Myocyte Enhancer Factor 2A (MEF2A) Defines Oxytocin-Induced Morphological Effects and Regulates Mitochondrial Function in Neurons

 , and
, and 
Abstract
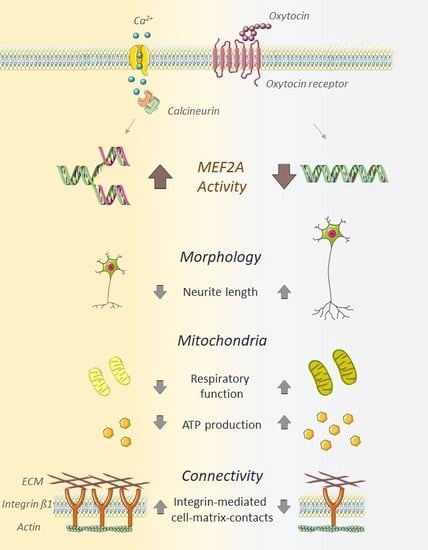
:
1. Introduction
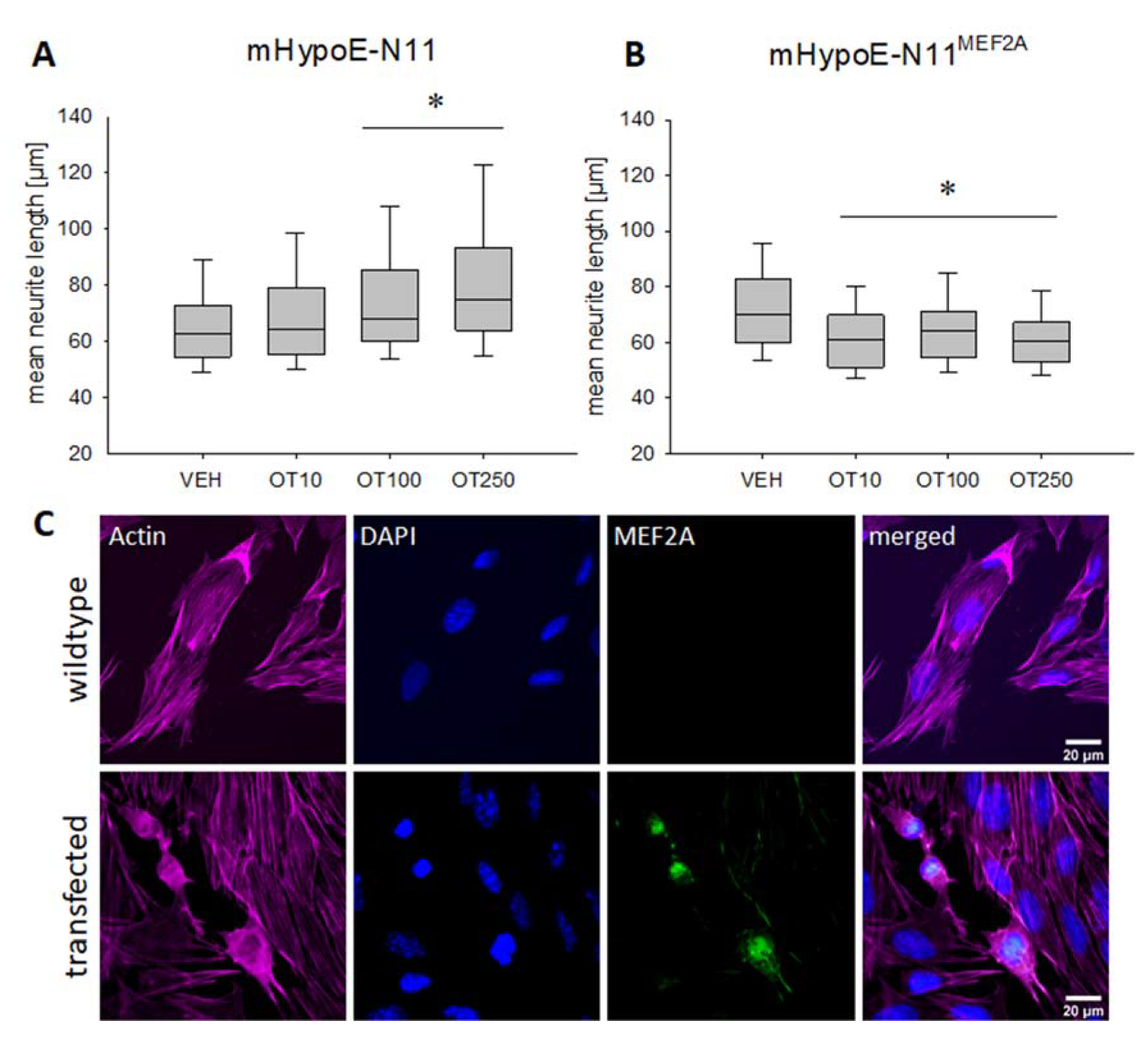
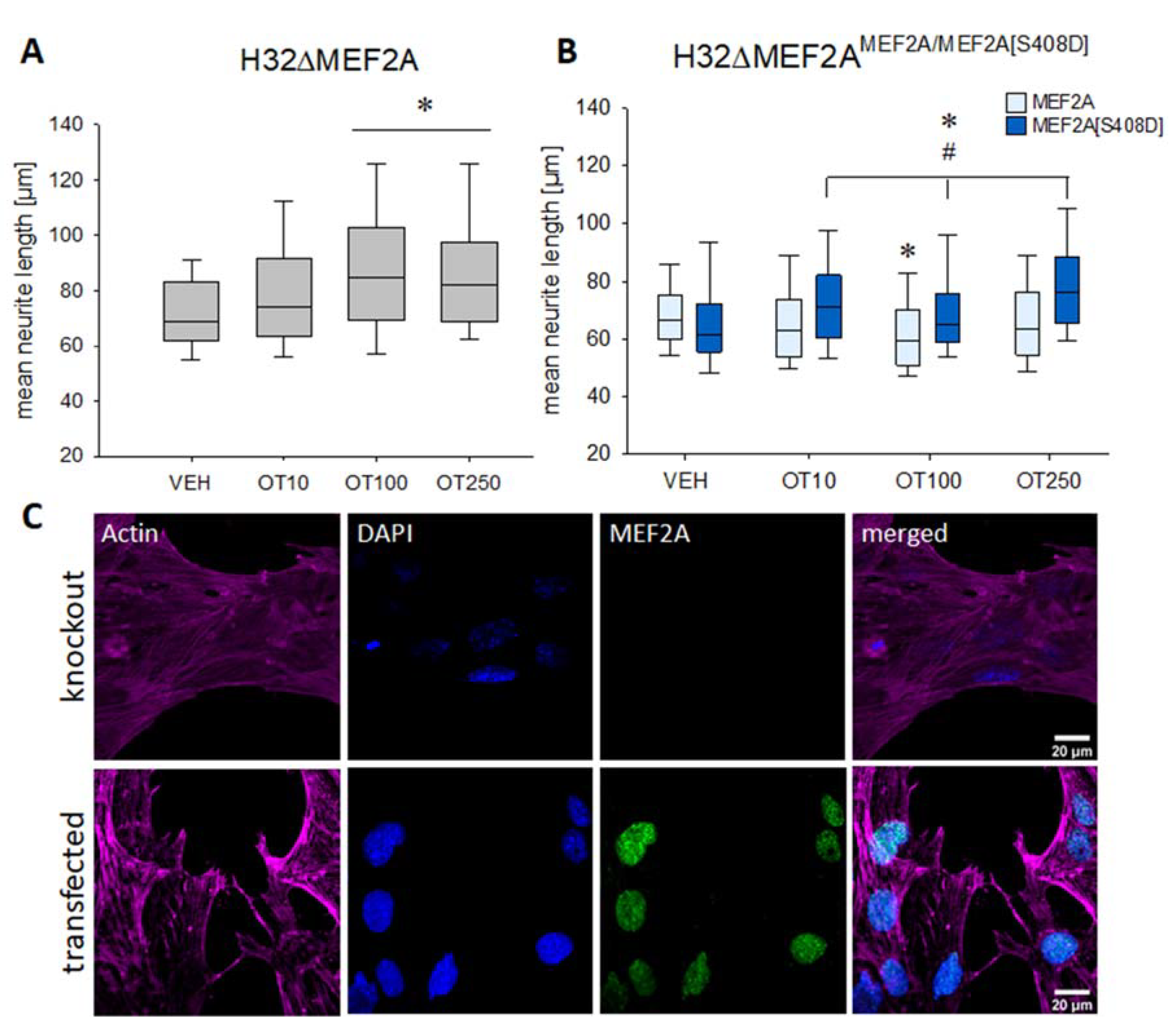
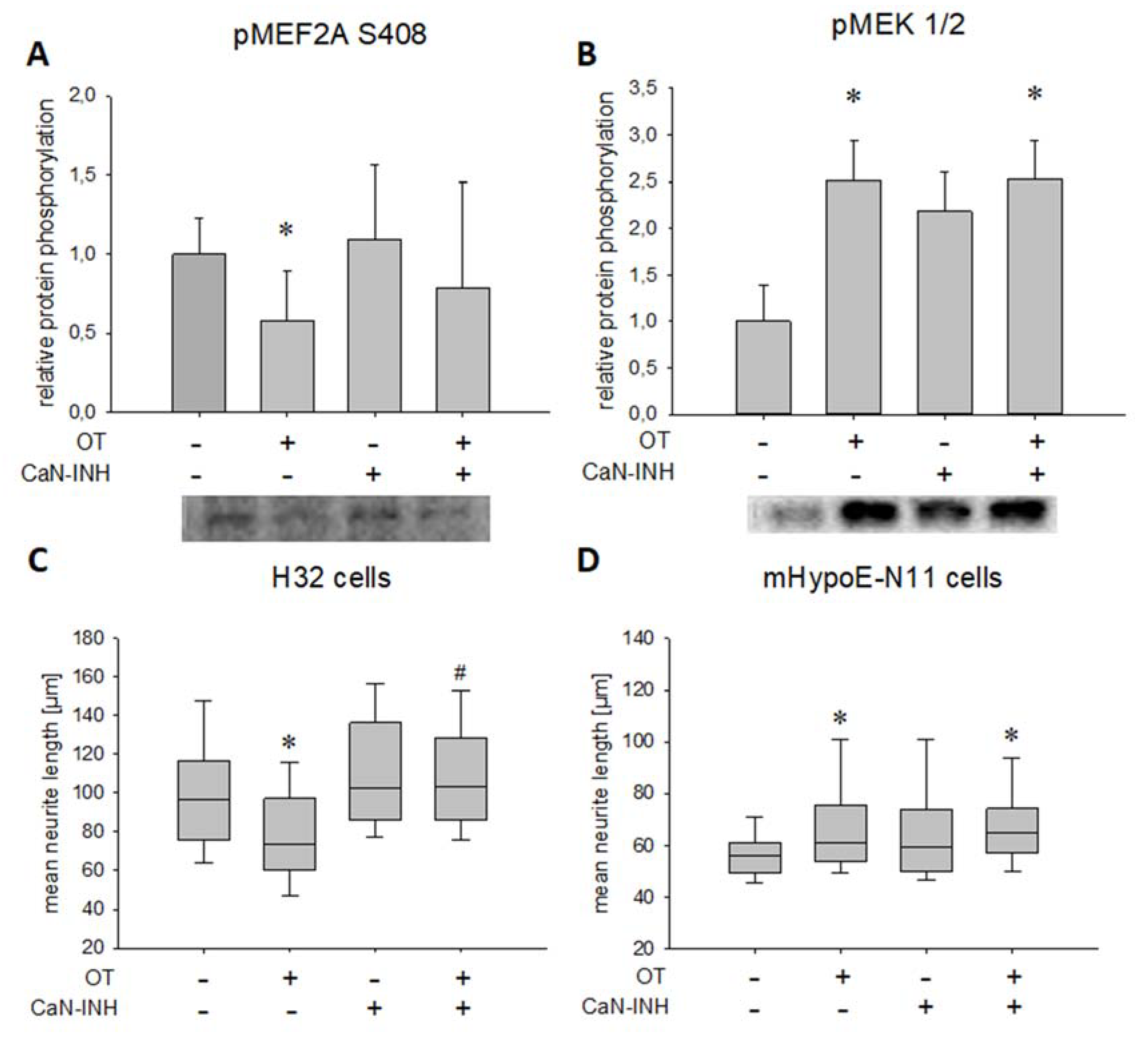
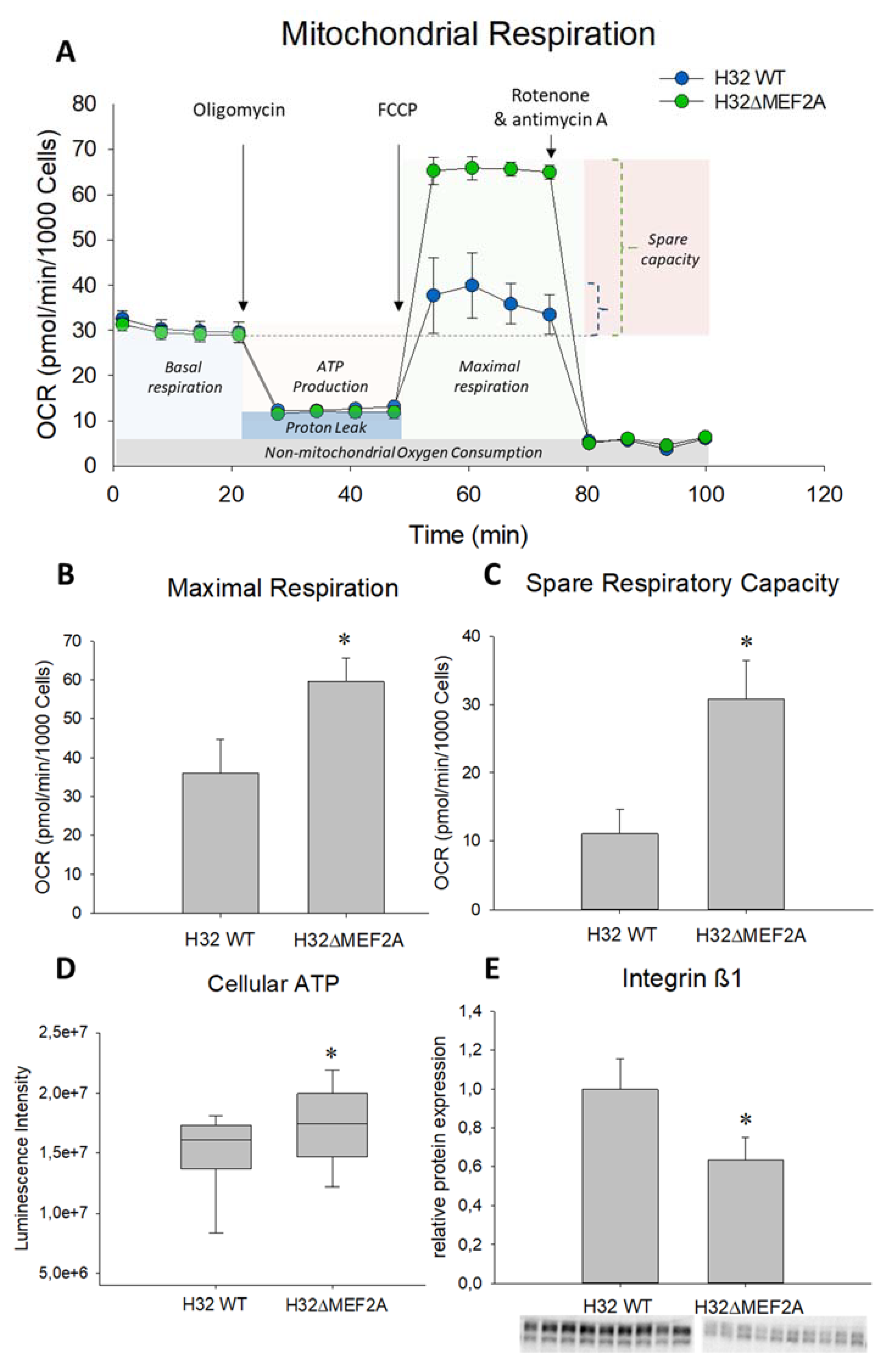
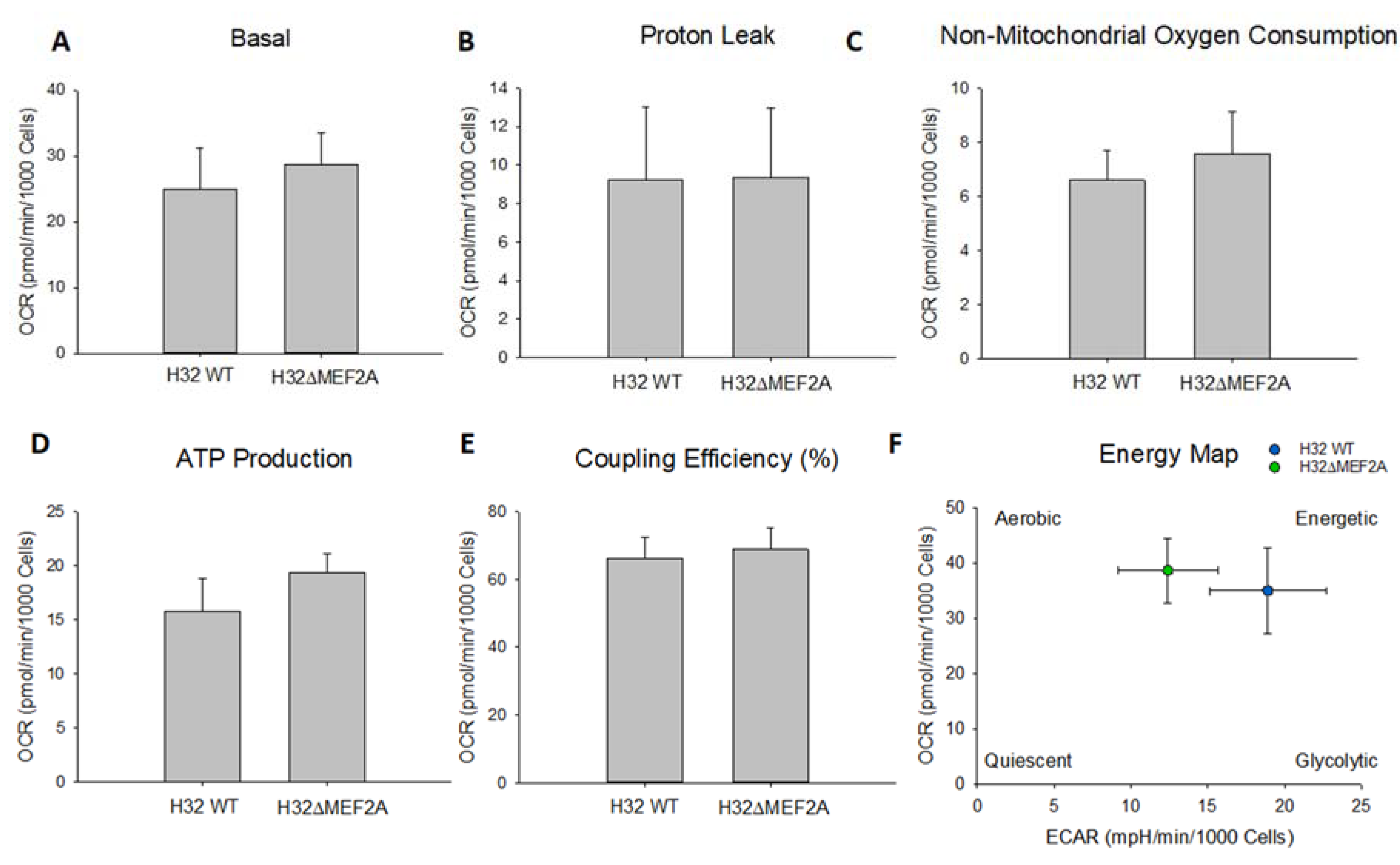
2. Results
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. CRISPR-Cas9 Mediated Knockout of MEF2A
4.3. Transfection of H32 and mHypoE-N11 Cells with MEF2A Overexpression Plasmids
4.4. Cell Stimulations
4.5. Protein Isolation
4.6. Western Blotting
4.7. Immunofluorescence
4.8. Morphological Characterization
4.9. Cell Viability Assay
4.10. Mitochondrial Respiration Analysis
4.11. CellTiter-Glo 2.0 Assay
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MEF2A | Myocyte enhancer factor 2A |
| OT | Oxytocin |
| CaN | Calcineurin |
| MAPK | Mitogen-activated protein kinase |
Appendix A



References
- Jurek, B.; Neumann, I.D. The Oxytocin Receptor: From Intracellular Signaling to Behavior. Physiol. Rev. 2018, 98, 1805–1908. [Google Scholar] [CrossRef] [PubMed]
- Gimpl, G.; Fahrenholz, F. The oxytocin receptor system: Structure, function, and regulation. Physiol. Rev. 2001, 81, 629–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, K.J.; Garner, J.P.; Libove, R.A.; Hyde, S.A.; Hornbeak, K.B.; Carson, D.S.; Liao, C.P.; Phillips, J.M.; Hallmayer, J.F.; Hardan, A.Y. Plasma oxytocin concentrations and OXTR polymorphisms predict social impairments in children with and without autism spectrum disorder. Proc. Natl. Acad. Sci. USA 2014, 111, 12258–12263. [Google Scholar] [CrossRef] [Green Version]
- Alaerts, K.; Bernaerts, S.; Vanaudenaerde, B.; Daniels, N.; Wenderoth, N. Amygdala-Hippocampal Connectivity Is Associated with Endogenous Levels of Oxytocin and Can Be Altered by Exogenously Administered Oxytocin in Adults with Autism. Biol. Psychiatry Cognit. Neurosci. Neuroimag. 2019, 4, 655–663. [Google Scholar] [CrossRef]
- Yuen, K.W.; Garner, J.P.; Carson, D.S.; Keller, J.; Lembke, A.; Hyde, S.A.; Kenna, H.A.; Tennakoon, L.; Schatzberg, A.F.; Parker, K.J. Plasma oxytocin concentrations are lower in depressed vs. healthy control women and are independent of cortisol. J. Psychiatry Res. 2014, 51, 30–36. [Google Scholar] [CrossRef]
