Dissecting the Genetic Architecture of Aphanomyces Root Rot Resistance in Lentil by QTL Mapping and Genome-Wide Association Study



 and
and
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Data Analysis
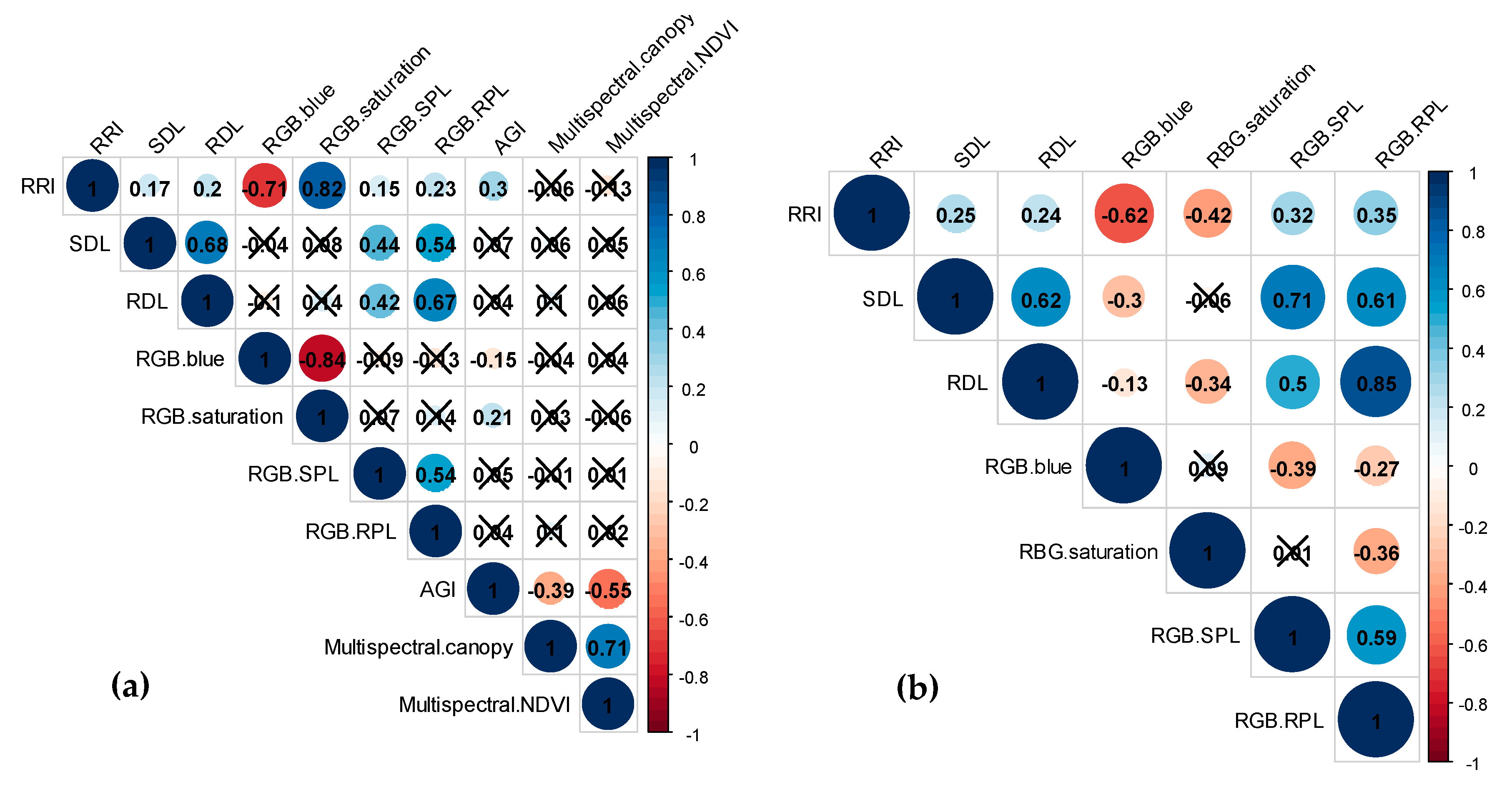
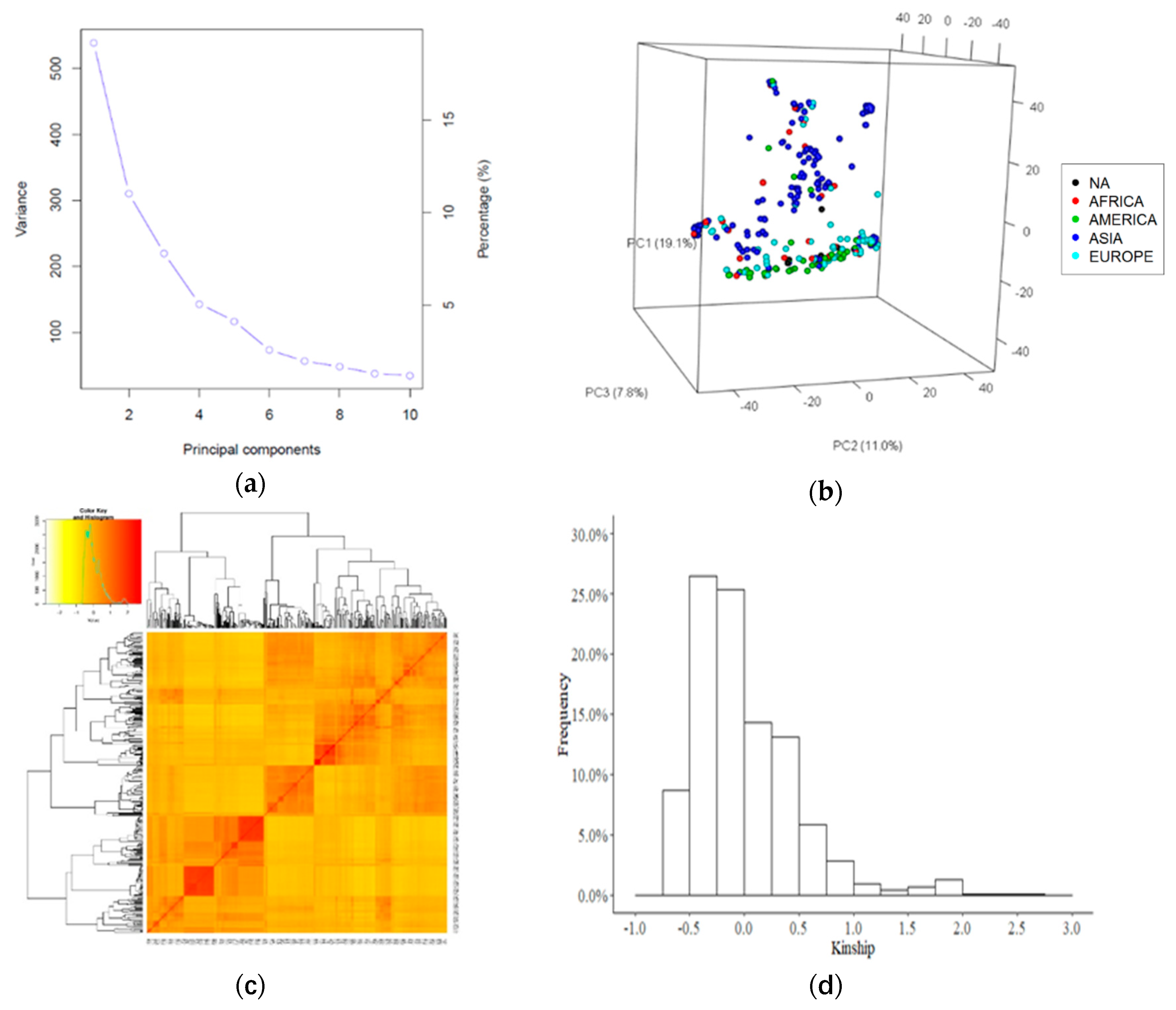
2.2. Genotypic Data Analysis
2.3. QTL Mapping
2.4. Genome-Wide Association Study
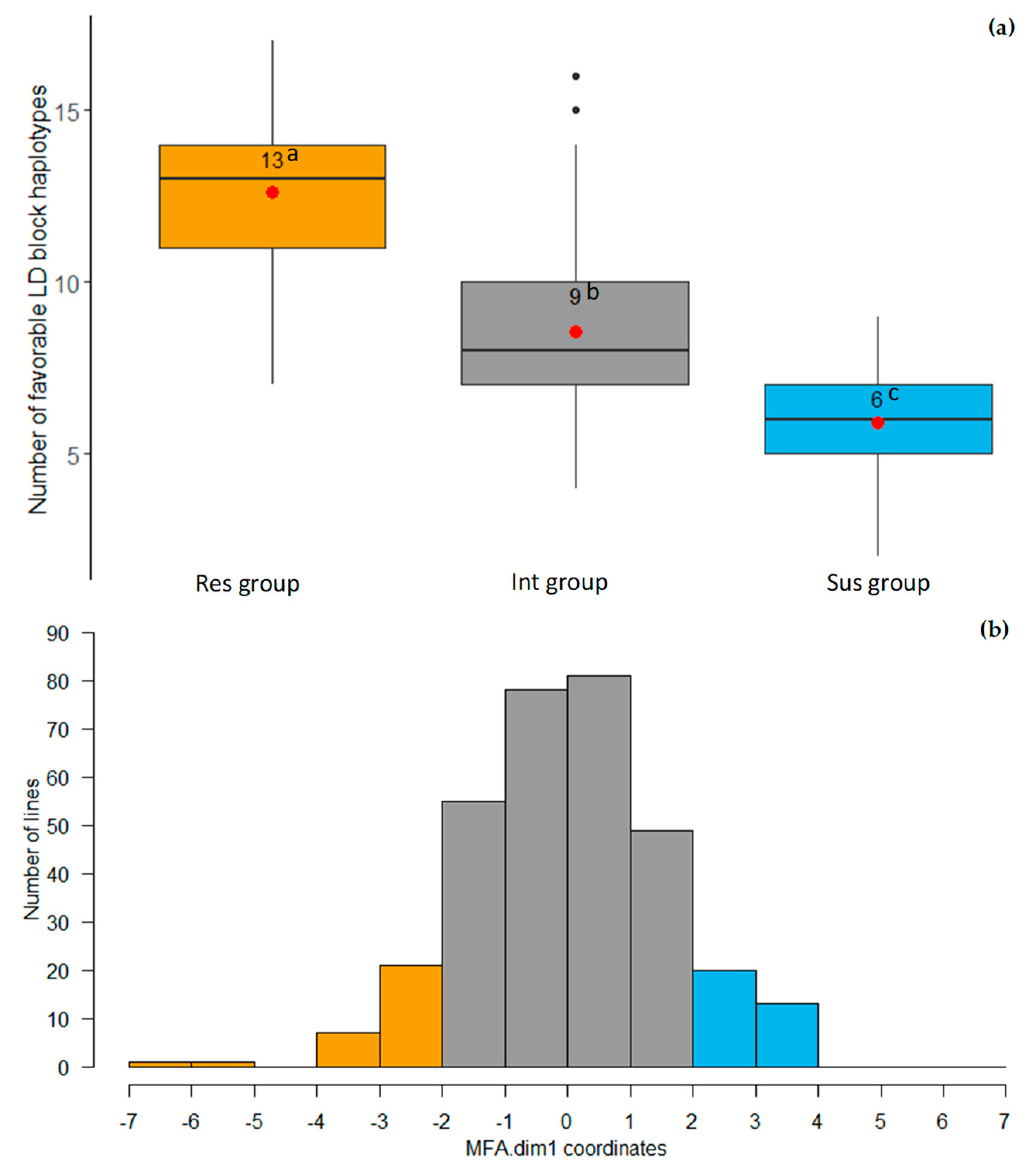
2.5. LD Block Haplotypes
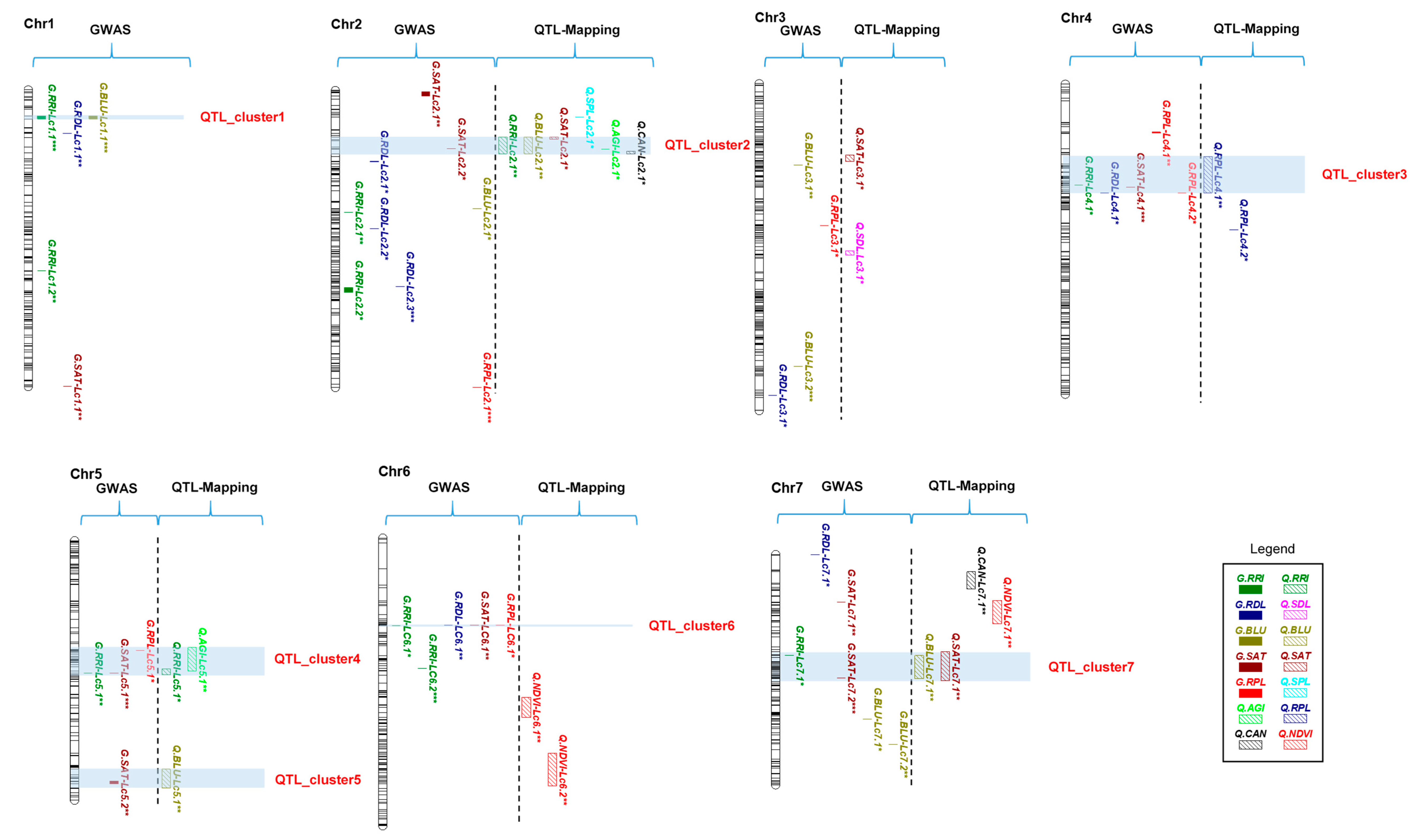
2.6. QTL Clusters
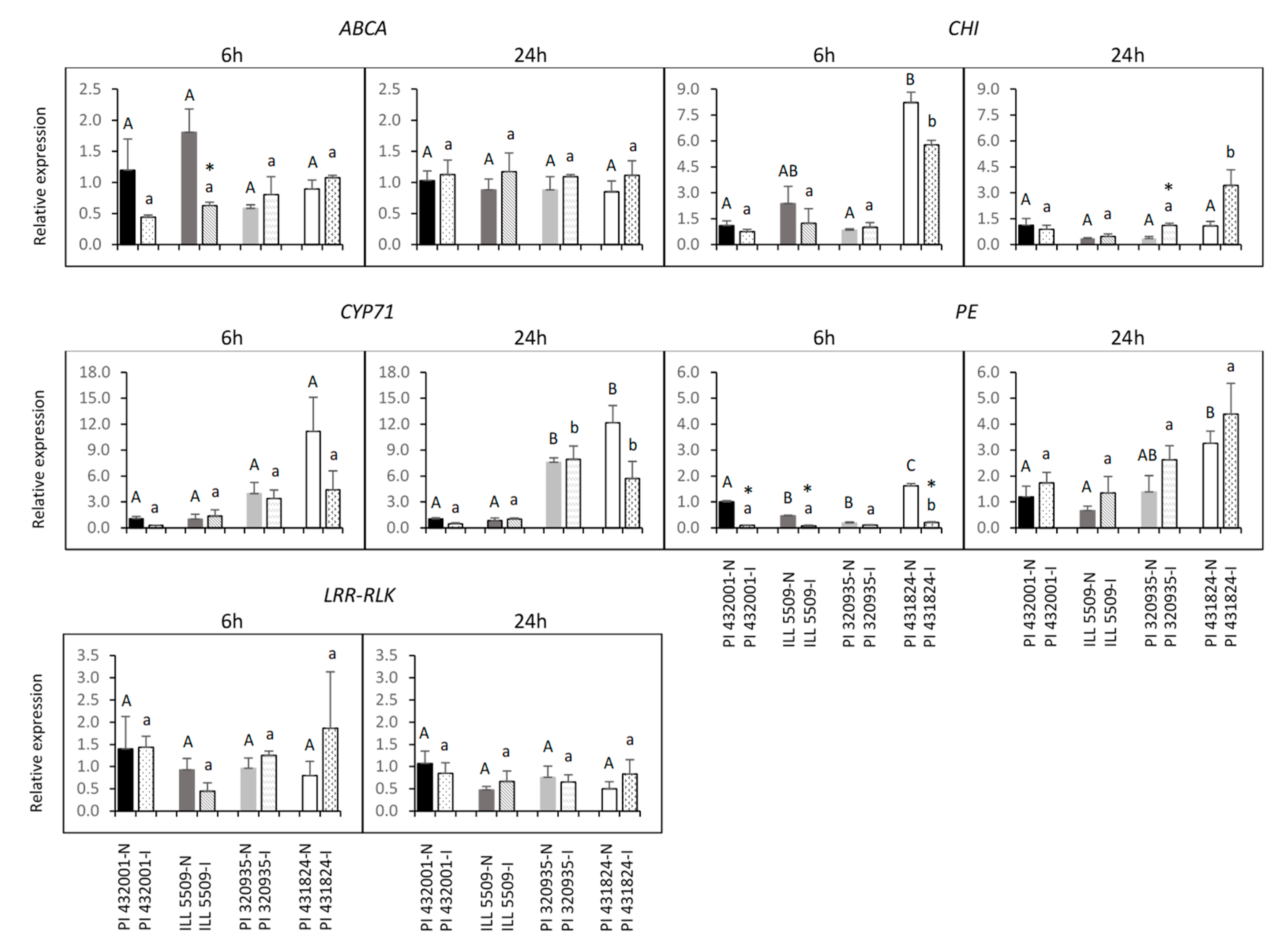
2.7. Prediction of Candidate Genes and Expression Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Inoculation Precedure
4.3. Traditional and Image-Based Phenotyping under Controlled Condition
4.4. Traditional and Image-Based Phenotyping under Field Condition
4.5. Genotyping
4.6. Statistical Analysis of Phenotypic Data
4.7. Linkage Map Construction and QTL Mapping
