Loricrin: Past, Present, and Future


Abstract
:1. Introduction and Overview
Bricks and Mortar: Which Matters More?
2. Epidermal Differentiation
2.1. Cornified Cell Envelopes: Just an Insoluble Matter?
2.2. Loricrin: A Major Cell Envelope (CE) Constituent
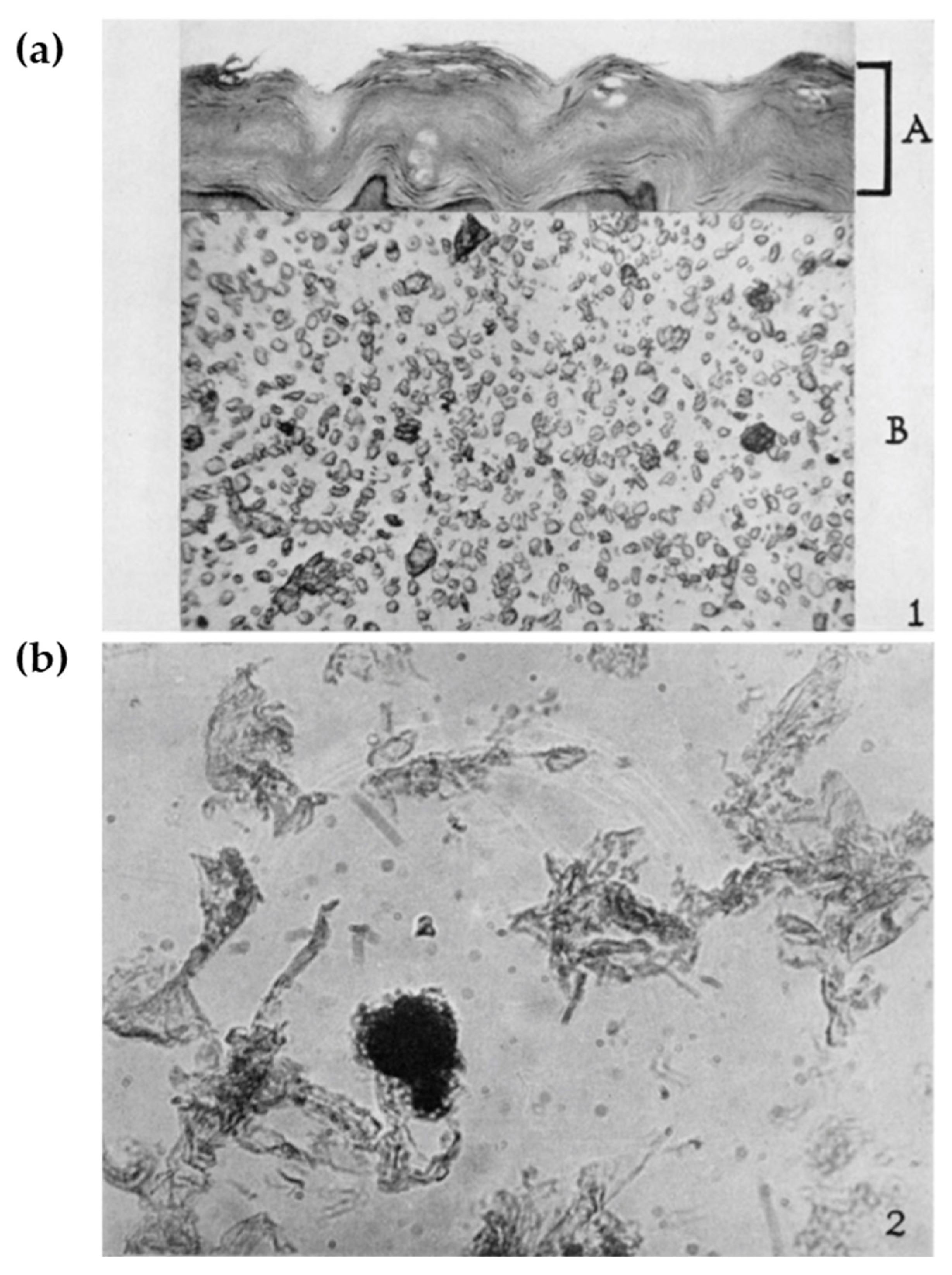
2.3. Tissue Expression of Loricrin: Along with the Air–Liquid Interface
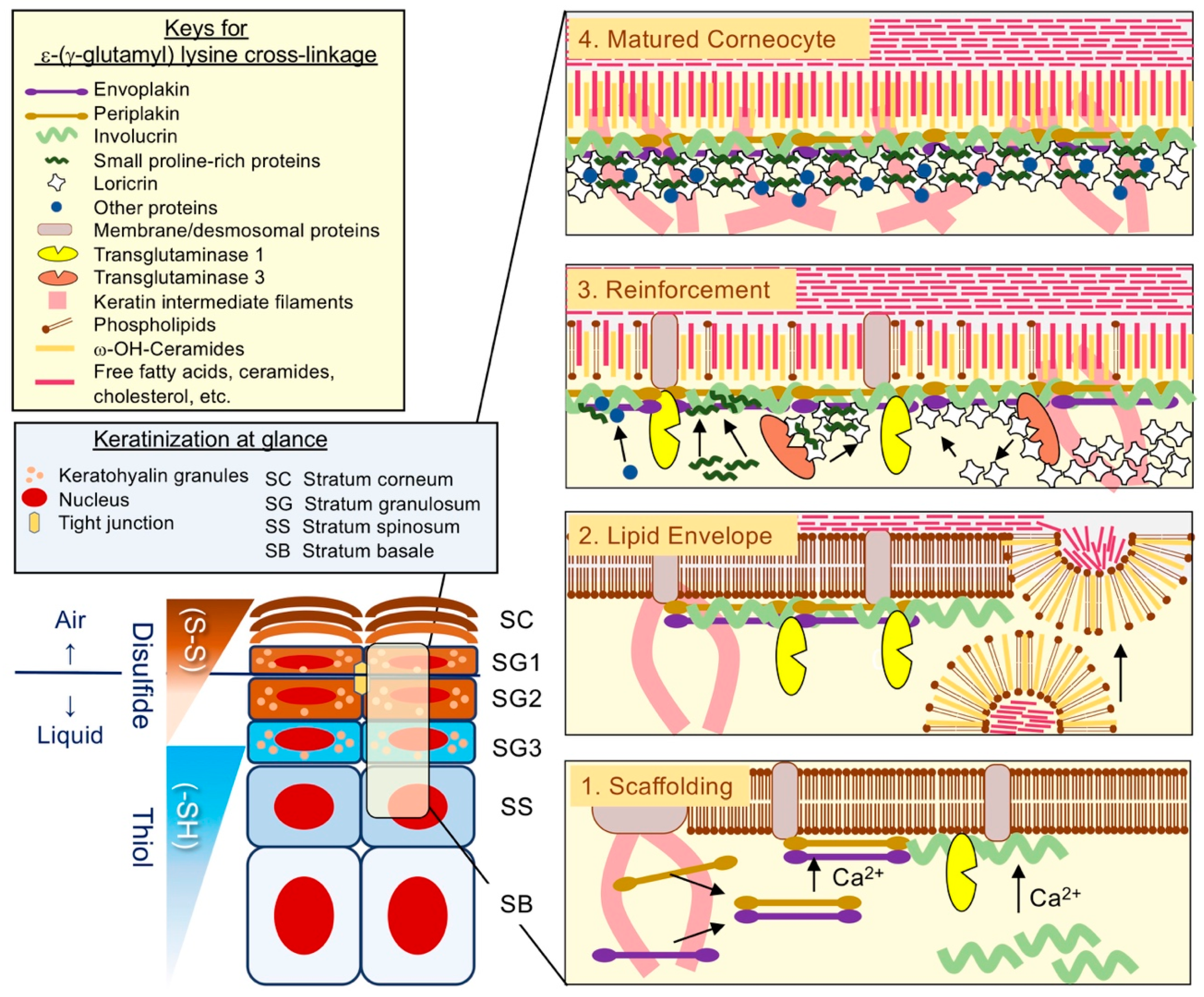
2.4. The Assembly of CE: Building the Brick Wall
3. Lessons from Mouse Models
3.1. Vohwinkel Syndrome Transgenic Mouse: Who Done It?
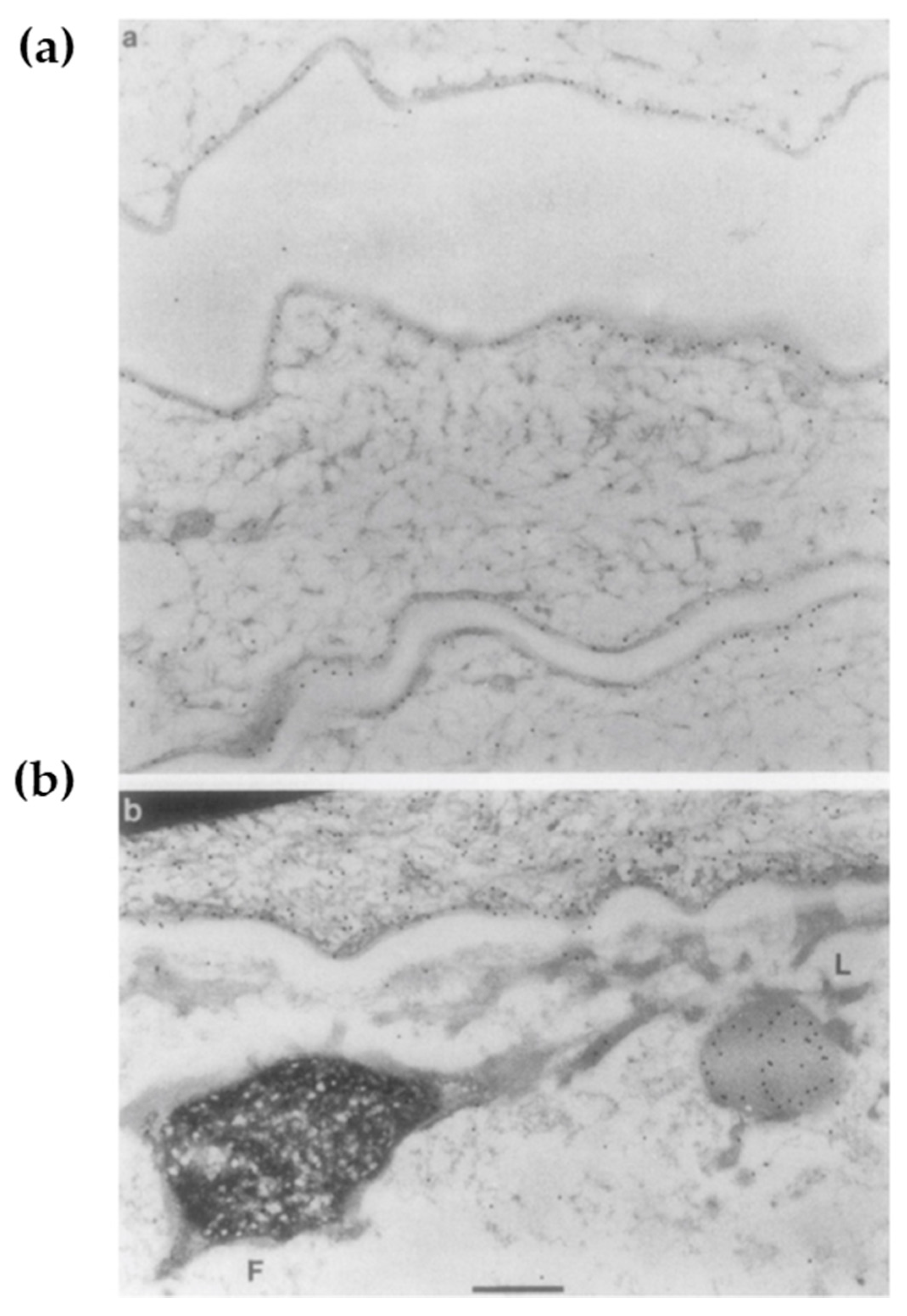
3.2. Loricrin Knockout Mouse: Dispensable but Indispensable
3.2.1. Dispensability for the Lipid-Based Permeability Barrier
3.2.2. Alternative Reinforcement Proteins Small Proline-Rich Proteins (SPRR)/Late Cornified Cell Envelope (LCE)
3.2.3. Indispensability for Cornification
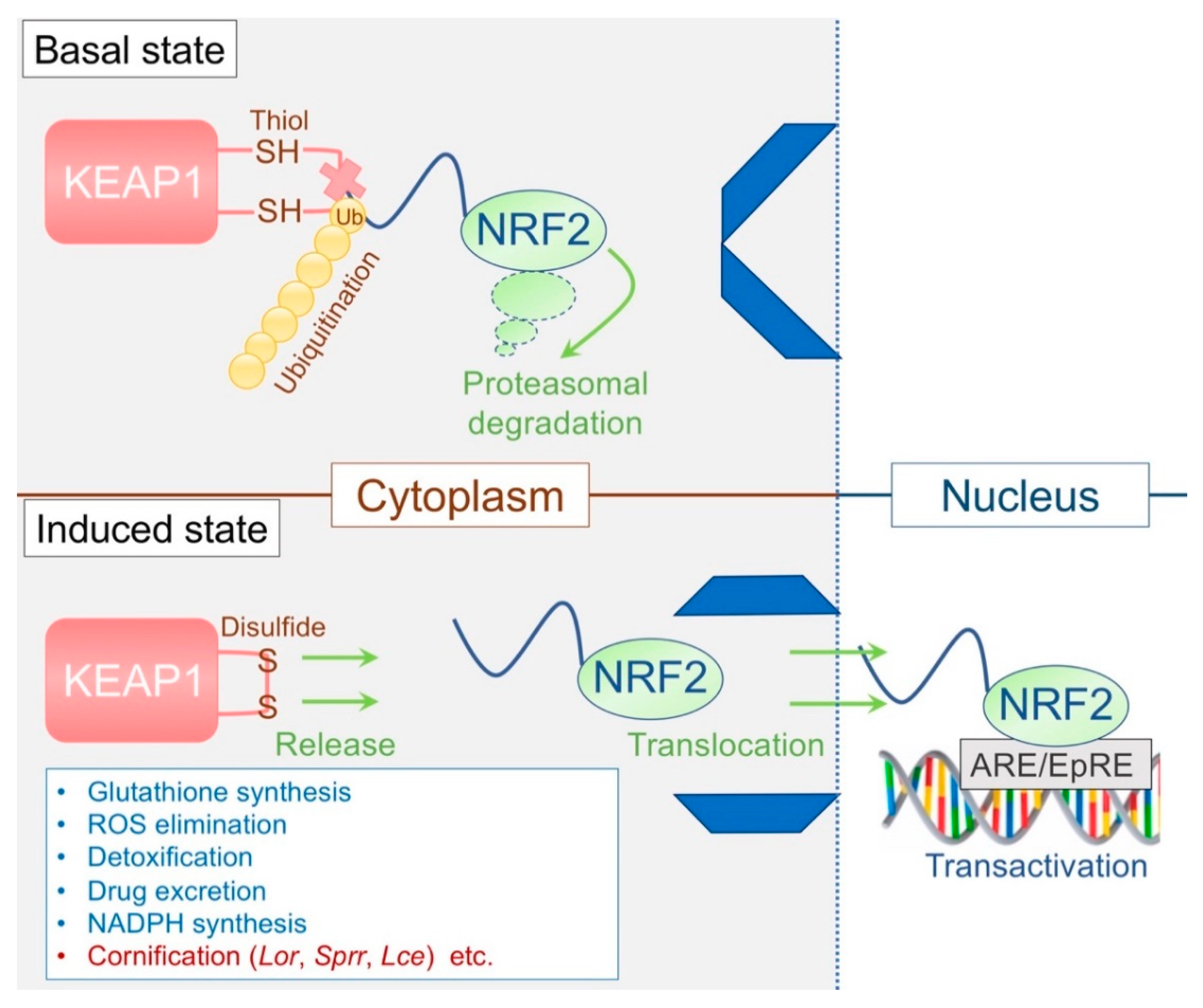
3.3. The KEAP1/NRF2 System: The Epidermal Keeper and Striker
3.3.1. The Epidermal Thiol Gradient and Cornification
3.3.2. Disrupted Epidermal Thiol Gradient and Recovery
3.3.3. Dominant-Negative NF-E2-related factor 2 (Nrf2) Mice
4. Epidermal Microenvironment and Immune Homeostasis
4.1. Lessons from Filaggrin-Deficiency: Flaky or Leaky
4.2. The F-Granule and L-Granule: “La Raison D’êTre” of the Epidermis
4.3. Atopy: Imprinted Cutaneous Immunological Memory?
4.4. Immunoanatomy of the Epithelium: It Is Not What It Is Made Of, but the Reaction on the Surface That Matters
4.5. Metabolic Regulation of the Epidermal Barrier: The Epidermal “A and D”
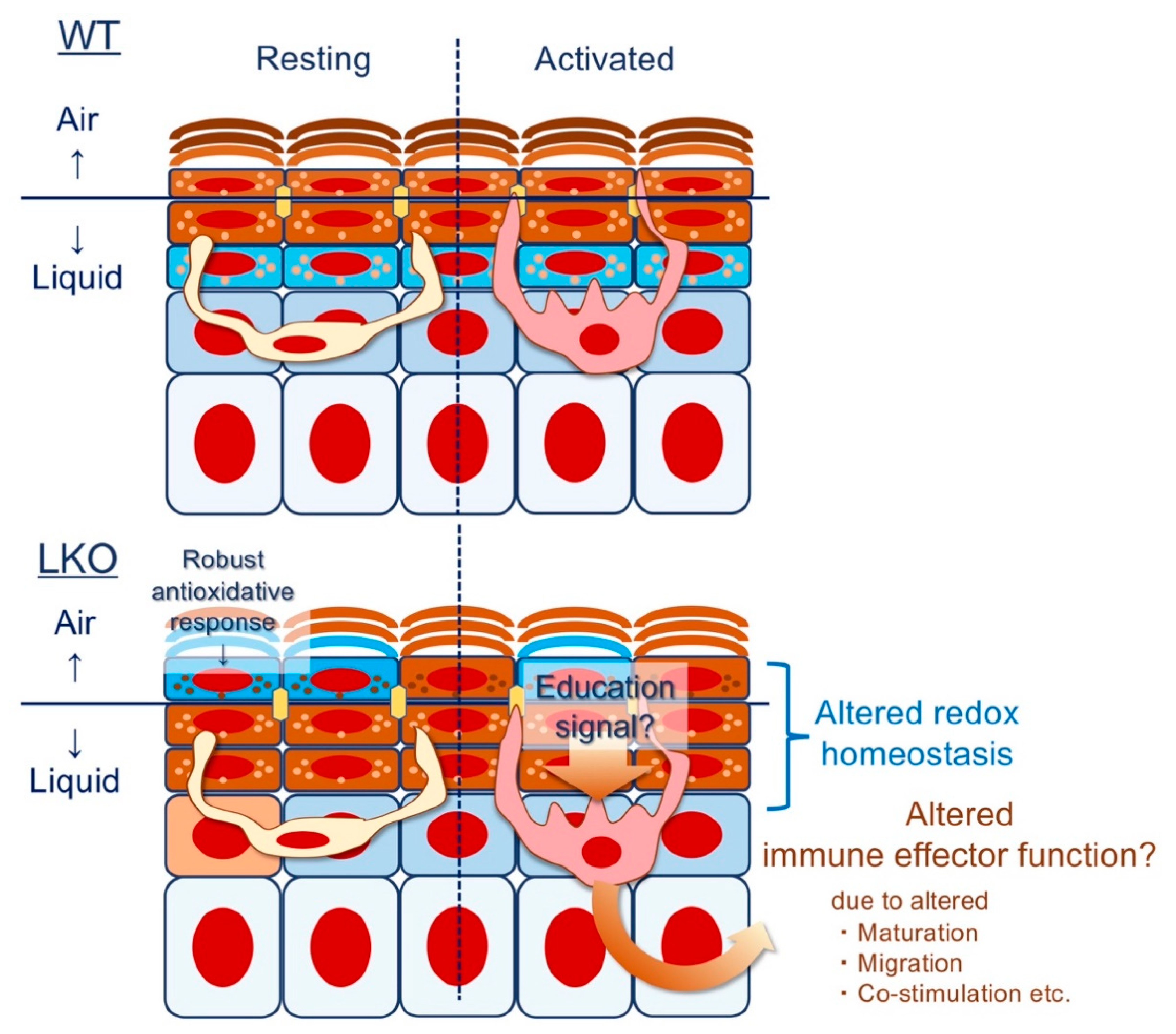
4.6. Epithelial Imprinting of Immunological Memory: Does Lor Instruct the Langerhans Cell (LC)?
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SC | stratum corneum |
| CE | cell envelopes |
| LG | lamellar granule |
| SG | stratum granulosum |
| ARCI | autosomal recessive congenital ichthyosis |
| LOF | loss-of-function |
| ABCA12 | ATP binding cassette subfamily A member 12 |
| TGM1 | transglutaminase 1 |
| LI | lamellar ichthyosis |
| IVL | involucrin |
| LOR | loricrin |
| EDC | epidermal differentiation complex |
| KIF | keratin intermediate filaments |
| mTECs | medullary thymic epithelial cells |
| FLG | filaggrin |
| KG | keratohyalin granules |
| SPRRs | small proline-rich proteins |
| VS | Vohwinkel syndrome |
| NLS | nuclear localization signal |
| LKO | LOR-knockout |
| LCE | late cornified cell envelope |
| UVB | ultraviolet B |
| KEAP1 | Kelch-like erythroid cell-derived protein with cap´n´collar homology-associated protein 1 |
| NRF2 | NF-E2-related factor 2 |
| ARE | antioxidant responsive element |
| EpRE | electrophile responsive element |
| LC | Langerhans cell |
| AD | atopic dermatitis |
| ACD | allergic contact dermatitis |
| CASP14 | caspase-14 |
| VAD | vitamin A deficiency |
| VDR | vitamin D receptor |
| RXRs | retinoid X receptors |
| PMNs | polymorphonuclear neutrophils |
References
- Strasser, B.; Mlitz, V.; Hermann, M.; Rice, R.H.; Eigenheer, R.A.; Alibardi, L.; Tschachler, E.; Eckhart, L. Evolutionary origin and diversification of epidermal barrier proteins in amniotes. Mol. Biol. Evol. 2014, 31, 3194–3205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Amagai, M. Dissecting the formation, structure and barrier function of the stratum corneum. Int. Immunol. 2015, 27, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Kalinin, A.; Marekov, L.N.; Steinert, P.M. Assembly of the epidermal cornified cell envelope. J. Cell Sci. 2001, 114, 3069–3070. [Google Scholar] [PubMed]
- Nemes, Z.; Steinert, P.M. Bricks and mortar of the epidermal barrier. Exp. Mol. Med. 1999, 31, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, T.; Akiyama, M. Inherited ichthyosis: Non-syndromic forms. J. Dermatol. 2016, 43, 242–251. [Google Scholar] [CrossRef]
- Djian, P.; Easley, K.; Green, H. Targeted ablation of the murine involucrin gene. J. Cell Biol. 2000, 151, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Koch, P.J.; de Viragh, P.A.; Scharer, E.; Bundman, D.; Longley, M.A.; Bickenbach, J.; Kawachi, Y.; Suga, Y.; Zhou, Z.; Huber, M.; et al. Lessons from loricrin-deficient mice: Compensatory mechanisms maintaining skin barrier function in the absence of a major cornified envelope protein. J. Cell Biol. 2000, 151, 389–400. [Google Scholar] [CrossRef]
- Backendorf, C.; Hohl, D. A common origin for cornified envelope proteins? Nat. Genet. 1992, 2, 91. [Google Scholar] [CrossRef]
- Mischke, D.; Korge, B.P.; Marenholz, I.; Volz, A.; Ziegler, A. Genes encoding structural proteins of epidermal cornification and S100 calcium-binding proteins form a gene complex (“epidermal differentiation complex”) on human chromosome 1q21. J. Investig. Dermatol. 1996, 106, 989–992. [Google Scholar] [CrossRef] [Green Version]