- Carson, D.S.; Berquist, S.W.; Trujillo, T.H.; Garner, J.P.; Hannah, S.L.; Hyde, S.A.; Sumiyoshi, R.D.; Jackson, L.P.; Moss, J.K.; Strehlow, M.C.; et al. Cerebrospinal fluid and plasma oxytocin concentrations are positively correlated and negatively predict anxiety in children. Mol. Psychiatry 2015, 20, 1085–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer-Lindenberg, A.; Domes, G.; Kirsch, P.; Heinrichs, M. Oxytocin and vasopressin in the human brain: Social neuropeptides for translational medicine. Nat. Rev. Neurosci. 2011, 12, 524–538. [Google Scholar] [CrossRef]
- Lukas, M.; Toth, I.; Reber, S.O.; Slattery, D.A.; Veenema, A.H.; Neumann, I.D. The neuropeptide oxytocin facilitates pro-social behavior and prevents social avoidance in rats and mice. Neuropsychopharmacology 2011, 36, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.S.; Barth, M.E.; Johnson, S.L.; Gotlib, I.H.; Johnson, S.C. Oxytocin Receptor (OXTR) Polymorphisms and Attachment in Human Infants. Front. Psychol. 2011, 2, 200. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.S.; Kumsta, R.; von Dawans, B.; Monakhov, M.; Ebstein, R.P.; Heinrichs, M. Common oxytocin receptor gene (OXTR) polymorphism and social support interact to reduce stress in humans. Proc. Natl. Acad. Sci. USA 2011, 108, 19937–19942. [Google Scholar] [CrossRef] [Green Version]
- Choe, H.K.; Reed, M.D.; Benavidez, N.; Montgomery, D.; Soares, N.; Yim, Y.S.; Choi, G.B. Oxytocin Mediates Entrainment of Sensory Stimuli to Social Cues of Opposing Valence. Neuron 2015, 87, 152–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clipperton-Allen, A.E.; Lee, A.W.; Reyes, A.; Devidze, N.; Phan, A.; Pfaff, D.W.; Choleris, E. Oxytocin, vasopressin and estrogen receptor gene expression in relation to social recognition in female mice. Physiol. Behav. 2012, 105, 915–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, M.; Anderson, G.; Betancort Medina, S.R.; Seo, M.; Ojala, J.O. Integrating Autism Spectrum Disorder Pathophysiology: Mitochondria, Vitamin A, CD38, Oxytocin, Serotonin and Melatonergic Alterations in the Placenta and Gut. Curr. Pharm. Des. 2019, 25, 4405–4420. [Google Scholar] [CrossRef] [PubMed]