4.8. LD, Population Structure, and GWAS
4.9. Haplotype Analysis
4.10. Prediction of Candidate Genes and Expression Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ARR | Aphanomyces root rot |
| LD | Linkage disequilibrium |
| GBS | Genotyping by sequencing |
| SNP | Single nucleotide polymorphism |
| QTL | Quantitative trait loci |
| LSP | Lentil single plant-derived |
| ICARDA | International Center for Agricultural Research in the Dry Areas |
| RIL | Recombinant inbred line |
| RGB | Red-Green-Blue |
| RRI | Root rot index |
| AGI | Above ground index |
| SDL | Shoot dry weight loss per plant |
| RDL | Root dry weight loss per plant |
| RGB.SPL | Number of pixels loss per plant in shoot |
| RGB.RPL | Number of pixels loss per plant in root |
| RGB.blue | Average intensity of blue channel |
| RGB.saturation | Standard deviation of saturation channel |
| Multispectral.NDVI | Standard deviation of normalized difference vegetation index |
| Multispectral.canopy | Canopy area |
| MAF | Minor allele frequency |
| ANOVA | Analysis of variance |
| PCA | Principle component analysis |
| PCs | Principle components |
| GAPIT | Genome Association and Prediction Integrated Tool |
| MTAs | Marker-trait associations |
| MFA | Multiple factor analysis |
| qRT-PCR | Quantitative reverse transcriptase-polymerase chain reaction |
| hpi | Hours post inoculation |
| dpi | Days post inoculation |
| LRR-RLK | Leucine Rich Repeat Receptor-like Kinase |
| CYP71 | Cytochrome P450 family 71 protein |
| ABCA | ABC transporter A family protein |
| PE | Pectin esterase |
| CHI | Chalcone-flavanone isomerase family protein |
| SAM | Sequence Alignment Map |
| BAM | Binary Alignment Map |
References
- FAO, FAOSTAT. Available online: http://www.fao.org/faostat/en/#data (accessed on 4 January 2018).