- Huebner, A.J.; Dai, D.; Morasso, M.; Schmidt, E.E.; Schafer, M.; Werner, S.; Roop, D.R. Amniotic fluid activates the nrf2/keap1 pathway to repair an epidermal barrier defect in utero. Dev. Cell 2012, 23, 1238–1246. [Google Scholar] [CrossRef] [Green Version]
- Ishitsuka, Y.; Huebner, A.J.; Rice, R.H.; Koch, P.J.; Speransky, V.V.; Steven, A.C.; Roop, D.R. Lce1 Family Members Are Nrf2-Target Genes that Are Induced to Compensate for the Loss of Loricrin. J. Investig. Dermatol. 2016, 136, 1656–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Bouameur, J.E.; Bar, J.; Rice, R.H.; Hornig-Do, H.T.; Roop, D.R.; Schwarz, N.; Brodesser, S.; Thiering, S.; Leube, R.E.; et al. A keratin scaffold regulates epidermal barrier formation, mitochondrial lipid composition, and activity. J. Cell Biol. 2015, 211, 1057–1075. [Google Scholar] [CrossRef] [PubMed]
- Matoltsy, A.G.; Balsamo, C.A. A study of the components of the cornified epithelium of human skin. J. Biophys. Biochem. Cytol. 1955, 1, 339–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, R.H.; Green, H. The cornified envelope of terminally differentiated human epidermal keratinocytes consists of cross-linked protein. Cell 1977, 11, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Rice, R.H.; Green, H. Presence in human epidermal cells of a soluble protein precursor of the cross-linked envelope: Activation of the cross-linking by calcium ions. Cell 1979, 18, 681–694. [Google Scholar] [CrossRef]
- Green, H. Terminal differentiation of cultured human epidermal cells. Cell 1977, 11, 405–416. [Google Scholar] [CrossRef]
- Eckert, R.L.; Green, H. Structure and evolution of the human involucrin gene. Cell 1986, 46, 583–589. [Google Scholar] [CrossRef]
- Mehrel, T.; Hohl, D.; Rothnagel, J.A.; Longley, M.A.; Bundman, D.; Cheng, C.; Lichti, U.; Bisher, M.E.; Steven, A.C.; Steinert, P.M.; et al. Identification of a major keratinocyte cell envelope protein, loricrin. Cell 1990, 61, 1103–1112. [Google Scholar] [CrossRef]
- Hohl, D.; Mehrel, T.; Lichti, U.; Turner, M.L.; Roop, D.R.; Steinert, P.M. Characterization of human loricrin. Structure and function of a new class of epidermal cell envelope proteins. J. Biol. Chem. 1991, 266, 6626–6636. [Google Scholar]
- Hohl, D.; Ruf Olano, B.; de Viragh, P.A.; Huber, M.; Detrisac, C.J.; Schnyder, U.W.; Roop, D.R. Expression patterns of loricrin in various species and tissues. Differentiation 1993, 54, 25–34. [Google Scholar] [CrossRef]
- Steinert, P.M.; Cantieri, J.S.; Teller, D.C.; Lonsdale-Eccles, J.D.; Dale, B.A. Characterization of a class of cationic proteins that specifically interact with intermediate filaments. Proc. Natl. Acad. Sci. USA 1981, 78, 4097–4101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dale, B.A.; Holbrook, K.A.; Steinert, P.M. Assembly of stratum corneum basic protein and keratin filaments in macrofibrils. Nature 1978, 276, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Steinert, P.M.; Marekov, L.N. The proteins elafin, filaggrin, keratin intermediate filaments, loricrin, and small proline-rich proteins 1 and 2 are isodipeptide cross-linked components of the human epidermal cornified cell envelope. J. Biol. Chem. 1995, 270, 17702–17711. [Google Scholar] [CrossRef] [Green Version]
- Michel, S.; Schmidt, R.; Robinson, S.M.; Shroot, B.; Reichert, U. Identification and subcellular distribution of cornified envelope precursor proteins in the transformed human keratinocyte line SV-K14. J. Investig. Dermatol. 1987, 88, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Nemes, Z.; Marekov, L.N.; Fesus, L.; Steinert, P.M. A novel function for transglutaminase 1: Attachment of long-chain omega-hydroxyceramides to involucrin by ester bond formation. Proc. Natl. Acad. Sci. USA 1999, 96, 8402–8407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuramoto, N.; Takizawa, T.; Takizawa, T.; Matsuki, M.; Morioka, H.; Robinson, J.M.; Yamanishi, K. Development of ichthyosiform skin compensates for defective permeability barrier function in mice lacking transglutaminase 1. J. Clin. Investig. 2002, 109, 243–250. [Google Scholar] [CrossRef]
- Sevilla, L.M.; Nachat, R.; Groot, K.R.; Klement, J.F.; Uitto, J.; Djian, P.; Maatta, A.; Watt, F.M. Mice deficient in involucrin, envoplakin, and periplakin have a defective epidermal barrier. J. Cell Biol. 2007, 179, 1599–1612. [Google Scholar] [CrossRef] [Green Version]