- Falougy, H.E.; Filova, B.; Ostatnikova, D.; Bacova, Z.; Bakos, J. Neuronal morphology alterations in autism and possible role of oxytocin. Endocr. Regul. 2019, 53, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Cabezas, M.A.; Barbas, H.; Zikopoulos, B. Parallel Development of Chromatin Patterns, Neuron Morphology, and Connections: Potential for Disruption in Autism. Front. Neuroanat. 2018, 12, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.M.; Chakrabarti, B.; Roberts, D.; Lai, M.C.; Suckling, J.; Baron-Cohen, S. From molecules to neural morphology: Understanding neuroinflammation in autism spectrum condition. Mol. Autism 2016, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Bringas, M.E.; Carvajal-Flores, F.N.; Lopez-Ramirez, T.A.; Atzori, M.; Flores, G. Rearrangement of the dendritic morphology in limbic regions and altered exploratory behavior in a rat model of autism spectrum disorder. Neuroscience 2013, 241, 170–187. [Google Scholar] [CrossRef]
- Jurek, B.; Slattery, D.A.; Hiraoka, Y.; Liu, Y.; Nishimori, K.; Aguilera, G.; Neumann, I.D.; van den Burg, E.H. Oxytocin Regulates Stress-Induced Crf Gene Transcription through CREB-Regulated Transcription Coactivator 3. J. Neurosci. 2015, 35, 12248–12260. [Google Scholar] [CrossRef] [Green Version]
- Zaslavsky, K.; Zhang, W.B.; McCready, F.P.; Rodrigues, D.C.; Deneault, E.; Loo, C.; Zhao, M.; Ross, P.J.; El Hajjar, J.; Romm, A.; et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat. Neurosci. 2019, 22, 556–564. [Google Scholar] [CrossRef]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [Green Version]
- Flavell, S.W.; Cowan, C.W.; Kim, T.K.; Greer, P.L.; Lin, Y.; Paradis, S.; Griffith, E.C.; Hu, L.S.; Chen, C.; Greenberg, M.E. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 2006, 311, 1008–1012. [Google Scholar] [CrossRef] [Green Version]
- Shalizi, A.; Gaudilliere, B.; Yuan, Z.; Stegmuller, J.; Shirogane, T.; Ge, Q.; Tan, Y.; Schulman, B.; Harper, J.W.; Bonni, A. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science 2006, 311, 1012–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrow, E.M.; Yoo, S.Y.; Flavell, S.W.; Kim, T.K.; Lin, Y.; Hill, R.S.; Mukaddes, N.M.; Balkhy, S.; Gascon, G.; Hashmi, A.; et al. Identifying autism loci and genes by tracing recent shared ancestry. Science 2008, 321, 218–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.; Berger, I.; Winter, J.; Jurek, B. Oxytocin alters the morphology of hypothalamic neurons via the transcription factor myocyte enhancer factor 2A (MEF-2A). Mol. Cell. Endocrinol. 