation, and Tumor Immunity: With a Focus on Macrophages and Natural Killer Cells. Cells 2019, 8, 1140. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Jing, Z.; Cheng, G. MicroRNAs: New regulators of Toll-like receptor signalling pathways. Biomed. Res. Int. 2014, 2014, 945169. [Google Scholar] [CrossRef] [Green Version]
- Momen-Heravi, F.; Bala, S. miRNA regulation of innate immunity. J. Leukoc. Biol. 2018, 103, 1205–1217. [Google Scholar] [CrossRef]
- Wu, H.; Neilson, J.R.; Kumar, P.; Manocha, M.; Shankar, P.; Sharp, P.A.; Manjunath, N. miRNA profiling of naive, effector and memory CD8 T cells. PLoS ONE 2007, 2, e1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lind, E.F.; Elford, A.R.; Ohashi, P.S. Micro-RNA 155 is required for optimal CD8+ T cell responses to acute viral and intracellular bacterial challenges. J. Immunol. 2013, 190, 1210–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gracias, D.T.; Stelekati, E.; Hope, J.L.; Boesteanu, A.C.; Doering, T.A.; Norton, J.; Mueller, Y.M.; Fraietta, J.A.; Wherry, E.J.; Turner, M. The microRNA miR-155 controls CD8+ T cell responses by regulating interferon signaling. Nat. Immunol. 2013, 14, 593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, R.M.; Kahn, D.; Gibson, W.S.J.; Round, J.L.; Scholz, R.L.; Chaudhuri, A.A.; Kahn, M.E.; Rao, D.S.; Baltimore, D. MicroRNA-155 Promotes Autoimmune Inflammation by Enhancing Inflammatory T Cell Development. Immunity 2010, 33, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.-F.; Thai, T.-H.; Calado, D.P.; Chaudhry, A.; Kubo, M.; Tanaka, K.; Loeb, G.B.; Lee, H.; Yoshimura, A.; Rajewsky, K.; et al. Foxp3-Dependent MicroRNA155 Confers Competitive Fitness to Regulatory T Cells by Targeting SOCS1 Protein. Immunity 2009, 30, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.-F.; Boldin, M.P.; Chaudhry, A.; Lin, L.-L.; Taganov, K.D.; Hanada, T.; Yoshimura, A.; Baltimore, D.; Rudensky, A.Y. Function of miR-146a in Controlling Treg Cell-Mediated Regulation of Th1 Responses. Cell 2010, 142, 914–929. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Wang, X.; Choi, I.Y.; Wang, Y.-C.; Liu, S.; Pham, A.T.; Moon, H.; Smith, D.J.; Rao, D.S.; Boldin, M.P.; et al. miR-146a modulates autoreactive Th17 cell differentiation and regulates organ-specific autoimmunity. J. Clin. Investig. 2017, 127, 3702–3716. [Google Scholar] [CrossRef] [Green Version]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Xi, Z.; Liu, R.; Yin, W.; Sui, Z.; Ren, B.; Miller, H.; Gong, Q.; Liu, C. The Function of MicroRNAs in B-Cell Development, Lymphoma, and Their Potential in Clinical Practice. Front. Immunol. 2018, 9, 936. [Google Scholar] [CrossRef]
- Lai, M.; Gonzalez-Martin, A.; Cooper, A.B.; Oda, H.; Jin, H.Y.; Shepherd, J.; He, L.; Zhu, J.; Nemazee, D.; Xiao, C. Regulation of B-cell development and tolerance by different members of the miR-17 approximately 92 family microRNAs. Nat. Commun. 2016, 7, 12207. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Calado, D.P.; Galler, G.; Thai, T.H.; Patterson, H.C.; Wang, J.; Rajewsky, N.; Bender, T.P.; Rajewsky, K. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell 2007, 131, 146–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, K.Y.; Owens, K.S.; Rogers, J.H.; Mullenix, J.; Velu, C.S.; Grimes, H.L.; Dahl, R. MIR-23A microRNA cluster inhibits B-cell development. Exp. Hematol. 