- Maestrini, E.; Monaco, A.P.; McGrath, J.A.; Ishida-Yamamoto, A.; Camisa, C.; Hovnanian, A.; Weeks, D.E.; Lathrop, M.; Uitto, J.; Christiano, A.M. A molecular defect in loricrin, the major component of the cornified cell envelope, underlies Vohwinkel’s syndrome. Nat. Genet. 1996, 13, 70–77. [Google Scholar] [CrossRef]
- Ishida-Yamamoto, A.; McGrath, J.A.; Lam, H.; Iizuka, H.; Friedman, R.A.; Christiano, A.M. The molecular pathology of progressive symmetric erythrokeratoderma: A frameshift mutation in the loricrin gene and perturbations in the cornified cell envelope. Am. J. Hum. Genet. 1997, 61, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Steven, A.C.; Bisher, M.E.; Roop, D.R.; Steinert, P.M. Biosynthetic pathways of filaggrin and loricrin—Two major proteins expressed by terminally differentiated epidermal keratinocytes. J. Struct. Biol. 1990, 104, 150–162. [Google Scholar] [CrossRef]
- Suga, Y.; Jarnik, M.; Attar, P.S.; Longley, M.A.; Bundman, D.; Steven, A.C.; Koch, P.J.; Roop, D.R. Transgenic mice expressing a mutant form of loricrin reveal the molecular basis of the skin diseases, Vohwinkel syndrome and progressive symmetric erythrokeratoderma. J. Cell Biol. 2000, 151, 401–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardman, M.J.; Sisi, P.; Banbury, D.N.; Byrne, C. Patterned acquisition of skin barrier function during development. Development 1998, 125, 1541–1552. [Google Scholar] [PubMed]
- Bickenbach, J.R.; Greer, J.M.; Bundman, D.S.; Rothnagel, J.A.; Roop, D.R. Loricrin expression is coordinated with other epidermal proteins and the appearance of lipid lamellar granules in development. J. Investig. Dermatol. 1995, 104, 405–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, C.R.; Aho, S.; Bosko, C.A. Filaggrin—Revisited. Int. J. Cosmet. Sci. 2013, 35, 412–423. [Google Scholar] [CrossRef]
- Steinert, P.M. The complexity and redundancy of epithelial barrier function. J. Cell Biol. 2000, 151, F5–F8. [Google Scholar] [CrossRef]
- Gibbs, S.; Fijneman, R.; Wiegant, J.; van Kessel, A.G.; van De Putte, P.; Backendorf, C. Molecular characterization and evolution of the SPRR family of keratinocyte differentiation markers encoding small proline-rich proteins. Genomics 1993, 16, 630–637. [Google Scholar] [CrossRef]
- Jackson, B.; Tilli, C.M.; Hardman, M.J.; Avilion, A.A.; MacLeod, M.C.; Ashcroft, G.S.; Byrne, C. Late cornified envelope family in differentiating epithelia—Response to calcium and ultraviolet irradiation. J. Investig. Dermatol. 2005, 124, 1062–1070. [Google Scholar] [CrossRef]
- Vermeij, W.P.; Backendorf, C. Skin cornification proteins provide global link between ROS detoxification and cell migration during wound healing. PLoS ONE 2010, 5, e11957. [Google Scholar] [CrossRef] [Green Version]
- de Cid, R.; Riveira-Munoz, E.; Zeeuwen, P.L.; Robarge, J.; Liao, W.; Dannhauser, E.N.; Giardina, E.; Stuart, P.E.; Nair, R.; Helms, C.; et al. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat. Genet. 2009, 41, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Vermeij, W.P.; Alia, A.; Backendorf, C. ROS quenching potential of the epidermal cornified cell envelope. J. Investig. Dermatol. 2011, 131, 1435–1441. [Google Scholar] [CrossRef] [Green Version]
- Steinert, P.M.; Candi, E.; Kartasova, T.; Marekov, L. Small proline-rich proteins are cross-bridging proteins in the cornified cell envelopes of stratified squamous epithelia. J. Struct. Biol. 1998, 122, 76–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, M.P. The structure and function of proline-rich regions in proteins. Biochem. J. 1994, 297 Pt 2, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Basmanav, F.B.U.; Cau, L.; Tafazzoli, A.; Mechin, M.C.; Wolf, S.; Romano, M.T.; Valentin, F.; Wiegmann, H.; Huchenq, A.; Kandil, R.; et al. Mutations in Three Genes Encoding Proteins Involved in Hair Shaft Formation Cause Uncombable Hair Syndrome. Am. J. Hum. Genet. 2016, 99, 1292–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candi, E.; Melino, G.; Mei, G.; Tarcsa, E.; Chung, S.I.; Marekov, L.N.; Steinert, P.M. Biochemical, structural, and transglutaminase substrate properties of human loricrin, the major epidermal cornified cell envelope protein. J. Biol. Chem. 1995, 270, 26382–26390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, R.H.; Bradshaw, K.M.; Durbin-Johnson, B.P.; Rocke, D.M.; Eigenheer, R.A.; Phinney, B.S.; Schmuth, M.; Gruber, R. Distinguishing ichthyoses by protein profiling. PLoS ONE 2013, 8, e75355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, R.H.; Durbin-Johnson, B.P.; Ishitsuka, Y.; Salemi, M.; Phinney, B.S.; Rocke, D.M.; Roop, D.R. Proteomic Analysis of Loricrin Knockout Mouse Epidermis. J. Proteome Res. 2016, 15, 2560–2566. [Google Scholar] [CrossRef] [PubMed]