2018, 477, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Sanborn, B.M. Hormonal signaling and signal pathway crosstalk in the control of myometrial calcium dynamics. Semin. Cell Dev. Biol. 2007, 18, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Van den Burg, E.H.; Stindl, J.; Grund, T.; Neumann, I.D.; Strauss, O. Oxytocin Stimulates Extracellular Ca2+ Influx Through TRPV2 Channels in Hypothalamic Neurons to Exert Its Anxiolytic Effects. Neuropsychopharmacology 2015, 40, 2938–2947. [Google Scholar] [CrossRef] [Green Version]
- Ying, L.; Becard, M.; Lyell, D.; Han, X.; Shortliffe, L.; Husted, C.I.; Alvira, C.M.; Cornfield, D.N. The transient receptor potential vanilloid 4 channel modulates uterine tone during pregnancy. Sci. Transl. Med. 2015, 7, 319ra204. [Google Scholar] [CrossRef] [Green Version]
- Zatkova, M.; Bacova, Z.; Puerta, F.; Lestanova, Z.; Alanazi, M.; Kiss, A.; Reichova, A.; Castejon, A.M.; Ostatnikova, D.; Bakos, J. Projection length stimulated by oxytocin is modulated by the inhibition of calcium signaling in U-87MG cells. J. Neural. Transm. 2018, 125, 1847–1856. [Google Scholar] [CrossRef]
- Pont, J.N.; McArdle, C.A.; Lopez Bernal, A. Oxytocin-stimulated NFAT transcriptional activation in human myometrial cells. Mol. Endocrinol. 2012, 26, 1743–1756. [Google Scholar] [CrossRef] [Green Version]
- Descazeaud, V.; Mestre, E.; Marquet, P.; Essig, M. Calcineurin regulation of cytoskeleton organization: A new paradigm to analyse the effects of calcineurin inhibitors on the kidney. J. Cell. Mol. Med. 2012, 16, 218–227. [Google Scholar] [CrossRef]
- Xiong, T.Q.; Chen, L.M.; Tan, B.H.; Guo, C.Y.; Li, Y.N.; Zhang, Y.F.; Li, S.L.; Zhao, H.; Li, Y.C. The effects of calcineurin inhibitor FK506 on actin cytoskeleton, neuronal survival and glial reactions after pilocarpine-induced status epilepticus in mice. Epilepsy Res. 2018, 140, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Lautermilch, N.J.; Spitzer, N.C. Regulation of calcineurin by growth cone calcium waves controls neurite extension. J. Neurosci. 2000, 20, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Lestanova, Z.; Bacova, Z.; Kiss, A.; Havranek, T.; Strbak, V.; Bakos, J. Oxytocin Increases Neurite Length and Expression of Cytoskeletal Proteins Associated with Neuronal Growth. J. Mol. Neurosci. 2016, 59, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Bakos, J.; Srancikova, A.; Havranek, T.; Bacova, Z. Molecular Mechanisms of Oxytocin Signaling at the Synaptic Connection. Neural Plast. 2018, 2018, 4864107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zatkova, M.; Reichova, A.; Bacova, Z.; Bakos, J. Activation of the Oxytocin Receptor Modulates the Expression of Synaptic Adhesion Molecules in a Cell-Specific Manner. J. Mol. Neurosci. 2019, 68, 171–180. [Google Scholar] [CrossRef]