- Ogutcen, E.; Ramsay, L.; von Wettberg, E.B.; Bett, K.E. Capturing variation in Lens (Fabaceae): Development and utility of an exome capture array for lentil. Appl. Plant Sci. 2018, 6, 1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaulin, E.; Jacquet, C.; Bottin, A.; Dumas, B. Root rot disease of legumes caused by Aphanomyces euteiches. Mol. Plant Pathol. 2007, 8, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Malvick, D.K.; Grau, C.R.; Percich, J.A. Characterization of Aphanomyces euteiches strains based on pathogenicity tests and random amplified polymorphic DNA analyses. Mycol. Res. 1998, 102, 465–475. [Google Scholar] [CrossRef]
- Tofte, J.; Smith, R.; Grau, C. Reaction of red clover to Aphanomyces euteiches. Plant Dis 1992, 76, 39–42. [Google Scholar] [CrossRef]
- Wu, L.; Chang, K.F.; Conner, R.L.; Strelkov, S.; Fredua-Agyeman, R.; Hwang, S.F.; Feindel, D. Aphanomyces euteiches: A threat to Canadian field pea production. Engineering 2018, 4, 542–551. [Google Scholar] [CrossRef]
- Moussart, A.; Even, M.; Tivoli, B. Reaction of genotypes from several species of grain and forage legumes to infection with a French pea isolate of the oomycete Aphanomyces euteiches. Eur. J. Plant Pathol. 2008, 122, 321–333. [Google Scholar] [CrossRef]
- Malvick, D.; Percich, J. Identification of Pisum sativum germ plasm with resistance to root rot caused by multiple strains of Aphanomyces euteiches. Plant Dis. 1999, 83, 51–54. [Google Scholar] [CrossRef] [Green Version]
- Levenfors, J.P.; Wikström, M.; Persson, L.; Gerhardson, B. Pathogenicity of Aphanomyces spp. from different leguminous crops in Sweden. Eur. J. Plant Pathol. 2003, 109, 535–543. [Google Scholar] [CrossRef]
- Kraft, J.; Tuck, J.; Kaiser, W. Assessment of lentil PI lines as source of relative resistance to common root rot, 1985. Biol. Cult. Tests Cont. Plant Dis. 1987, 2, 12. [Google Scholar]
- Vandemark, G.; Porter, L. First report of lentil root rot caused by Aphanomyces euteiches in Idaho. Plant Dis. 2010, 94, 480. [Google Scholar] [CrossRef]
- Banniza, S.; Bhadauria, V.; Peluola, C.; Armstrong-Cho, C.; Morrall, R. First report of Aphanomyces euteiches in Saskatchewan. Can. Plant Dis. Surv. 2013, 93, 163–164. [Google Scholar]
- Gossen, B.D.; Conner, R.L.; Chang, K.F.; Pasche, J.S.; McLaren, D.L.; Henriquez, M.A.; Chatterton, S.; Hwang, S.F. Identifying and managing root rot of pulses on the northern great plains. Plant Dis. 2016, 100, 1965–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterton, S.; Harding, M.W.; Bowness, R.; Mclaren, D.L.; Banniza, S.; Gossen, B.D. Importance and causal agents of root rot on field pea and lentil on the Canadian prairies, 2014–2017. Can. J. Plant Pathol. 2019, 41, 98–114. [Google Scholar] [CrossRef]
- Wu, L. Occurrence and management of root rot of field pea caused by Aphanomyces euteiches. Master Thesis, University of Alberta, Edmonton, AB, Canada, 2018. [Google Scholar]
- Papavizas, G.C.; Ayers, W.A. Aphanomyces species and their root diseases in pea and sugarbeet: A review. US Dep. Agric. Agric. Res. Serv. Tech. Bull. 1974, 1485, 158. [Google Scholar]
- McGee, R.J.; Coyne, C.J.; Pilet-Nayel, M.L.; Moussart, A.; Tivoli, B.; Baranger, A.; Hamon, C.; Vandemark, G.; McPhee, K. Registration of pea germplasm lines partially resistant to Aphanomyces root rot for breeding fresh or freezer pea and dry pea types. J. Plant Regist. 2012, 6, 203–207. [Google Scholar] [CrossRef]
- Wicker, E.; Rouxel, F. Specific behaviour of french Aphanomyces euteiches drechs. Populations for virulence and aggressiveness on pea, related to isolates from Europe, America and New Zealand. Eur. J. Plant Pathol. 2001, 107, 919–929. [Google Scholar] [CrossRef]
- Desgroux, A.; L’anthoëne, V.; Roux-Duparque, M.; Rivière, J.P.; Aubert, G.; Tayeh, N.; Moussart, A.; Mangin, P.; Vetel, P.; Piriou, C. McGee, R.J.; Coyne, C.J.; Burstin, J.; Baranger, A.; Manzanares-Dauleux, M.; Bourion, V.; Pilet-Nayel, M. Genome-wide association mapping of partial resistance to Aphanomyces euteiches in pea. BMC Genomics 2016, 17, 124. [Google Scholar] [CrossRef]
- Bett, K.E.; Cook, D.R. KnowPulse. Available online: https://knowpulse.usask.ca/ (accessed on 15 August 2018).