- Ishitsuka, Y.; Roop, D.R. Loricrin Confers Photoprotective Function against UVB in Corneocytes. J. Investig. Dermatol. 2018, 138, 2684–2687. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.I.; Kobayashi, A.; Wakabayashi, N.; Kim, S.G.; Yamamoto, M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2046–2051. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.C.; Tattersall, D.; Norgett, E.E.; O’Toole, E.A.; Kelsell, D.P. Premature terminal differentiation and a reduction in specific proteases associated with loss of ABCA12 in Harlequin ichthyosis. Am. J. Pathol. 2009, 174, 970–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, Y.; Akiyama, M.; Sugiyama-Nakagiri, Y.; Sakai, K.; Goto, M.; McMillan, J.R.; Ota, M.; Sawamura, D.; Shimizu, H. Expression of the keratinocyte lipid transporter ABCA12 in developing and reconstituted human epidermis. Am. J. Pathol. 2007, 171, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.; Farwanah, H.; Willrodt, A.H.; Huebner, A.J.; Sandhoff, K.; Roop, D.; Hohl, D.; Bloch, W.; Werner, S. Nrf2 links epidermal barrier function with antioxidant defense. EMBO Mol. Med. 2012, 4, 364–379. [Google Scholar] [CrossRef]
- Schafer, M.; Dutsch, S.; auf dem Keller, U.; Navid, F.; Schwarz, A.; Johnson, D.A.; Johnson, J.A.; Werner, S. Nrf2 establishes a glutathione-mediated gradient of UVB cytoprotection in the epidermis. Genes Dev. 2010, 24, 1045–1058. [Google Scholar] [CrossRef] [Green Version]
- Kawachi, Y.; Xu, X.; Taguchi, S.; Sakurai, H.; Nakamura, Y.; Ishii, Y.; Fujisawa, Y.; Furuta, J.; Takahashi, T.; Itoh, K.; et al. Attenuation of UVB-induced sunburn reaction and oxidative DNA damage with no alterations in UVB-induced skin carcinogenesis in Nrf2 gene-deficient mice. J. Investig. Dermatol. 2008, 128, 1773–1779. [Google Scholar] [CrossRef] [Green Version]
- Magin, T.M. A keaper and a striker maintain epidermal homeostasis. Nat. Genet. 2003, 35, 202–204. [Google Scholar] [CrossRef]
- Johnson, D.A.; Andrews, G.K.; Xu, W.; Johnson, J.A. Activation of the antioxidant response element in primary cortical neuronal cultures derived from transgenic reporter mice. J. Neurochem. 2002, 81, 1233–1241. [Google Scholar] [CrossRef]
- Auf dem Keller, U.; Huber, M.; Beyer, T.A.; Kumin, A.; Siemes, C.; Braun, S.; Bugnon, P.; Mitropoulos, V.; Johnson, D.A.; Johnson, J.A.; et al. Nrf transcription factors in keratinocytes are essential for skin tumor prevention but not for wound healing. Mol. Cell. Biol. 2006, 26, 3773–3784. [Google Scholar] [CrossRef] [Green Version]
- Imakado, S.; Bickenbach, J.R.; Bundman, D.S.; Rothnagel, J.A.; Attar, P.S.; Wang, X.J.; Walczak, V.R.; Wisniewski, S.; Pote, J.; Gordon, J.S.; et al. Targeting expression of a dominant-negative retinoic acid receptor mutant in the epidermis of transgenic mice results in loss of barrier function. Genes Dev. 1995, 9, 317–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attar, P.S.; Wertz, P.W.; McArthur, M.; Imakado, S.; Bickenbach, J.R.; Roop, D.R. Inhibition of retinoid signaling in transgenic mice alters lipid processing and disrupts epidermal barrier function. Mol. Endocrinol. 1997, 11, 792–800. [Google Scholar] [CrossRef]
- Kumar, S.; Sandell, L.L.; Trainor, P.A.; Koentgen, F.; Duester, G. Alcohol and aldehyde dehydrogenases: Retinoid metabolic effects in mouse knockout models. Biochim. Biophys. Acta 2012, 1821, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Havran, W.L.; Chien, Y.H.; Allison, J.P. Recognition of self antigens by skin-derived T cells with invariant gamma delta antigen receptors. Science 1991, 252, 1430–1432. [Google Scholar] [CrossRef] [PubMed]
- Jameson, J.; Ugarte, K.; Chen, N.; Yachi, P.; Fuchs, E.; Boismenu, R.; Havran, W.L. A role for skin gammadelta T cells in wound repair. Science 2002, 296, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Girardi, M.; Oppenheim, D.E.; Steele, C.R.; Lewis, J.M.; Glusac, E.; Filler, R.; Hobby, P.; Sutton, B.; Tigelaar, R.E.; Hayday, A.C. Regulation of cutaneous malignancy by gammadelta T cells. Science 2001, 294, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, K.; Witherden, D.A.; Havran, W.L. All hands on DE(T)C: Epithelial-resident gammadelta T cells respond to tissue injury. Cell. Immunol. 2015, 296, 57–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez de Aguero, M.; Vocanson, M.; Hacini-Rachinel, F.; Taillardet, M.; Sparwasser, T.; Kissenpfennig, A.; Malissen, B.; Kaiserlian, D.; Dubois, B. Langerhans cells protect from allergic contact dermatitis in mice by tolerizing CD8(+) T cells and activating Foxp3(+) regulatory T cells. J. Clin. Investig. 2012, 122, 1700–1711. [Google Scholar] [CrossRef]