- Blume, A.; Bosch, O.J.; Miklos, S.; Torner, L.; Wales, L.; Waldherr, M.; Neumann, I.D. Oxytocin reduces anxiety via ERK1/2 activation: Local effect within the rat hypothalamic paraventricular nucleus. Eur. J. Neurosci. 2008, 27, 1947–1956. [Google Scholar] [CrossRef]
- Jurek, B.; Slattery, D.A.; Maloumby, R.; Hillerer, K.; Koszinowski, S.; Neumann, I.D.; van den Burg, E.H. Differential contribution of hypothalamic MAPK activity to anxiety-like behaviour in virgin and lactating rats. PLoS ONE 2012, 7, e37060. [Google Scholar] [CrossRef]
- Martinetz, S.; Meinung, C.P.; Jurek, B.; von Schack, D.; van den Burg, E.H.; Slattery, D.A.; Neumann, I.D. De Novo Protein Synthesis Mediated by the Eukaryotic Elongation Factor 2 Is Required for the Anxiolytic Effect of Oxytocin. Biol. Psychiatry 2019, 85, 802–811. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Olson, E.N. MEF2: A central regulator of diverse developmental programs. Development 2007, 134, 4131–4140. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef] [Green Version]
- Naya, F.J.; Black, B.L.; Wu, H.; Bassel-Duby, R.; Richardson, J.A.; Hill, J.A.; Olson, E.N. Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat. Med. 2002, 8, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- She, H.; Yang, Q.; Shepherd, K.; Smith, Y.; Miller, G.; Testa, C.; Mao, Z. Direct regulation of complex I by mitochondrial MEF2D is disrupted in a mouse model of Parkinson disease and in human patients. J. Clin. Invest. 2011, 121, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front. Physiol. 2014, 5, 150. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, M.F.; Elwell, C.; Johnson, M.H. Mitochondrial Dysfunction in Autism Spectrum Disorders. Autism Open Access 2016, 6, 1000190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiore, R.; Khudayberdiev, S.; Christensen, M.; Siegel, G.; Flavell, S.W.; Kim, T.K.; Greenberg, M.E.; Schratt, G. Mef2-mediated transcription of the miR379-410 cluster regulates activity-dependent dendritogenesis by fine-tuning Pumilio2 protein levels. EMBO J. 2009, 28, 697–710. [Google Scholar] [CrossRef]