- Chen, C.; Mitchell, S.E.; Elshire, R.J.; Buckler, E.S.; El-Kassaby, Y.A. Mining conifers’ mega-genome using rapid and efficient multiplexed high-throughput genotyping-by-sequencing (GBS) SNP discovery platform. Tree Genet. Genomes 2013, 9, 1537–1544. [Google Scholar] [CrossRef]
- Ju, M.; Zhou, Z.; Mu, C.; Zhang, X.; Gao, J.; Liang, Y.; Chen, J.; Wu, Y.; Li, X.; Wang, S.; et al. Dissecting the genetic architecture of Fusarium verticillioides seed rot resistance in maize by combining QTL mapping and genome-wide association analysis. Sci. Rep. 2017, 7, 46446. [Google Scholar] [CrossRef]
- Liu, W.; Naruoka, Y.; Miller, K.; Garland-Campbell, K.A.; Carter, A.H. Characterizing and validating stripe rust resistance loci in US Pacific Northwest winter wheat accessions (Triticum aestivum L.) by genome-wide association and linkage mapping. The Plant Genome 2018, 11, 170087. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Han, Y.; Li, Y.; Liu, D.; Sun, M.; Zhao, Y.; Lv, C.; Li, D.; Yang, Z.; Huang, L.; et al. Loci and candidate gene identification for resistance to Sclerotinia sclerotiorum in soybean (Glycine max L. Merr.) via association and linkage maps. Plant J. 2015, 82, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: a review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araus, J.L.; Cairns, J.E. Field high-throughput phenotyping: the new crop breeding frontier. Trends P. Sci. 2014, 19, 52–61. [Google Scholar] [CrossRef]
- Hamon, C.; Coyne, C.J.; McGee, R.J.; Lesné, A.; Esnault, R.; Mangin, P.; Hervé, M.; Le Goff, I.; Deniot, G.; Roux-Duparque, M.; et al. QTL meta-analysis provides a comprehensive view of loci controlling partial resistance to Aphanomyces euteiches in four sources of resistance in pea. BMC Plant Biol. 2013, 13, 1. [Google Scholar] [CrossRef] [Green Version]
- Desgroux, A.; Baudais, V.N.; Aubert, V.; Le Roy, G.; de Larambergue, H.; Miteul, H.; Aubert, G.; Boutet, G.; Duc, G.; Baranger, A.; et al. Comparative Genome-Wide-Association Mapping identifies common loci controlling root system architecture and resistance to Aphanomyces euteiches in pea. Front. Plant Sci. 2018, 8, 2195. [Google Scholar] [CrossRef] [Green Version]
- Bonhomme, M.; André, O.; Badis, Y.; Ronfort, J.; Burgarella, C.; Chantret, N.; Prosperi, J.M.; Briskine, R.; Mudge, J.; Debellé, F.; et al. High-density genome-wide association mapping implicates an F-box encoding gene in Medicago truncatula resistance to Aphanomyces euteiches. New Phytol. 2014, 201, 1328–1342. [Google Scholar] [CrossRef]
- Badis, Y.; Bonhomme, M.; Lafitte, C.; Huguet, S.; Balzergue, S.; Dumas, B.; Jacquet, C. Transcriptome analysis highlights preformed defences and signalling pathways controlled by the prAe1 quantitative trait locus (QTL), conferring partial resistance to Aphanomyces euteiches in Medicago truncatula. Mol. Plant. Pathol. 2015, 16, 973–986. [Google Scholar] [CrossRef]
- Sharpe, A.G.; Ramsay, L.; Sanderson, L.A.; Fedoruk, M.J.; Clarke, W.E.; Li, R.; Kagale, S.; Vijayan, P.; Vandenberg, A.; Bett, K.E. Ancient orphan crop joins modern era: gene-based SNP discovery and mapping in lentil. BMC Genomics 2013, 14, 192. [Google Scholar] [CrossRef]
- Wong, M.M.; Gujaria-Verma, N.; Ramsay, L.; Yuan, H.Y.; Caron, C.; Diapari, M.; Vandenberg, A.; Bett, K.E. Classification and characterization of species within the genus Lens using genotyping-by-sequencing (GBS). PLoS ONE 2015, 10, e0122025. [Google Scholar] [CrossRef] [Green Version]
- Idrissi, O.; Udupa, S.M.; De Keyser, E.; McGee, R.J.; Coyne, C.J.; Saha, G.C.; Muehlbauer, F.J.; Van Damme, P.; De Riek, J. Identification of quantitative trait loci controlling root and shoot traits associated with drought tolerance in a lentil (Lens culinaris Medik.) recombinant inbred line population. Front. Plant Sci. 2016, 7, 1174. [Google Scholar] [CrossRef] [Green Version]
- Aldemir, S.; Ateş, D.; Temel, H.Y.; Yağmur, B.; Alsaleh, A.; Kahriman, A.; Özkan, H.; Vandenberg, A.; Tanyolac, M.B. QTLs for iron concentration in seeds of the cultivated lentil (Lens culinaris Medic.) via genotyping by sequencing. Turk. J. Agric. and Forest 2017, 41, 243–255. [Google Scholar] [CrossRef] [Green Version]
- Bhadauria, V.; Ramsay, L.; Bett, K.E.; Banniza, S. QTL mapping reveals genetic determinants of fungal disease resistance in the wild lentil species Lens ervoides. Sci. Rep. 2017, 7, 3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilet-Nayel, M.; Muehlbauer, F.; McGee, R.; Kraft, J.; Baranger, A.; Coyne, C. Quantitative trait loci for partial resistance to Aphanomyces root rot in pea. Theor. Appl. Genet. 2002, 106, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Shehata, M.; Davis, D.; Pfleger, F. Breeding for resistance to Aphanomyces euteiches root rot and Rhizoctonia solani stem rot in peas. J. Am. Soc. Hortic. Sci. 1983, 108, 1080–1085. [Google Scholar]
- Pilet-Nayel, M.; Muehlbauer, F.; McGee, R.; Kraft, J.; Baranger, A.; Coyne, C. Consistent quantitative trait loci in pea for partial resistance to Aphanomyces euteiches isolates from the United States and France. Phytopathology 2005, 95, 1287–1293. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Wang, S.; Zhou, Z.; Wang, S.; Dong, C.; Mu, C.; Song, Y.; Ma, P.; Li, C.; Wang, Z.; et al. Linkage mapping and GWAS reveal candidate genes conferring thermotolerance of seed-set in maize. J. Exp. Bot. 2019, 70, 4849–4864. [Google Scholar] [CrossRef]
- Maccaferri, M.; El-Feki, W.; Nazemi, G.; Salvi, S.; Canè, M.A.; Colalongo, M.C.; Stefanelli, S.; Tuberosa, R. Prioritizing quantitative trait loci for root system architecture in tetraploid wheat. J. Exp. Bot. 2016, 67, 1161–1178. [Google Scholar] [CrossRef]