2010, 38, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Yang, J.; Yuan, R.; Peng, J.; Liu, L.; Guo, X. Effects of miR-181a on the biological function of multiple myeloma. Oncol. Rep. 2019, 42, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, M.; Louka, M.L.; Khairy, E.; Tash, F.; Ali-Labib, R.; El-Habashy, S. MicroRNA-181a and its target Smad 7 as potential biomarkers for tracking child acute lymphoblastic leukemia. Gene 2017, 628, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Qi, J.; Sun, X.; Wang, W.; Wei, G.; Wu, Y.; Gao, Q.; Zheng, J. MicroRNA-181a promotes cell proliferation and inhibits apoptosis in gastric cancer by targeting RASSF1A. Oncol. Rep. 2018, 40, 1959–1970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, H.; Zhao, M.; Lu, Q. Identifying the differentially expressed microRNAs in autoimmunity: A systemic review and meta-analysis. Autoimmunity 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Lu, Q. Genetic and epigenetic influences on the loss of tolerance in autoimmunity. Cell Mol. Immunol. 2018, 15, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Martin, A.; Adams, B.D.; Lai, M.; Shepherd, J.; Salvador-Bernaldez, M.; Salvador, J.M.; Lu, J.; Nemazee, D.; Xiao, C. The microRNA miR-148a functions as a critical regulator of B cell tolerance and autoimmunity. Nat. Immunol. 2016, 17, 433–440. [Google Scholar] [CrossRef]
- Alsaleh, G.; François, A.; Philippe, L.; Gong, Y.-Z.; Bahram, S.; Cetin, S.; Pfeffer, S.; Gottenberg, J.-E.; Wachsmann, D.; Georgel, P.; et al. MiR-30a-3p Negatively Regulates BAFF Synthesis in Systemic Sclerosis and Rheumatoid Arthritis Fibroblasts. PLoS ONE 2014, 9, e111266. [Google Scholar] [CrossRef]
- Gumkowska-Sroka, O.; Jagoda, K.; Owczarek, A.; Helbig, G.; Giemza-Stoklosa, J.; Kotyla, P.J. Cytometric Characterization of Main Immunocompetent Cells in Patients with Systemic Sclerosis: Relationship with Disease Activity and Type of Immunosuppressive Treatment. J. Clin. Med. 2019, 8, 625. [Google Scholar] [CrossRef] [Green Version]
- Stypinska, B.; Wajda, A.; Walczuk, E.; Olesinska, M.; Lewandowska, A.; Walczyk, M.; Paradowska-Gorycka, A. The Serum Cell-Free microRNA Expression Profile in MCTD, SLE, SSc, and RA Patients. J. Clin. Med. 2020, 9, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honarpisheh, M.; Kohler, P.; von Rauchhaupt, E.; Lech, M. The Involvement of MicroRNAs in Modulation of Innate and Adaptive Immunity in Systemic Lupus Erythematosus and Lupus Nephritis. J. Immunol. Res. 2018, 2018, 4126106. [Google Scholar] [PubMed] [Green Version]
- Lai, N.S.; Koo, M.; Yu, C.L.; Lu, M.C. Immunopathogenesis of systemic lupus erythematosus and rheumatoid arthritis: The role of aberrant expression of non-coding RNAs in T cells. Clin. Exp. Immunol. 2017, 187, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, I.K.Y.; Chow, J.X.; Lau, C.S.; Chan, V.S.F. MicroRNA-mediated immune regulation in rheumatic diseases. Cancer Lett. 2018, 431, 201–212. [Google Scholar] [CrossRef]
- Le, X.; Yu, X.; Shen, N. Novel insights of microRNAs in the development of systemic lupus erythematosus. Curr. Opin. Rheumatol. 2017, 29, 450–457. [Google Scholar] [CrossRef]
- Nalewajska, M.; Gurazda, K.; Styczynska-Kowalska, E.; Marchelek-Mysliwiec, M.; Pawlik, A.; Dziedziejko, V. The Role of MicroRNAs in Selected Forms of Glomerulonephritis. Int. J. Mol. Sci. 2019, 20, 5050. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Liang, M.; Hou, X.; Zhang, Y.; Zhang, H.; Guo, Z.; Jinyu, J.; Feng, Z.; Mei, Z. The role of microRNA-16 in the pathogenesis of autoimmune diseases: A comprehensive review. Biomed. Pharmacother. 2019, 112, 108583. [Google Scholar] [CrossRef]