- Rosenblum, M.D.; Gratz, I.K.; Paw, J.S.; Lee, K.; Marshak-Rothstein, A.; Abbas, A.K. Response to self antigen imprints regulatory memory in tissues. Nature 2011, 480, 538–542. [Google Scholar] [CrossRef]
- Ali, N.; Zirak, B.; Rodriguez, R.S.; Pauli, M.L.; Truong, H.A.; Lai, K.; Ahn, R.; Corbin, K.; Lowe, M.M.; Scharschmidt, T.C.; et al. Regulatory T Cells in Skin Facilitate Epithelial Stem Cell Differentiation. Cell 2017, 169, 1119–1129. [Google Scholar] [CrossRef] [Green Version]
- Scharschmidt, T.C.; Vasquez, K.S.; Truong, H.A.; Gearty, S.V.; Pauli, M.L.; Nosbaum, A.; Gratz, I.K.; Otto, M.; Moon, J.J.; Liese, J.; et al. A Wave of Regulatory T Cells into Neonatal Skin Mediates Tolerance to Commensal Microbes. Immunity 2015, 43, 1011–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, O.J.; Linehan, J.L.; Shih, H.Y.; Bouladoux, N.; Han, S.J.; Smelkinson, M.; Sen, S.K.; Byrd, A.L.; Enamorado, M.; Yao, C.; et al. Commensal-specific T cell plasticity promotes rapid tissue adaptation to injury. Science 2019, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukhari, S.; Mertz, A.F.; Naik, S. Eavesdropping on the conversation between immune cells and the skin epithelium. Int. Immunol. 2019, 31, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Jarnik, M.; de Viragh, P.A.; Scharer, E.; Bundman, D.; Simon, M.N.; Roop, D.R.; Steven, A.C. Quasi-normal cornified cell envelopes in loricrin knockout mice imply the existence of a loricrin backup system. J. Investig. Dermatol. 2002, 118, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Eyerich, K.; Brown, S.J.; Perez White, B.E.; Tanaka, R.J.; Bissonette, R.; Dhar, S.; Bieber, T.; Hijnen, D.J.; Guttman-Yassky, E.; Irvine, A.; et al. Human and computational models of atopic dermatitis: A review and perspectives by an expert panel of the International Eczema Council. J. Allergy Clin. Immunol. 2019, 143, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Smith, F.J.; Irvine, A.D.; Terron-Kwiatkowski, A.; Sandilands, A.; Campbell, L.E.; Zhao, Y.; Liao, H.; Evans, A.T.; Goudie, D.R.; Lewis-Jones, S.; et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat. Genet. 2006, 38, 337–342. [Google Scholar] [CrossRef]
- Palmer, C.N.; Irvine, A.D.; Terron-Kwiatkowski, A.; Zhao, Y.; Liao, H.; Lee, S.P.; Goudie, D.R.; Sandilands, A.; Campbell, L.E.; Smith, F.J.; et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 2006, 38, 441–446. [Google Scholar] [CrossRef]
- Paller, A.S.; Spergel, J.M.; Mina-Osorio, P.; Irvine, A.D. The atopic march and atopic multimorbidity: Many trajectories, many pathways. J. Allergy Clin. Immunol. 2019, 143, 46–55. [Google Scholar] [CrossRef]
- Lack, G.; Fox, D.; Northstone, K.; Golding, J. Factors associated with the development of peanut allergy in childhood. N. Engl. J. Med. 2003, 348, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Weidinger, S.; O’Sullivan, M.; Illig, T.; Baurecht, H.; Depner, M.; Rodriguez, E.; Ruether, A.; Klopp, N.; Vogelberg, C.; Weiland, S.K.; et al. Filaggrin mutations, atopic eczema, hay fever, and asthma in children. J. Allergy Clin. Immunol. 2008, 121, 1203–1209. [Google Scholar] [CrossRef]
- Venkataraman, D.; Soto-Ramirez, N.; Kurukulaaratchy, R.J.; Holloway, J.W.; Karmaus, W.; Ewart, S.L.; Arshad, S.H.; Erlewyn-Lajeunesse, M. Filaggrin loss-of-function mutations are associated with food allergy in childhood and adolescence. J. Allergy Clin. Immunol. 2014, 134, 876–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. OMIM.org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019, 47, D1038–D1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irvine, A.D.; McLean, W.H. Breaking the (un)sound barrier: Filaggrin is a major gene for atopic dermatitis. J. Investig. Dermatol. 2006, 126, 1200–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, A.; Nagao, K.; Yokouchi, M.; Sasaki, H.; Amagai, M. External antigen uptake by Langerhans cells with reorganization of epidermal tight junction barriers. J. Exp. Med. 2009, 206, 2937–2946. [Google Scholar] [CrossRef] [Green Version]
- Lack, G. Update on risk factors for food allergy. J. Allergy Clin. Immunol. 2012, 129, 1187–1197. [Google Scholar] [CrossRef]
- Dupont, C.; Kalach, N.; Soulaines, P.; Legoue-Morillon, S.; Piloquet, H.; Benhamou, P.H. Cow’s milk epicutaneous immunotherapy in children: A pilot trial of safety, acceptability, and impact on allergic reactivity. J. Allergy Clin. Immunol. 2010, 125, 1165–1167. [Google Scholar] [CrossRef]
- Dioszeghy, V.; Mondoulet, L.; Dhelft, V.; Ligouis, M.; Puteaux, E.; Benhamou, P.H.; Dupont, C. Epicutaneous immunotherapy results in rapid allergen uptake by dendritic cells through intact skin and downregulates the allergen-specific response in sensitized mice. J. Immunol. 2011, 186, 5629–5637. [Google Scholar] [CrossRef]