- Sallam, A.; Arbaoui, M.; El-Esawi, M.; Abshire, N.; Martsch, R. Identification and verification of QTL associated with frost tolerance using linkage mapping and GWAS in winter faba bean. Front. Plant Sci. 2016, 7, 1098. [Google Scholar] [CrossRef] [Green Version]
- Sural, S.; Qian, G.; Pramanik, S. Segmentation and histogram generation using the HSV color space for image retrieval, Proceedings of the International Conference on Image Processing, Rochester, NY, USA, 22-25 Sep 2002; IEEE: Piscataway, NJ, USA, 2002. [Google Scholar]
- Kou, Y.J.; Wang, S.P. Broad-spectrum and durability: understanding of quantitative disease resistance. Curr. Opin. Plant Biol. 2010, 13, 181–185. [Google Scholar] [CrossRef]
- Poland, J.A.; Balint-Kurti, P.J.; Wisser, R.J.; Pratt, R.C.; Nelson, R.J. Shades of gray: the world of quantitative disease resistance. Trends Plant Sci. 2009, 14, 21–29. [Google Scholar] [CrossRef]
- Michelmore, R.W.; Christopoulou, M.; Caldwell, K.S. Impacts of resistance gene genetics, function, and evolution on a durable future. Annu. Rev. Phytopathol. 2013, 51, 291–319. [Google Scholar] [CrossRef] [PubMed]
- Theodoulou, F.L.; Kerr, I.D. ABC transporter research: going strong 40 years on. Biochem. Soc. Trans. 2015, 43, 1033–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Çakır, B.; Kılıçkaya, O. Whole-genome survey of the putative ATP-binding cassette transporter family genes in Vitis vinifera. PLoS ONE 2013, 8, e78860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, A.; Shitan, N.; Sato, S.; Nakamura, Y.; Tabata, S.; Yazaki, K. Genome-wide analysis of ATP-binding cassette (ABC) proteins in a model legume plant, Lotus japonicus: comparison with Arabidopsis ABC protein family. DNA Res. 2006, 13, 205–228. [Google Scholar] [CrossRef] [Green Version]
- Ofori, P.A.; Mizuno, A.; Suzuki, M.; Martinoia, E.; Reuscher, S.; Aoki, K.; Shibata, D.; Otagaki, S.; Matsumoto, S.; Shiratake, K. Genome-wide analysis of ATP binding cassette (ABC) transporters in tomato. PLoS ONE 2018, 13, e0200854. [Google Scholar] [CrossRef]
- Lionetti, V.; Cervone, F.; Bellincampi, D. Methyl esterification of pectin plays a role during plant-pathogen interactions and affects plant resistance to diseases. J. Pant Physiol. 2012, 169, 1623–1630. [Google Scholar] [CrossRef]
- Pham, A.T.; McNally, K.; Abdel-Haleem, H.; Boerma, H.R.; Li, Z. Fine mapping and identification of candidate genes controlling the resistance to southern root-knot nematode in PI 96354. Theor. Appl. Genet. 2013, 126, 1825–1838. [Google Scholar] [CrossRef]
- Liu, N.; Sun, Y.; Pei, Y.; Zhang, X.; Wang, P.; Li, X.; Li, F.; Hou, Y. A pectin methylesterase inhibitor enhances resistance to Verticillium wilt. Plant Physiol. 2018, 176, 2202–2220. [Google Scholar] [CrossRef] [Green Version]
- Volpi, C.; Janni, M.; Lionetti, V.; Bellincampi, D.; Favaron, F.; D’Ovidio, R. The ectopic expression of a pectin methyl esterase inhibitor increases pectin methyl esterification and limits fungal diseases in wheat. Mol. Plant Microbe Interact. 2011, 24, 1012–1019. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, V.; Raiola, A.; Camardella, L.; Giovane, A.; Obel, N.; Pauly, M.; Favaron, F.; Cervone, F.; Bellincampi, D. Overexpression of pectin methylesterase inhibitors in Arabidopsis restricts fungal infection by Botrytis cinerea. Plant Physiol. 2007, 143, 1871–1880. [Google Scholar] [CrossRef] [Green Version]
- Dai, G.; Nicole, M.; Andary, C.; Martinez, C.; Bresson, E.; Boher, B.; Daniel, J.F.; Geiger, J.P. Flavonoids accumulate in cell walls, middle lamellae and callose-rich papillae during an incompatible interaction between Xanthomonas campestris pv. malvacearum and cotton. Physiol. Mol. Plant Pathol. 1996, 49, 285–306. [Google Scholar] [CrossRef]
- Skadhauge, B.; Thomsen, K.K.; Von Wettstein, D. The role of the barley testa layer and its flavonoid content in resistance to Fusarium infections. Hereditas 1997, 126, 147–160. [Google Scholar] [CrossRef]
- Mihail, J.D.; Rush, C.M.; Singleton, L.L. Methods for Research on Soilborne Phytopathogenic Fungi, 2nd ed.; The American Phytopathological Society Press: St. Paul, MN, USA, 1992. [Google Scholar]
- Marzougui, A.; Ma, Y.; Zhang, C.; McGee, R.; Coyne, C.; Main, D.; Sankaran, S. Advanced imaging for quantitative evaluation of Aphanomyces root rot resistance in lentil. Front. Plant Sci. 2019, 10, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyne, C.J.; Porter, L.D.; Boutet, G.; Ma, Y.; McGee, R.J.; Lesné, A.; Baranger, A.; Pilet-Nayel, M.L. Confirmation of Fusarium root rot resistance QTL Fsp-Ps 2.1 of pea under controlled conditions. BMC Plant Biol. 2019, 19, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamon, C.; Baranger, A.; Coyne, C.J.; McGee, R.J.; Le Goff, I.; L’Anthoene, V.; Esnault, R.; Riviere, J.P.; Klein, A.; Mangin, P.; et al. New consistent QTL in pea associated with partial resistance to Aphanomyces euteiches in multiple French and American environments. Theor. Appl. Genet. 2011, 123, 261–281. [Google Scholar] [CrossRef] [PubMed]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [Green Version]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H. Stacks: building and genotyping loci de novo from short-read sequences. G3: Genes Genom. Genet. 2011, 1, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Browning, S.R. Genotype imputation with millions of reference samples. Am. J. Hum. Genet. 2016, 98, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S-PLUS, 2nd ed.; Springer-Verlag: New York City, NY, USA, 2013. [Google Scholar]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using lme4. arXiv 2014, arXiv:1406.5823. [Google Scholar]