- Pfeiffer, B.E.; Zang, T.; Wilkerson, J.R.; Taniguchi, M.; Maksimova, M.A.; Smith, L.N.; Cowan, C.W.; Huber, K.M. Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron 2010, 66, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Brusco, J.; Haas, K. Interactions between mitochondria and the transcription factor myocyte enhancer factor 2 (MEF2) regulate neuronal structural and functional plasticity and metaplasticity. J. Physiol. 2015, 593, 3471–3481. [Google Scholar] [CrossRef] [Green Version]
- Mugele, K.; Kugler, H.; Spiess, J. Immortalization of a fetal rat brain cell line that expresses corticotropin-releasing factor mRNA. DNA Cell Biol. 1993, 12, 119–126. [Google Scholar] [CrossRef]
- Bikbaev, A.; Frischknecht, R.; Heine, M. Brain extracellular matrix retains connectivity in neuronal networks. Sci. Rep. 2015, 5, 14527. [Google Scholar] [CrossRef]
- Chklovskii, D.B. Synaptic connectivity and neuronal morphology: Two sides of the same coin. Neuron 2004, 43, 609–617. [Google Scholar]
- Tomaselli, K.; Doherty, P.; Emmett, C.; Damsky, C.; Walsh, F.; Reichardt, L. Expression of beta 1 integrins in sensory neurons of the dorsal root ganglion and their functions in neurite outgrowth on two laminin isoforms. J. Neurosci. 1993, 13, 4880–4888. [Google Scholar] [CrossRef]
- Hurlemann, R. Oxytocin-Augmented Psychotherapy: Beware of Context. Neuropsychopharmacology 2017, 42, 377. [Google Scholar] [CrossRef] [PubMed]
- Kosfeld, M.; Heinrichs, M.; Zak, P.J.; Fischbacher, U.; Fehr, E. Oxytocin increases trust in humans. Nature 2005, 435, 673–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tost, H.; Kolachana, B.; Hakimi, S.; Lemaitre, H.; Verchinski, B.A.; Mattay, V.S.; Weinberger, D.R.; Meyer-Lindenberg, A. A common allele in the oxytocin receptor gene (OXTR) impacts prosocial temperament and human hypothalamic-limbic structure and function. Proc. Natl. Acad. Sci. USA 2010, 107, 13936–13941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yatawara, C.J.; Einfeld, S.L.; Hickie, I.B.; Davenport, T.A.; Guastella, A.J. The effect of oxytocin nasal spray on social interaction deficits observed in young children with autism: A randomized clinical crossover trial. Mol. Psychiatry 2016, 21, 1225–1231. [Google Scholar] [CrossRef] [Green Version]