- Senti, G.; von Moos, S.; Tay, F.; Graf, N.; Johansen, P.; Kundig, T.M. Determinants of efficacy and safety in epicutaneous allergen immunotherapy: Summary of three clinical trials. Allergy 2015, 70, 707–710. [Google Scholar] [CrossRef] [Green Version]
- Marrakchi, S.; Guigue, P.; Renshaw, B.R.; Puel, A.; Pei, X.Y.; Fraitag, S.; Zribi, J.; Bal, E.; Cluzeau, C.; Chrabieh, M.; et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 2011, 365, 620–628. [Google Scholar] [CrossRef]
- Tortola, L.; Rosenwald, E.; Abel, B.; Blumberg, H.; Schafer, M.; Coyle, A.J.; Renauld, J.C.; Werner, S.; Kisielow, J.; Kopf, M. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J. Clin. Investig. 2012, 122, 3965–3976. [Google Scholar] [CrossRef]
- Imai, Y.; Yasuda, K.; Sakaguchi, Y.; Haneda, T.; Mizutani, H.; Yoshimoto, T.; Nakanishi, K.; Yamanishi, K. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 13921–13926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.F.; Dudda, J.C.; Bachtanian, E.; Lembo, A.; Liller, S.; Durr, C.; Heimesaat, M.M.; Bereswill, S.; Fejer, G.; Vassileva, R.; et al. Toll-like receptor and IL-12 signaling control susceptibility to contact hypersensitivity. J. Exp. Med. 2008, 205, 2151–2162. [Google Scholar] [CrossRef] [PubMed]
- Natsuga, K.; Cipolat, S.; Watt, F.M. Increased Bacterial Load and Expression of Antimicrobial Peptides in Skin of Barrier-Deficient Mice with Reduced Cancer Susceptibility. J. Investig. Dermatol. 2016, 136, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lay, K.; Yuan, S.; Gur-Cohen, S.; Miao, Y.; Han, T.; Naik, S.; Pasolli, H.A.; Larsen, S.B.; Fuchs, E. Stem cells repurpose proliferation to contain a breach in their niche barrier. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Dainichi, T.; Kitoh, A.; Otsuka, A.; Nakajima, S.; Nomura, T.; Kaplan, D.H.; Kabashima, K. The epithelial immune microenvironment (EIME) in atopic dermatitis and psoriasis. Nat. Immunol. 2018, 19, 1286–1298. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Matzinger, P.; Kamala, T. Tissue-based class control: The other side of tolerance. Nat. Rev. Immunol. 2011, 11, 221–230. [Google Scholar] [CrossRef]
- Denecker, G.; Hoste, E.; Gilbert, B.; Hochepied, T.; Ovaere, P.; Lippens, S.; Van den Broecke, C.; Van Damme, P.; D’Herde, K.; Hachem, J.P.; et al. Caspase-14 protects against epidermal UVB photodamage and water loss. Nat. Cell Biol. 2007, 9, 666–674. [Google Scholar] [CrossRef]
- Lavker, R.M.; Matoltsy, A.G. Formation of horny cells: The fate of cell organelles and differentiation products in ruminal epithelium. J. Cell Biol. 1970, 44, 501–512. [Google Scholar] [CrossRef]
- Hoste, E.; Kemperman, P.; Devos, M.; Denecker, G.; Kezic, S.; Yau, N.; Gilbert, B.; Lippens, S.; De Groote, P.; Roelandt, R.; et al. Caspase-14 is required for filaggrin degradation to natural moisturizing factors in the skin. J. Investig. Dermatol. 2011, 131, 2233–2241. [Google Scholar] [CrossRef] [Green Version]
- Matsuki, M.; Yamashita, F.; Ishida-Yamamoto, A.; Yamada, K.; Kinoshita, C.; Fushiki, S.; Ueda, E.; Morishima, Y.; Tabata, K.; Yasuno, H.; et al. Defective stratum corneum and early neonatal death in mice lacking the gene for transglutaminase 1 (keratinocyte transglutaminase). Proc. Natl. Acad. Sci. USA 1998, 95, 1044–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicotera, P.; Melino, G. Caspase-14 and epidermis maturation. Nat. Cell Biol. 2007, 9, 621–622. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.; Kirfel, J.; Bussow, H.; Vidal, M.; Magin, T.M. Complete cytolysis and neonatal lethality in keratin 5 knockout mice reveal its fundamental role in skin integrity and in epidermolysis bullosa simplex. Mol. Biol. Cell 2001, 12, 1775–1789. [Google Scholar] [CrossRef] [Green Version]
- Wallace, L.; Roberts-Thompson, L.; Reichelt, J. Deletion of K1/K10 does not impair epidermal stratification but affects desmosomal structure and nuclear integrity. J. Cell Sci. 2012, 125, 1750–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, H.; Nagao, K.; Kubo, A.; Hata, T.; Shimizu, A.; Mizuno, H.; Yamada, T.; Amagai, M. Altered stratum corneum barrier and enhanced percutaneous immune responses in filaggrin-null mice. J. Allergy Clin. Immunol. 2012, 129, 1538–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, Y.; Zhuang, D.Z.; Han, R.; Isaac, G.; Tobin, J.J.; McKee, M.; Welti, R.; Brissette, J.L.; Fitzgerald, M.L.; Freeman, M.W. ABCA12 maintains the epidermal lipid permeability barrier by facilitating formation of ceramide linoleic esters. J. Biol. Chem. 2008, 283, 36624–36635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, I.; Hacking, D.F.; Hilton, A.A.; Mukhamedova, N.; Meikle, P.J.; Ellis, S.; Satterley, K.; Collinge, J.E.; de Graaf, C.A.; Bahlo, M.; et al. A mouse model of harlequin ichthyosis delineates a key role for Abca12 in lipid homeostasis. PLoS Genet. 2008, 4, e1000192. [Google Scholar] [CrossRef]