- Team, R.C. R: A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, Austria. Available online: https://www.R-project.org/ (accessed on 20 January 2019).
- Kosambi, D.D. The estimation of map distances from recombination values. Annals. Eugenics. 1943, 12, 172–175. [Google Scholar] [CrossRef]
- Margarido, G.; Souza, A.; Garcia, A. OneMap: software for genetic mapping in outcrossing species. Hereditas 2007, 144, 78–79. [Google Scholar] [CrossRef]
- Wang, S. Windows QTL Cartographer 2.5; Department of Statistics, North Carolina State University: Raleigh, NC, USA, 2007. [Google Scholar]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2004, 21, 263–265. [Google Scholar] [CrossRef] [Green Version]
- Hill, W.; Weir, B. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 1988, 33, 54–78. [Google Scholar] [CrossRef]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Wang, N.; Akey, J.M.; Zhang, K.; Chakraborty, R.; Jin, L. Distribution of recombination crossovers and the origin of haplotype blocks: the interplay of population history, recombination, and mutation. Am. J. Hum. Genet. 2002, 71, 1227–1234. [Google Scholar] [CrossRef] [Green Version]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: an R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. In Bioinformatics Methods and Protocols, 2nd ed.; Springer: New York City, NY, USA, 2000; pp. 365–386. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Saha, G.C.; Vandemark, G.J. Stability of expression of reference genes among different lentil (Lens culinaris) genotypes subjected to cold stress, white mold disease, and Aphanomyces root rot. Plant Mol. Biol. Rep. 2013, 31, 1109–1115. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Trait a | Number of Lines | Number of Observations | Min b | Max b | Mean b | SE b | Skew c | Kurtosis c | Normality Test d,f | G Effect e,f | R Effect e,f | H2 e |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RIL | RRI | 189 | 1564 | 0 | 5 | 1.54 | 0.04 | 0.19 | 0.30 | ns | *** | ns | 0.24 |
| SDL | 189 | 551 | 1 | 4 | 1.69 | 0.03 | 0.76 | 0.70 | *** | *** | * | 0.10 | |
| RDL | 189 | 554 | 1 | 4 | 1.9 | 0.04 | 0.71 | 1.02 | *** | *** | * | 0.13 | |
| RGB.blue | 189 | 1563 | 131.19 | 197.79 | 171.2 | 0.27 | −0.21 | −0.18 | ns | *** | *** | 0.52 | |
| RGB.saturation | 189 | 1563 | 0.05 | 0.17 | 0.10 | 0.00 | 0.30 | −0.24 | ns | *** | ns | 0.47 | |
| RGB.SPL | 189 | 541 | 1 | 5 | 1.54 | 0.03 | 0.58 | −0.13 | *** | *** | ns | 0.17 | |
| RGB.RPL | 189 | 541 | 1 | 5 | 1.65 | 0.03 | 0.72 | −0.02 | *** | *** | ns | 0.21 | |
| AGI | 173 | 505 | 0.31 | 4.81 | 2.58 | 0.03 | 0.09 | −0.20 | ns | *** | * | 0.13 | |
| Multispectral.NDVI | 173 | 497 | 0.01 | 0.24 | 0.12 | 0.00 | 0.06 | −0.11 | ns | ** | ns | 0.13 | |
| Multispectral.canopy | 173 | 497 | −0.19 | 0.93 | 0.33 | 0.01 | −0.04 | 0.25 | ns | * | * | 0.05 | |
| Association | RRI | 326 | 3052 | 0 | 5 | 3.35 | 0.01 | −1.05 | 2.07 | *** | *** | *** | 0.73 |
| SDL | 326 | 2910 | 1 | 5 | 2.19 | 0.02 | 0.28 | −0.81 | ns | *** | *** | 0.53 | |
| RDL | 326 | 2911 | 1 | 5 | 2.11 | 0.02 | 0.62 | −0.62 | *** | *** | *** | 0.50 | |
| RGB.blue | 326 | 3052 | 50.53 | 119.67 | 79.82 | 0.20 | 0.41 | −0.06 | *** | *** | *** | 0.62 | |
| RGB.saturation | 326 | 3052 | 0.31 | 0.43 | 0.38 | 0.00 | −0.20 | 0.12 | ** | *** | *** | 0.73 | |
| RGB.SPL | 326 | 2895 | 1 | 5 | 2.57 | 0.02 | −0.08 | −0.78 | * | *** | *** | 0.50 | |
| RGB.RPL | 326 | 2895 | 1 | 5 | 2.52 | 0.02 | 0.16 | −0.93 | ns | *** | *** | 0.54 |
| Trait | QTL a | Environment b | Closest Marker | Chr | Position | LOD c | R2 | CI d | Parental Allele e |
|---|---|---|---|---|---|---|---|---|---|
| RRI | Q.RRI-Lc2.1 | CC | LcChr2-10483056 | 2 | 10483056 | 3.2** | 6.2% | 22.8−31.0 | K191-2 |
| Q.RRI -Lc5.1 | CC | LcChr5-229370222 | 5 | 229370222 | 2.7* | 5.3% | 62.0−64.8 | K192-1 | |
| SDL | Q.SDL-Lc3.1 | CC | LcChr3-124302109 | 3 | 124302109 | 2.8* | 5.9% | 81.8−83.9 | K192-1 |
| RGB.blue | Q.BLU-Lc2.1 | CC | LcChr2-10483056 | 2 | 10483056 | 3.3** | 6.5% | 22.8−30.8 | K192-1 |
| Q.BLU-Lc5.1 | CC | LcChr5-257437930 | 5 | 257437930 | 3.5** | 6.9% | 110.6−119.7 | K192-1 | |
| Q.BLU-Lc7.1 | CC | LcChr7-93158569 | 7 | 93158569 | 3.1** | 6.0% | 48.8−60.0 | K192-1 | |
| RGB.saturation | Q.SAT-Lc2.1 | CC | LcChr2-8058084 | 2 | 8058084 | 2.7* | 5.2% | 22.8−23.8 | K191-2 |
| Q.SAT-Lc3.1 | CC | LcChr3-65935857 | 3 | 65935857 | 2.7* | 5.7% | 35.0−38.1 | K192-1 | |
| Q.SAT-Lc7.1 | CC | LcChr7-93158569 | 7 | 93158569 | 4.2** | 8.1% | 47.0−61.0 | K191-2 | |
| RGB.SPL | Q.SPL-Lc2.1 | CC | LcChr2-4851535 | 2 | 4851535 | 2.6* | 5.3% | 13.1−13.1 | K191-2 |
| RGB.RPL | Q.RPL-Lc4.1 | CC | LcChr4-83603021 | 4 | 83603021 | 4.8** | 9.5% | 35.5−53.2 | K192-1 |
| Q.RPL-Lc4.2 | CC | LcChr4-175357959 | 4 | 175357959 | 2.8* | 5.7% | 71.44−71.44 | K191-2 | |
| AGI | Q.AGI-Lc2.1 | Field | LcChr2-10483056 | 2 | 10483056 | 2.7* | 5.6% | 28.7−28.8 | K191-2 |
| Q.AGI-Lc5.1 | Field | LcChr5-229370222 | 5 | 229370222 | 3.4** | 7.0% | 51.7−63.1 | K192-1 | |
| Multispectral.canopy | Q.CAN-Lc2.1 | Field | LcChr2-10483056 | 2 | 10483056 | 2.6* | 5.7% | 29.8−31.0 | K192-1 |
| Q.CAN-Lc7.1 | Field | LcChr7-61352757 | 7 | 61352757 | 3.2** | 6.8% | 8.3−16.7 | K192-1 | |
| Multispectral.NDVI | Q.NDVI-Lc6.1 | Field | LcChr6-170967409 | 6 | 170967409 | 4.1** | 8.8% | 77.0−86.8 | K191-2 |
| Q.NDVI-Lc6.2 | Field | LcChr6-196641316 | 6 | 196641316 | 5.6** | 12.1% | 104.1−120.0 | K191-2 | |
| Q.NDVI-Lc7.1 | Field | LcChr7-63933214 | 7 | 63933214 | 3.7** | 7.8% | 22.2−33.6 | K192-1 |
| Trait | QTLa | Trait-Associated Marker | Chr | Position | CI b | Number of Markers c | p-Value d | MAF e | R2 | Favorable Allele f |