- Vinuesa, C.G.; Rigby, R.J.; Yu, D. Logic and extent of miRNA-mediated control of autoimmune gene expression. Int. Rev. Immunol. 2009, 28, 112–138. [Google Scholar] [CrossRef]
- Linnemann, B. Antiphospholipid syndrome—An update. Vasa 2018, 47, 451–464. [Google Scholar] [CrossRef]
- Giemza-Stoklosa, J.; Islam, M.A.; Kotyla, P.J. Hyperferritinaemia: An Iron Sword of Autoimmunity. Curr. Pharm. Des. 2019, 25, 2909–2918. [Google Scholar] [CrossRef]
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.; PG, D.E.G.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Keeling, D.; Mackie, I.; Moore, G.W.; Greer, I.A.; Greaves, M. Guidelines on the investigation and management of antiphospholipid syndrome. Br. J. Haematol. 2012, 157, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Ugolini-Lopes, M.R.; Criado, P.R.; Parsi, K.; Kucukkaya, R.D.; Amigo, M.-C.; Tektonidou, M.G.; Andrade, D. Treatment of Non-criteria Manifestations in Antiphospholipid Syndrome. In Antiphospholipid Syndrome: Current Research Highlights and Clinical Insights; Erkan, D., Lockshin, M.D., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 247–266. [Google Scholar] [CrossRef]
- Islam, M.A.; Alam, F.; Wong, K.K. Comorbid association of antiphospholipid antibodies and migraine: A systematic review and meta-analysis. Autoimmun. Rev. 2017, 16, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Alam, F.; Kamal, M.A.; Wong, K.K.; Sasongko, T.H.; Gan, S.H. ‘Non-Criteria’ Neurologic Manifestations of Antiphospholipid Syndrome: A Hidden Kingdom to be Discovered. CNS Neurol. Disord. Drug. Targets. 2016, 15, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Alam, F.; Sasongko, T.H.; Gan, S.H. Antiphospholipid antibody-mediated thrombotic mechanisms in antiphospholipid syndrome: Towards pathophysiology-based treatment. Curr. Pharm. Des. 2016, 22, 4451–4469. [Google Scholar] [CrossRef]
- Sammaritano, L.R. Antiphospholipid syndrome. Best. Pract. Res. Clin. Rheumatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Erkan, D. Diagnosis and management of the antiphospholipid syndrome. N. Engl. J. Med. 2018, 378, 2010–2021. [Google Scholar] [CrossRef]
- Sciascia, S.; Sanna, G.; Khamashta, M.A.; Cuadrado, M.J.; Erkan, D.; Andreoli, L.; Bertolaccini, M.L. The estimated frequency of antiphospholipid antibodies in young adults with cerebrovascular events: A systematic review. Ann. Rheum. Dis. 2015, 74, 2028–2033. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.A. Antiphospholipid antibodies and antiphospholipid syndrome in cancer: Uninvited guests in troubled times. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Islam, M.A.; Alam, S.S.; Kundu, S.; Prodhan, A.; Khandker, S.S.; Reshetnyak, T.; Kotyla, P.J.; Hassan, R.; Hossan, T. Prevalence of antiphospholipid antibodies in Behcet’s disease: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0227836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yelnik, C.M.; Urbanski, G.; Drumez, E.; Sobanski, V.; Maillard, H.; Lanteri, A.; Morell-Dubois, S.; Caron, C.; Dubucquoi, S.; Launay, D.; et al. Persistent triple antiphospholipid antibody positivity as a strong risk factor of first thrombosis, in a long-term follow-up study of patients without history of thrombosis or obstetrical morbidity. Lupus 2017, 26, 163–169. [Google Scholar] [CrossRef]