- Squier, C.A.; Kremer, M.J. Biology of oral mucosa and esophagus. J. Natl. Cancer Inst. Monogr. 2001, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Thompson, I.O.; van der Bijl, P.; van Wyk, C.W.; van Eyk, A.D. A comparative light-microscopic, electron-microscopic and chemical study of human vaginal and buccal epithelium. Arch. Oral Biol. 2001, 46, 1091–1098. [Google Scholar] [CrossRef]
- Smith, S.A.; Dale, B.A. Immunologic localization of filaggrin in human oral epithelia and correlation with keratinization. J. Investig. Dermatol. 1986, 86, 168–172. [Google Scholar] [CrossRef]
- Oh, J.W.; Chung, O.; Cho, Y.S.; MacGregor, G.R.; Plikus, M.V. Gene loss in keratinization programs accompanies adaptation of cetacean skin to aquatic lifestyle. Exp. Dermatol. 2015, 24, 572–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strasser, B.; Mlitz, V.; Fischer, H.; Tschachler, E.; Eckhart, L. Comparative genomics reveals conservation of filaggrin and loss of caspase-14 in dolphins. Exp. Dermatol. 2015, 24, 365–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spergel, J.M.; Mizoguchi, E.; Brewer, J.P.; Martin, T.R.; Bhan, A.K.; Geha, R.S. Epicutaneous sensitization with protein antigen induces localized allergic dermatitis and hyperresponsiveness to methacholine after single exposure to aerosolized antigen in mice. J. Clin. Investig. 1998, 101, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Goubier, A.; Dubois, B.; Gheit, H.; Joubert, G.; Villard-Truc, F.; Asselin-Paturel, C.; Trinchieri, G.; Kaiserlian, D. Plasmacytoid dendritic cells mediate oral tolerance. Immunity 2008, 29, 464–475. [Google Scholar] [CrossRef] [Green Version]
- Iijima, M.; Katz, S.I. Specific immunologic tolerance to dinitrofluorobenzene following topical application of dinitrothiocyanobenzene: Modulation by suppressor T cells. J. Investig. Dermatol. 1983, 81, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Pickard, C.; Louafi, F.; McGuire, C.; Lowings, K.; Kumar, P.; Cooper, H.; Dearman, R.J.; Cumberbatch, M.; Kimber, I.; Healy, E.; et al. The cutaneous biochemical redox barrier: A component of the innate immune defenses against sensitization by highly reactive environmental xenobiotics. J. Immunol. 2009, 183, 7576–7584. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Barajas, B.; Wang, M.; Nel, A.E. Nrf2 activation by sulforaphane restores the age-related decrease of T(H)1 immunity: Role of dendritic cells. J. Allergy Clin. Immunol. 2008, 121, 1255–1261. [Google Scholar] [CrossRef] [Green Version]
- Hovav, A.H. Dendritic cells of the oral mucosa. Mucosal Immunol. 2014, 7, 27–37. [Google Scholar] [CrossRef]
- Novak, N.; Haberstok, J.; Bieber, T.; Allam, J.P. The immune privilege of the oral mucosa. Trends Mol. Med. 2008, 14, 191–198. [Google Scholar] [CrossRef]
- Law, S.; Wertz, P.W.; Swartzendruber, D.C.; Squier, C.A. Regional variation in content, composition and organization of porcine epithelial barrier lipids revealed by thin-layer chromatography and transmission electron microscopy. Arch. Oral Biol. 1995, 40, 1085–1091. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Wesemann, D.R.; Nagler, C.R. The Microbiome, Timing, and Barrier Function in the Context of Allergic Disease. Immunity 2016, 44, 728–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, G.L.V.; Leite, A.Z.; Higuchi, B.S.; Gonzaga, M.I.; Mariano, V.S. Intestinal dysbiosis and probiotic applications in autoimmune diseases. Immunology 2017, 152, 1–12. [Google Scholar] [CrossRef]
- Chen, Y.E.; Fischbach, M.A.; Belkaid, Y. Skin microbiota-host interactions. Nature 2018, 553, 427–436. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Sadlack, B.; Merz, H.; Schorle, H.; Schimpl, A.; Feller, A.C.; Horak, I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 1993, 75, 253–261. [Google Scholar] [CrossRef]
- Kuhn, R.; Lohler, J.; Rennick, D.; Rajewsky, K.; Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef]
- Sellon, R.K.; Tonkonogy, S.; Schultz, M.; Dieleman, L.A.; Grenther, W.; Balish, E.; Rennick, D.M.; Sartor, R.B. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 1998, 66, 5224–5231. [Google Scholar] [CrossRef] [Green Version]
- Dianda, L.; Hanby, A.M.; Wright, N.A.; Sebesteny, A.; Hayday, A.C.; Owen, M.J. T cell receptor-alpha beta-deficient mice fail to develop colitis in the absence of a microbial environment. Am. J. Pathol. 1997, 150, 91–97. [Google Scholar]