|---|---|---|---|---|---|---|---|---|---|---|
| RRI | G.RRI-Lc.1.1 | 1569_6 | 1 | 72094185 | 71880185-72308185 | 2 | 3.4 × 10−10 *** | 24% | 10.7% | G/A |
| G.RRI-Lc.1.2 | 869_19 | 1 | 271625013 | 271384013-271839013 | 3 | 6 × 10−5 ** | 18% | 3.9% | G/A | |
| G.RRI-Lc.2.1 | 1827_75 | 2 | 102752889 | 102361889-103143889 | 7 | 1.4 × 10−5 ** | 39% | 19.3% | T/C | |
| G.RRI-Lc.2.2 | 2799_53 | 2 | 283548685 | 283157685-283939685 | 2 | 1.7 × 10−4 * | 27% | 13.4% | G/T | |
| G.RRI-Lc.4.1 | 5009_13 | 4 | 109679991 | 109524991-109834991 | 6 | 2.6 × 10−4 * | 25% | 1.4% | G/A | |
| G.RRI-Lc.5.1 | 7154_47 | 5 | 225981924 | 225823924-226139924 | 2 | 2.7 × 10−5 ** | 20% | 10.0% | T/A | |
| G.RRI-Lc.6.1 | 9084_41 | 6 | 51508203 | 50746203-52270203 | 4 | 3.6 × 10−4 * | 5% | 3.1% | C/T | |
| G.RRI-Lc.6.2 | 8442_65 | 6 | 155091517 | 154329517-155853517 | 4 | 9.2 × 10−10 *** | 49% | 2.3% | A/C | |
| G.RRI-Lc.7.1 | 9286_7 | 7 | 101144016 | 101016016-101272016 | 1 | 3.8 × 10−4 * | 6% | 8.4% | A/G | |
| RDL | G.RDL-Lc.1.1 | 1604_28 | 1 | 75421049 | 75207049-75635049 | 2 | 4.6 × 10−5 ** | 11% | 5.5% | G/T |
| G.RDL-Lc.2.1 | 2065_7 | 2 | 14688732 | 14369732-15007732 | 4 | 1.3 × 10−4 * | 6% | 6.5% | G/A | |
| G.RDL-Lc.2.2 | 2066_11 | 2 | 146997456 | 146678456-147316456 | 1 | 1.6 × 10−4 * | 17% | 4.9% | C/T | |
| G.RDL-Lc.2.3 | 2770_18 | 2 | 279028234 | 278709234-279347234 | 1 | 8.4 × 10−7 *** | 24% | 2.4% | C/T | |
| G.RDL-Lc.3.1 | 4445_35 | 3 | 192079718 | 191596718-192562718 | 5 | 4.1 × 10−4 * | 14% | 2.3% | G/T | |
| G.RDL-Lc.4.1 | 5190_31 | 4 | 145662855 | 145507855-145817855 | 1 | 2.1 × 10−4 * | 37% | 3.3% | A/T | |
| G.RDL-Lc.6.1 | 9009_9 | 6 | 40654874 | 39892874-41416874 | 3 | 5 × 10−6 ** | 45% | 4.2% | A/G | |
| G.RDL-Lc.7.1 | 10617_29 | 7 | 9677731 | 9549731-9805731 | 1 | 2.3 × 10−4 * | 31% | 6.4% | C/T | |
| RGB.saturation | G.SAT-Lc1.1 | 1323_60 | 1 | 329974767 | 329760767-330188767 | 4 | 5.1 × 10−5 ** | 29% | 4.2% | G/A |
| G.SAT-Lc2.1 | 1913_55 | 2 | 1176134 | 857134-1495134 | 7 | 5 × 10−5 ** | 48% | 8.3% | G/A | |
| G.SAT-Lc2.2 | 3450_21 | 2 | 9515161 | 9196161-9834161 | 8 | 2.1 × 10−4 * | 17% | 21.0% | A/T | |
| G.SAT-Lc4.1 | 5069_30 | 4 | 123435206 | 123280206-123590206 | 2 | 2 × 10−7 *** | 36% | 1.7% | T/C | |
| G.SAT-Lc5.1 | 7154_47 | 5 | 225981924 | 225823924-226139924 | 2 | 8.4 × 10−9 *** | 20% | 3.5% | T/A | |
| G.SAT-Lc5.2 | 7541_42 | 5 | 258022703 | 257864703-258180703 | 5 | 2.3 × 10−6 ** | 13% | 1.7% | T/C | |
| G.SAT-Lc6.1 | 9009_9 | 6 | 40654874 | 39892874-41416874 | 3 | 2.6 × 10−6 ** | 45% | 1.9% | A/G | |
| G.SAT-Lc7.1 | 10394_17 | 7 | 62649254 | 62521254-62777254 | 3 | 3.3 × 10−5 ** | 25% | 3.8% | C/T | |
| G.SAT-Lc7.2 | 9492_47 | 7 | 157790680 | 157662680-157918680 | 3 | 5.6 × 10−7 *** | 39% | 21.4% | A/T | |
| RGB.blue | G.BLU-Lc1.1 | 1569_6 | 1 | 72094185 | 71880185-72308185 | 2 | 1.1 × 10−6 *** | 24% | 13.7% | G/A |
| G.BLU-Lc2.1 | 3136_31 | 2 | 41405986 | 41086986-41724986 | 1 | 3.2 × 10−4 * | 39% | 3.2% | T/C | |
| G.BLU-Lc3.1 | 4814_43 | 3 | 75993537 | 75510537-76476537 | 3 | 2.4 × 10−6 ** | 40% | 5.9% | T/A | |
| G.BLU-Lc3.2 | 4403_39 | 3 | 189225788 | 188742788-189708788 | 4 | 6.1 × 10−7 *** | 27% | 2.1% | A/C | |
| G.BLU-Lc7.1 | 9616_9 | 7 | 197564498 | 197436498-197692498 | 4 | 1.6 × 10−4 * | 47% | 2.6% | G/A | |
| G.BLU-Lc7.2 | 10127_27 | 7 | 245915689 | 245787689-246043689 | 2 | 7.9 × 10−5 ** | 22% | 8.6% | G/T | |
| RGB.RPL | G.RPL-Lc2.1 | 3028_63 | 2 | 309553121 | 309234121-309872121 | 6 | 2.1 × 10−6 *** | 13% | 7.1% | C/T |
| G.RPL-Lc3.1 | 3562_32 | 3 | 110087057 | 109604057-110570057 | 2 | 4.8 × 10−4 * | 12% | 7.7% | C/T | |
| G.RPL-Lc4.1 | 6453_18 | 4 | 9587251 | 9432251-9742251 | 2 | 1.9 × 10−5 ** | 25% | 16.2% | T/C | |
| G.RPL-Lc4.2 | 5190_31 | 4 | 145662855 | 145507855-145817855 | 1 | 8.9 × 10−4 * | 37% | 1.9% | A/T | |
| G.RPL-Lc5.1 | 6649_58 | 5 | 127185901 | 127027901-127343901 | 1 | 1.5 × 10−4 * | 20% | 6.2% | T/C | |
| G.RPL-Lc6.1 | 9009_9 | 6 | 40654874 | 39892874-41416874 | 3 | 1.3 × 10−4 * | 45% | 7.8% | A/G |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Marzougui, A.; Coyne, C.J.; Sankaran, S.; Main, D.; Porter, L.D.; Mugabe, D.; Smitchger, J.A.; Zhang, C.; Amin, M.N.; et al. Dissecting the Genetic Architecture of Aphanomyces Root Rot Resistance in Lentil by QTL Mapping and Genome-Wide Association Study. Int. J. Mol. Sci. 2020, 21, 2129. https://doi.org/10.3390/ijms21062129
Ma Y, Marzougui A, Coyne CJ, Sankaran S, Main D, Porter LD, Mugabe D, Smitchger JA, Zhang C, Amin MN, et al. Dissecting the Genetic Architecture of Aphanomyces Root Rot Resistance in Lentil by QTL Mapping and Genome-Wide Association Study. International Journal of Molecular Sciences. 2020; 21(6):2129. https://doi.org/10.3390/ijms21062129
Chicago/Turabian StyleMa, Yu, Afef Marzougui, Clarice J. Coyne, Sindhuja Sankaran, Dorrie Main, Lyndon D. Porter, Deus Mugabe, Jamin A. Smitchger, Chongyuan Zhang, Md. Nurul Amin, and et al. 2020. "Dissecting the Genetic Architecture of Aphanomyces Root Rot Resistance in Lentil by QTL Mapping and Genome-Wide Association Study" International Journal of Molecular Sciences 21, no. 6: 2129. https://doi.org/10.3390/ijms21062129
APA StyleMa, Y., Marzougui, A., Coyne, C. J., Sankaran, S., Main, D., Porter, L. D., Mugabe, D., Smitchger, J. A., Zhang, C., Amin, M. N., Rasheed, N., Ficklin, S. P., & McGee, R. J. (2020). Dissecting the Genetic Architecture of Aphanomyces Root Rot Resistance in Lentil by QTL Mapping and Genome-Wide Association Study. International Journal of Molecular Sciences, 21(6), 2129. https://doi.org/10.3390/ijms21062129