- Islam, M.A.; Khandker, S.S.; Alam, F.; Kamal, M.A.; Gan, S.H. Genetic risk factors in thrombotic primary antiphospholipid syndrome: A systematic review with bioinformatic analyses. Autoimmun. Rev. 2018, 17, 226–243. [Google Scholar] [CrossRef]
- Exner, T.; Barber, S.; Kronenberg, H.; Rickard, K.A. Familial Association of the Lupus Anticoagulant. Br. J. Haematol. 1980, 45, 89–96. [Google Scholar] [CrossRef]
- Matthey, F.; Walshe, K.; Mackie, I.; Machin, S. Familial occurrence of the antiphospholipid syndrome. J. Clin. Pathol. 1989, 42, 495–497. [Google Scholar] [CrossRef] [Green Version]
- Jolidon, R.-M.; Knecht, H.; Humair, L.; de Torrente, A. Different clinical presentations of a lupus anticoagulant in the same family. Klin. Wochenschr. 1991, 69, 340–344. [Google Scholar] [CrossRef]
- Islam, M.A.; Wong, K.K.; Sasongko, T.H.; Gan, S.H.; Wong, J.S. Familial primary antiphospholipid syndrome: A report of co-occurrence in three Malaysian family members. Eur. J. Rheumatol. 2016, 3, 139–141. [Google Scholar] [CrossRef]
- Arnett, F.; Olsen, M.; Anderson, K.; Reveille, J. Molecular analysis of major histocompatibility complex alleles associated with the lupus anticoagulant. J. Clin. Investig. 1991, 87, 1490–1495. [Google Scholar] [CrossRef]
- Asherson, R.; Doherty, D.; Vergani, D.; Khamashta, M.; Hughes, G. Major histocompatibility complex associations with primary antiphospholipid syndrome. Arthritis. Rheum. 1992, 35, 124–125. [Google Scholar] [CrossRef]
- Caliz, R.; Atsumi, T.; Kondeatis, E.; Amengual, O.; Khamashta, M.; Vaughan, R.; Lanchbury, J.; Hughes, G. HLA class II gene polymorphisms in antiphospholipid syndrome: Haplotype analysis in 83 Caucasoid patients. Rheumatology 2001, 40, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Granados, J.; Vargas-Alarcon, G.; Drenkard, C.; Andrade, F.; Melin-Aldana, H.; Alcocer-Varela, J.; Alarcón-Segovia, D. Relationship of anticardiolipin antibodies and antiphospholipid syndrome to HLA-DR7 in Mexican patients with systemic lupus erythematosus (SLE). Lupus 1997, 6, 57–62. [Google Scholar] [CrossRef]
- Freitas, M.V.; Da Silva, L.; Deghaide, N.H.; Donadi, E.A.; Louzada-Júnior, P. Is HLA class II susceptibility to primary antiphospholipid syndrome different from susceptibility to secondary antiphospholipid syndrome? Lupus 2004, 13, 125–131. [Google Scholar] [CrossRef]
- Al Attia, H.; Santosh, A.; Al Farhan, M. Observations on class II antigens and genetic susceptibility to primary antiphospholipid (Hughes) syndrome in Arab patients. Clin. Exp. Rheumatol. 2008, 26, 506. [Google Scholar]
- Sebastiani, G.D.; Iuliano, A.; Cantarini, L.; Galeazzi, M. Genetic aspects of the antiphospholipid syndrome: An update. Autoimmun. Rev. 2016, 15, 433–439. [Google Scholar] [CrossRef]
- Berman, H.; Ugarte-Gil, M.; Espinosa, G.; Tàssies, D.; Monteagudo, J.; Reverter, J.; Cervera, R. Can inherited thrombophilia modulate the clinical phenotype of patients with antiphospholipid syndrome. Clin. Exp. Rheumatol. 2013, 31, 926–932. [Google Scholar]
- Teruel, R.; Perez-Sanchez, C.; Corral, J.; Herranz, M.T.; Perez-Andreu, V.; Saiz, E.; Garcia-Barbera, N.; Martinez-Martinez, I.; Roldan, V.; Vicente, V.; et al. Identification of miRNAs as potential modulators of tissue factor expression in patients with systemic lupus erythematosus and antiphospholipid syndrome. J. Thromb. Haemost. 2011, 9, 1985–1992. [Google Scholar] [CrossRef]