- Parker, K.J.; Oztan, O.; Libove, R.A.; Sumiyoshi, R.D.; Jackson, L.P.; Karhson, D.S.; Summers, J.E.; Hinman, K.E.; Motonaga, K.S.; Phillips, J.M.; et al. Intranasal oxytocin treatment for social deficits and biomarkers of response in children with autism. Proc. Natl. Acad. Sci. USA 2017, 114, 8119–8124. [Google Scholar] [CrossRef] [Green Version]
- Shalizi, A.; Bilimoria, P.M.; Stegmuller, J.; Gaudilliere, B.; Yang, Y.; Shuai, K.; Bonni, A. PIASx is a MEF2 SUMO E3 ligase that promotes postsynaptic dendritic morphogenesis. J. Neurosci. 2007, 27, 10037–10046. [Google Scholar] [CrossRef]
- Tomizawa, K.; Iga, N.; Lu, Y.F.; Moriwaki, A.; Matsushita, M.; Li, S.T.; Miyamoto, O.; Itano, T.; Matsui, H. Oxytocin improves long-lasting spatial memory during motherhood through MAP kinase cascade. Nat. Neurosci. 2003, 6, 384–390. [Google Scholar] [CrossRef]
- Zhou, C.J.; Yada, T.; Kohno, D.; Kikuyama, S.; Suzuki, R.; Mizushima, H.; Shioda, S. PACAP activates PKA, PKC and Ca(2+) signaling cascades in rat neuroepithelial cells. Peptides 2001, 22, 1111–1117. [Google Scholar] [CrossRef]
- Ichida, M.; Finkel, T. Ras regulates NFAT3 activity in cardiac myocytes. J. Biol. Chem. 2001, 276, 3524–3530. [Google Scholar] [CrossRef] [Green Version]
- Molkentin, J.D. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res. 2004, 63, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Cao, M.; Chang, C.W.; Wang, C.; Shi, X.; Zhan, X.; Birnbaum, S.G.; Bezprozvanny, I.; Huber, K.M.; Wu, J.I. Autism-Associated Chromatin Regulator Brg1/SmarcA4 Is Required for Synapse Development and Myocyte Enhancer Factor 2-Mediated Synapse Remodeling. Mol. Cell. Biol. 2016, 36, 70–83. [Google Scholar]
- Lilja, J.; Ivaska, J. Integrin activity in neuronal connectivity. J. Cell Sci. 2018, 131, jcs212803. [Google Scholar] [CrossRef] [Green Version]
- Estrella, N.L.; Desjardins, C.A.; Nocco, S.E.; Clark, A.L.; Maksimenko, Y.; Naya, F.J. MEF2 transcription factors regulate distinct gene programs in mammalian skeletal muscle differentiation. J. Biol. Chem. 2015, 290, 1256–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, E.; Werb, Z. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. J. Cell Biol. 2002, 158, 357–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cavanagh, E.M.; Ferder, M.; Inserra, F.; Ferder, L. Angiotensin II, mitochondria, cytoskeletal, and extracellular matrix connections: An integrating viewpoint. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H550–H558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolak-Suki, E.; Imsirovic, J.; Nishibori, Y.; Krishnan, R.; Suki, B. Regulation of Mitochondrial Structure and Dynamics by the Cytoskeleton and Mechanical Factors. Int. J. Mol. Sci. 2017, 18, 1812. [Google Scholar] [CrossRef]