- van den Hoogen, L.L.; Rossato, M.; Lopes, A.P.; Pandit, A.; Bekker, C.P.; Fritsch-Stork, R.D.; van Roon, J.A.; Radstake, T.R. microRNA downregulation in plasmacytoid dendritic cells in interferon-positive systemic lupus erythematosus and antiphospholipid syndrome. Rheumatology 2018, 57, 1669–1674. [Google Scholar] [CrossRef] [Green Version]
- Perez-Sanchez, C.; Aguirre, M.A.; Ruiz-Limon, P.; Barbarroja, N.; Jimenez-Gomez, Y.; de la Rosa, I.A.; Rodriguez-Ariza, A.; Collantes-Estevez, E.; Segui, P.; Velasco, F.; et al. Atherothrombosis-associated microRNAs in Antiphospholipid syndrome and Systemic Lupus Erythematosus patients. Sci. Rep. 2016, 6, 31375. [Google Scholar] [CrossRef]
- Zhou, H.; Wolberg, A.S.; Roubey, R.A. Characterization of monocyte tissue factor activity induced by IgG antiphospholipid antibodies and inhibition by dilazep. Blood 2004, 104, 2353–2358. [Google Scholar] [CrossRef]
- Reverter, J.-C.; Tàssies, D.; Font, J.; Monteagudo, J.; Escolar, G.S.; Ingelmo, M.; Ordinas, A. Hypercoagulable state in patients with antiphospholipid syndrome is related to high induced tissue factor expression on monocytes and to low free protein S. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1319–1326. [Google Scholar] [CrossRef]
- Kornberg, A.; Blank, M.; Kaufman, S.; Shoenfeld, Y. Induction of tissue factor-like activity in monocytes by anti-cardiolipin antibodies. J. Immunol. 1994, 153, 1328–1332. [Google Scholar]
- Dai, Y.; Huang, Y.-S.; Tang, M.; Lv, T.-Y.; Hu, C.-X.; Tan, Y.-H.; Xu, Z.-M.; Yin, Y.-B. Microarray analysis of microRNA expression in peripheral blood cells of systemic lupus erythematosus patients. Lupus 2007, 16, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Perez-Andreu, V.; Teruel, R.; Corral, J.; Roldan, V.; Garcia-Barbera, N.; Salloum-Asfar, S.; Gomez-Lechon, M.J.; Bourgeois, S.; Deloukas, P.; Wadelius, M.; et al. miR-133a regulates vitamin K 2,3-epoxide reductase complex subunit 1 (VKORC1), a key protein in the vitamin K cycle. Mol. Med. 2013, 18, 1466–1472. [Google Scholar] [CrossRef] [PubMed]
- Rieder, M.J.; Reiner, A.P.; Gage, B.F.; Nickerson, D.A.; Eby, C.S.; McLeod, H.L.; Blough, D.K.; Thummel, K.E.; Veenstra, D.L.; Rettie, A.E. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N. Engl. J. Med. 2005, 352, 2285–2293. [Google Scholar] [CrossRef] [Green Version]
- Stafford, D. The vitamin K cycle. J. Thromb. Haemost. 2005, 3, 1873–1878. [Google Scholar] [CrossRef]
- Yu, J.; Cao, X.; Zheng, Y.; Yan, L.; Wang, J. Abnormal expression of miR133a in patients with acute myocardial infarction following radical surgery for gastric cancer and the underlying mechanism. Mol. Med. Rep. 2018, 18, 5023–5029. [Google Scholar]
- Ray, M.; Gabunia, K.; Vrakas, C.N.; Herman, A.B.; Kako, F.; Kelemen, S.E.; Grisanti, L.A.; Autieri, M.V. Genetic Deletion of IL-19 (Interleukin-19) Exacerbates Atherogenesis in Il19(-/-)xLdlr(-/-) Double Knockout Mice by Dysregulation of mRNA Stability Protein HuR (Human Antigen R). Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1297–1308. [Google Scholar] [CrossRef] [Green Version]