- Perrino, B.A.; Ng, L.Y.; Soderling, T.R. Calcium regulation of calcineurin phosphatase activity by its B subunit and calmodulin. Role of the autoinhibitory domain. J. Biol. Chem. 1995, 270, 340–346. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | mHypoE-N11 (Mouse) | H32 (Rat) | ||||
|---|---|---|---|---|---|---|
| Manipulation | Wild-type | mHypoE-N11MEF2A | Wild-type | H32ΔMEF2A | H32ΔMEF2AMEF2A | H32ΔMEF2AMEF2A[S408D] (inactive) |
| Level of MEF2A | − | ++ | + | − | ++ | ++ |
| Antibody | Company and CAT Number | Dilution and Diluent | Secondary Antibody |
|---|---|---|---|
| MEF2A total | Acris AP06372PU-N | 1:2000 in 5% MP | HRP-coupled anti-rabbit |
| pMEF2A S408 | CusAb PA000728 | 1:5000 in 5% BSA | HRP-coupled anti-rabbit |
| pMEK1/2 4199 | Cell Signaling 9154 | 1:5000 in 5% BSA | HRP-coupled anti-rabbit |
| Integrin ß1 (N-20) | Santa Cruz sc-6622 | 1:1000 in 5% BSA | HRP-coupled anti-goat |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyer, M.; Kuffner, K.; Winter, J.; Neumann, I.D.; Wetzel, C.H.; Jurek, B. Myocyte Enhancer Factor 2A (MEF2A) Defines Oxytocin-Induced Morphological Effects and Regulates Mitochondrial Function in Neurons. Int. J. Mol. Sci. 2020, 21, 2200. https://doi.org/10.3390/ijms21062200
Meyer M, Kuffner K, Winter J, Neumann ID, Wetzel CH, Jurek B. Myocyte Enhancer Factor 2A (MEF2A) Defines Oxytocin-Induced Morphological Effects and Regulates Mitochondrial Function in Neurons. International Journal of Molecular Sciences. 2020; 21(6):2200. https://doi.org/10.3390/ijms21062200
Chicago/Turabian StyleMeyer, Magdalena, Kerstin Kuffner, Julia Winter, Inga D. Neumann, Christian H. Wetzel, and Benjamin Jurek. 2020. "Myocyte Enhancer Factor 2A (MEF2A) Defines Oxytocin-Induced Morphological Effects and Regulates Mitochondrial Function in Neurons" International Journal of Molecular Sciences 21, no. 6: 2200. https://doi.org/10.3390/ijms21062200
APA StyleMeyer, M., Kuffner, K., Winter, J., Neumann, I. D., Wetzel, C. H., & Jurek, B. (2020). Myocyte Enhancer Factor 2A (MEF2A) Defines Oxytocin-Induced Morphological Effects and Regulates Mitochondrial Function in Neurons. International Journal of Molecular Sciences, 21(6), 2200. https://doi.org/10.3390/ijms21062200