- Shemer, A.; Willis, R.; Gonzalez, E.B.; Romay-Penabad, Z.; Shovman, O.; Shoenfeld, Y.; Blank, M.; Amital, H. Oral administration of Domain-I of beta-2glycoprotein-I induces immunological tolerance in experimental murine antiphospholipid syndrome. J. Autoimmun. 2019, 99, 98–103. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Q.; Song, Y.; Lai, L.; Wang, J.; Yu, H.; Cao, X.; Wang, Q. MicroRNA-98 negatively regulates IL-10 production and endotoxin tolerance in macrophages after LPS stimulation. FEBS Lett. 2011, 585, 1963–1968. [Google Scholar] [CrossRef] [Green Version]
- Zandman-Goddard, G.; Pierangeli, S.S.; Gertel, S.; Blank, M. Tolerogenic dendritic cells specific for beta2-glycoprotein-I Domain-I, attenuate experimental antiphospholipid syndrome. J. Autoimmun. 2014, 54, 72–80. [Google Scholar] [CrossRef]
- Nakamachi, Y.; Kawano, S.; Takenokuchi, M.; Nishimura, K.; Sakai, Y.; Chin, T.; Saura, R.; Kurosaka, M.; Kumagai, S. MicroRNA-124a is a key regulator of proliferation and monocyte chemoattractant protein 1 secretion in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis. Rheum. 2009, 60, 1294–1304. [Google Scholar] [CrossRef]
- Liu, H.; Xiong, W.; Liu, F.; Lin, F.; He, J.; Liu, C.; Lin, Y.; Dong, S. MicroRNA-133b regulates the growth and migration of vascular smooth muscle cells by targeting matrix metallopeptidase 9. Pathol. Res. Pract. 2019, 215, 1083–1088. [Google Scholar] [CrossRef]
- Zheng, C.G.; Chen, B.Y.; Sun, R.H.; Mou, X.Z.; Han, F.; Li, Q.; Huang, H.J.; Liu, J.Q.; Tu, Y.X. miR-133b Downregulation Reduces Vulnerable Plaque Formation in Mice with AS through Inhibiting Macrophage Immune Responses. Mol. Ther. Nucleic. Acids. 2019, 16, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Yi, Y.; Jia, S.; Peng, X.; Yang, H.; Guo, R. The miR-145 rs353291 C allele increases susceptibility to atherosclerosis. Front. Biosci. 2020, 25, 577–592. [Google Scholar]
- Zhang, Y.N.; Xie, B.D.; Sun, L.; Chen, W.; Jiang, S.L.; Liu, W.; Bian, F.; Tian, H.; Li, R.K. Phenotypic switching of vascular smooth muscle cells in the ‘normal region’ of aorta from atherosclerosis patients is regulated by miR-145. J. Cell Mol. Med. 2016, 20, 1049–1061. [Google Scholar] [CrossRef] [Green Version]
- Su, L.-C.; Xu, W.-D.; Huang, A.-F. IRAK family in inflammatory autoimmune diseases. Autoimmun. Rev. 2020, 102461. [Google Scholar] [CrossRef]
- Cheng, H.S.; Njock, M.S.; Khyzha, N.; Dang, L.T.; Fish, J.E. Noncoding RNAs regulate NF-kappaB signaling to modulate blood vessel inflammation. Front. Genet. 2014, 5, 422. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Sheng, L.; Wang, H.; Xie, H.; Mu, Y.; Wang, T.; Yan, J. Anti-β2GPI/β2GPI stimulates activation of THP-1 cells through TLR4/MD-2/MyD88 and NF-κB signaling pathways. Thromb. Res. 2013, 132, 742–749. [Google Scholar] [CrossRef]
- Xie, H.; Kong, X.; Zhou, H.; Xie, Y.; Sheng, L.; Wang, T.; Xia, L.; Yan, J. TLR4 is involved in the pathogenic effects observed in a murine model of antiphospholipid syndrome. Clin. Immunol. 2015, 160, 198–210. [Google Scholar] [CrossRef]
- Xia, L.; Xie, H.; Yu, Y.; Zhou, H.; Wang, T.; Yan, J. The Effects of NF-kappaB and c-Jun/AP-1 on the Expression of Prothrombotic and Proinflammatory Molecules Induced by Anti-beta2GPI in Mouse. PLoS ONE 2016, 11, e0147958. [Google Scholar] [CrossRef]
- Ranjbar, R.; Hesari, A.; Ghasemi, F.; Sahebkar, A. Expression of microRNAs and IRAK1 pathway genes are altered in gastric cancer patients with Helicobacter pylori infection. J. Cell Biochem. 2018, 119, 7570–7576. [Google Scholar] [CrossRef]
- Venugopal, P.; Koshy, T.; Lavu, V.; Ranga Rao, S.; Ramasamy, S.; Hariharan, S.; Venkatesan, V. Differential expression of microRNAs let-7a, miR-125b, miR-100, and miR-21 and interaction with NF-kB pathway genes in periodontitis pathogenesis. J. Cell Physiol. 2018, 233, 5877–5884. [Google Scholar] [CrossRef]
- Donners, M.M.; Beckers, L.; Lievens, D.; Munnix, I.; Heemskerk, J.; Janssen, B.J.; Wijnands, E.; Cleutjens, J.; Zernecke, A.; Weber, C.; et al. The CD40-TRAF6 axis is the key regulator of the CD40/CD40L system in neointima formation and arterial remodeling. Blood 2008, 111, 4596–4604. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Xiong, J.; Takeuchi, M.; Kurama, T.; Goeddel, D.V. TRAF6 is a signal transducer for interleukin-1. Nature 1996, 383, 443–446. [Google Scholar] [CrossRef]
- Xia, L.; Zhou, H.; Hu, L.; Xie, H.; Wang, T.; Xu, Y.; Liu, J.; Zhang, X.; Yan, J. Both NF-κB and c-Jun/AP-1 involved in anti-β2GPI/β2GPI-induced tissue factor expression in monocytes. Thromb. Haemost. 2013, 109, 643–651. [Google Scholar] [CrossRef]
- Li, Y.; Yan, L.; Zhang, W.; Hu, N.; Chen, W.; Wang, H.; Kang, M.; Ou, H. Suppression of endothelial nitric oxide synthase expression and endothelial cell proliferation by an intronic 27-ntmiRNA and it’s a novel link to AP-1. Am. J. Transl. Res. 2015, 7, 285–297. [Google Scholar]
- Bao, C.X.; Zhang, D.X.; Wang, N.N.; Zhu, X.K.; Zhao, Q.; Sun, X.L. MicroRNA-335-5p suppresses lower extremity deep venous thrombosis by targeted inhibition of PAI-1 via the TLR4 signalingpathway. J. Cell Biochem. 2018, 119, 4692–4710. [Google Scholar] [CrossRef]
- Li, N.X.; Sun, J.W.; Yu, L.M. Evaluation of the circulating MicroRNA-495 and Stat3 as prognostic and predictive biomarkers for lower extremity deep venous thrombosis. J. Cell Biochem. 2018, 119, 5262–5273. [Google Scholar] [CrossRef]
- Chen, L.J.; Yang, L.; Cheng, X.; Xue, Y.K.; Chen, L.B. Overexpression of miR-24 Is Involved in the Formation of Hypocoagulation State after Severe Trauma by Inhibiting the Synthesis of Coagulation Factor X. Dis. Markers 2017, 2017, 3649693. [Google Scholar] [CrossRef]
- Gao, J.; Ma, X.; Zhang, Y.; Guo, M.; Shi, D. The role of microRNAs in prethrombotic status associated with coronary artery disease. Thromb. Haemost. 2017, 117, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Tan, M.; Xiang, Q.; Zhou, Z.; Yan, H. Thrombin-activated platelet-derived exosomes regulate endothelial cell expression of ICAM-1 via microRNA-223 during the thrombosis-inflammation response. Thromb. Res. 2017, 154, 96–105. [Google Scholar] [CrossRef]
- Sahu, A.; Jha, P.K.; Prabhakar, A.; Singh, H.D.; Gupta, N.; Chatterjee, T.; Tyagi, T.; Sharma, S.; Kumari, B.; Singh, S.; et al. MicroRNA-145 Impedes Thrombus Formation via Targeting Tissue Factor in Venous Thrombosis. EBioMedicine 2017, 26, 175–186. [Google Scholar] [CrossRef] [Green Version]


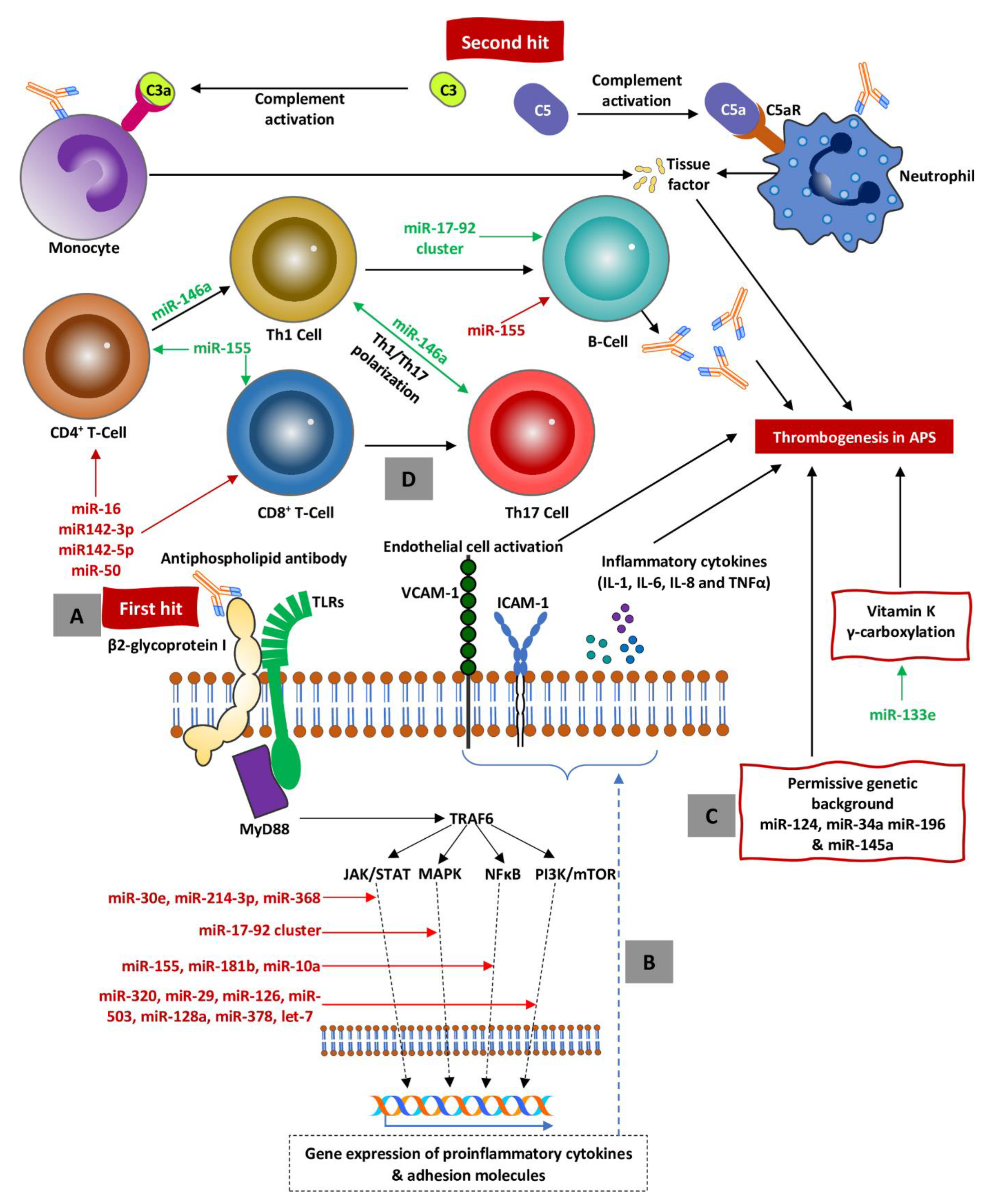{kind=link}
| miRNA | Type of Cells | Target (or Population Studied) | Activity | References |
|---|---|---|---|---|
| miR-19b | White blood cells | Tissue factor | Downregulated | [96] |
| miR-20a | ||||
| miR-296-5p | Plasma and supernatants | Tissue factor, Plasminogen activator inhibitor, Monocyte chemoattractant protein, Vascular endothelial growth factor | Upregulated | [16] |
| miR-133b | ||||
| miR-124-3p | ||||
| miR-206 | ||||
| miR-34a-5p | ||||
| miR-423-5p | ||||
| miR-122-5p | ||||
| miR-193a-5p | ||||
| miR-210-3p | ||||
| miR-192-5p | ||||
| miR-25-3p | ||||
| miR-204-5p | ||||
| miR-31-5p | ||||
| miR-205-5p | ||||
| miR-150-5p | ||||
| miR-196a-5p | ||||
| miR-885-5p | ||||
| miR-155-5p | ||||
| miR-373 | ||||
| miR-20a-5p | Downregulated | |||
| miR-30d-5p | ||||
| miR-24-3p | ||||
| miR-17-5p | ||||
| miR-30a-5p | ||||
| miR-19b-3p | ||||
| miR-191-5p | ||||
| miR-128-p | ||||
| miR-106b-5p | ||||
| miR-22-3p | ||||
| miR-26a-5p | ||||
| miR-26b-5p | ||||
| miR-376c-3p | ||||
| miR-222-3p | ||||
| miR-103a-3p | ||||
| miR-15a-5p | ||||
| miR-211-5p | ||||
| miR-145-5p | ||||
| miR-374a-5p | ||||
| miR-143-3p | ||||
| miR-125b | Dendritic cells | PAPS vs. HC | Downregulated | [97] |
| miR-127a | PAPS vs. HC | |||
| miR-150a | SLE+APS vs. HC | |||
| miR-181 a | PAPS vs. HC | |||
| miR-221a | PAPS vs. HC | |||
| miR-335 | PAPS vs. HC | |||
| miR-362 | SLE+APS vs. HC | |||
| miR-532 | SLE+APS vs. HC | |||
| miR-29a | SLE+APS vs. HC PAPS vs. HC | |||
| miR-196b | SLE+APS vs. HC | |||
| let-7g | PAPs vs. HC | |||
| miR-744 | PAPs vs. HC | |||
| miR-193b | SLE+APS vs. HC | |||
| let-7e | PAPs vs. HC | |||
| miR-30a-5p | PAPs vs. HC | |||
| miR-30d | SLE+APS vs. HC | |||
| miR-30e-3p | SLE+APS vs. HC PAPs vs. HC | |||
| mir590-3p | PAPS vs. HC | |||
| miR-126 | PAPS vs. HC | |||
| miR-1275 | SLE+APS vs. HC | |||
| miR-4443 | Neutrophils | SLE and APS vs. HC | Upregulated | [98] |
| miR-146b-5p | ||||
| miR-302d-3p | ||||
| miR-7-5p | ||||
| miR-193a-5p | ||||
| miR-320e | Downregulated | |||
| miR-346 | ||||
| miR-155-5p | ||||
| miR-22-3p | ||||
| miR-486-3p | ||||
| miR-15a-5p | ||||
| miR-144-3p | ||||
| miR-186-5p | ||||
| Let-7g-5p | ||||
| miR-151a-3p | ||||
| miR-32-5p | ||||
| miR-27b-3p | ||||
| miR-548aa | ||||
| miR-194-5p | ||||
| miR-4431 | ||||
| miR-21-5p | ||||
| miR-324-5p | ||||
| miR-374a-5p | ||||
| miR-132-3p | ||||
| miR-126-3p | ||||
| miR-450-5p | ||||
| miR-140-5p | ||||
| miR-494 | ||||
| miR-301a-3p | ||||
| miR-142-3p | ||||
| miR-92a-3p | ||||
| miR-30e-5p | ||||
| miR-590-5p | ||||
| miR-339-3p | ||||
| miR-630 | ||||
| miR-71-5p | ||||
| miR-106b-5p | ||||
| miR-1537 | ||||
| miR-197-3p | ||||
| miR-503 | ||||
| miR-582-3p | ||||
| miR-340-5p | ||||
| miR-27a-3p | ||||
| miR-26b-5p | ||||
| miR-338-3p | ||||
| miR-30b-5p | ||||
| miR-1260b | ||||
| miR-302b-3p | ||||
| miR-484 | ||||
| miR-532-5p | ||||
| miR-4454 | ||||
| miR-26a-5p | ||||
| miR-17-5p | ||||
| miR-299-3p | ||||
| miR-29b-3p | ||||
| miR-125-5p | ||||
| miR-875-5p | ||||
| miR-142-5p | ||||
| miR-363-3p |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotyla, P.J.; Islam, M.A. MicroRNA (miRNA): A New Dimension in the Pathogenesis of Antiphospholipid Syndrome (APS). Int. J. Mol. Sci. 2020, 21, 2076. https://doi.org/10.3390/ijms21062076
Kotyla PJ, Islam MA. MicroRNA (miRNA): A New Dimension in the Pathogenesis of Antiphospholipid Syndrome (APS). International Journal of Molecular Sciences. 2020; 21(6):2076. https://doi.org/10.3390/ijms21062076
Chicago/Turabian StyleKotyla, Przemysław J., and Md Asiful Islam. 2020. "MicroRNA (miRNA): A New Dimension in the Pathogenesis of Antiphospholipid Syndrome (APS)" International Journal of Molecular Sciences 21, no. 6: 2076. https://doi.org/10.3390/ijms21062076
APA StyleKotyla, P. J., & Islam, M. A. (2020). MicroRNA (miRNA): A New Dimension in the Pathogenesis of Antiphospholipid Syndrome (APS). International Journal of Molecular Sciences, 21(6), 2076. https://doi.org/10.3390/ijms21062076