- Strober, W.; Ehrhardt, R.O. Chronic intestinal inflammation: An unexpected outcome in cytokine or T cell receptor mutant mice. Cell 1993, 75, 203–205. [Google Scholar] [CrossRef]
- Geoghegan, J.A.; Irvine, A.D.; Foster, T.J. Staphylococcus aureus and Atopic Dermatitis: A Complex and Evolving Relationship. Trends Microbiol. 2018, 26, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Archer, N.K.; Dillen, C.A.; Wang, Y.; Ashbaugh, A.G.; Ortines, R.V.; Kao, T.; Lee, S.K.; Cai, S.S.; Miller, R.J.; et al. Staphylococcus aureus Epicutaneous Exposure Drives Skin Inflammation via IL-36-Mediated T Cell Responses. Cell Host Microbe 2017, 22, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Matsumoto, M.; Katayama, Y.; Oguma, R.; Wakabayashi, S.; Nygaard, T.; Saijo, S.; Inohara, N.; Otto, M.; Matsue, H.; et al. Staphylococcus aureus Virulent PSMalpha Peptides Induce Keratinocyte Alarmin Release to Orchestrate IL-17-Dependent Skin Inflammation. Cell Host Microbe 2017, 22, 667–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fay, J.C.; Wu, C.I. Hitchhiking under positive Darwinian selection. Genetics 2000, 155, 1405–1413. [Google Scholar] [PubMed]
- Lamont, R.J.; Hajishengallis, G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol. Med. 2015, 21, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; Granucci, F.; Kraehenbuhl, J.P.; Ricciardi-Castagnoli, P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001, 2, 361–367. [Google Scholar] [CrossRef]
- Hemmi, H.; Yoshino, M.; Yamazaki, H.; Naito, M.; Iyoda, T.; Omatsu, Y.; Shimoyama, S.; Letterio, J.J.; Nakabayashi, T.; Tagaya, H.; et al. Skin antigens in the steady state are trafficked to regional lymph nodes by transforming growth factor-beta1-dependent cells. Int. Immunol. 2001, 13, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Allan, R.S.; Waithman, J.; Bedoui, S.; Jones, C.M.; Villadangos, J.A.; Zhan, Y.; Lew, A.M.; Shortman, K.; Heath, W.R.; Carbone, F.R. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 2006, 25, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Henri, S.; Poulin, L.F.; Tamoutounour, S.; Ardouin, L.; Guilliams, M.; de Bovis, B.; Devilard, E.; Viret, C.; Azukizawa, H.; Kissenpfennig, A.; et al. CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J. Exp. Med. 2010, 207, 189–206. [Google Scholar] [CrossRef] [Green Version]
- Ohnmacht, C.; Pullner, A.; King, S.B.; Drexler, I.; Meier, S.; Brocker, T.; Voehringer, D. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J. Exp. Med. 2009, 206, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doebel, T.; Voisin, B.; Nagao, K. Langerhans Cells—The Macrophage in Dendritic Cell Clothing. Trends Immunol. 2017, 38, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Lucas, A.D. Protein kinase C transduces the signal for Langerhans’ cell migration from the epidermis. Immunology 1993, 79, 621–626. [Google Scholar]
- Kaplan, D.H.; Jenison, M.C.; Saeland, S.; Shlomchik, W.D.; Shlomchik, M.J. Epidermal langerhans cell-deficient mice develop enhanced contact hypersensitivity. Immunity 2005, 23, 611–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, T.; Honda, T.; Egawa, G.; Yamamoto, Y.; Ichijo, R.; Toyoshima, F.; Dainichi, T.; Kabashima, K. Transient elevation of cytoplasmic calcium ion concentration at a single cell level precedes morphological changes of epidermal keratinocytes during cornification. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yokouchi, M.; Atsugi, T.; Logtestijn, M.V.; Tanaka, R.J.; Kajimura, M.; Suematsu, M.; Furuse, M.; Amagai, M.; Kubo, A. Epidermal cell turnover across tight junctions based on Kelvin’s tetrakaidecahedron cell shape. eLife 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, H.; Taneda, A.; Kanaoka, Y.; Sekine, T. The histochemical distribution of protein bound sulfhydryl groups in human epidermis by the new staining method. J. Histochem. Cytochem. 1979, 27, 942–946. [Google Scholar] [CrossRef]
- Vahlquist, A.; Fischer, J.; Torma, H. Inherited Nonsyndromic Ichthyoses: An Update on Pathophysiology, Diagnosis and Treatment. Am. J. Clin. Dermatol. 2018, 19, 51–66. [Google Scholar] [CrossRef]
- Blume-Peytavi, U.; Fowler, J.; Kemeny, L.; Draelos, Z.; Cook-Bolden, F.; Dirschka, T.; Eichenfield, L.; Graeber, M.; Ahmad, F.; Alio Saenz, A.; et al. Long-term safety and efficacy of trifarotene 50 mug/g cream, a first-in-class RAR-gamma selective topical retinoid, in patients with moderate facial and truncal acne. J. Eur. Acad. Dermatol. Venereol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Liang, F.X.; Bosland, M.C.; Huang, H.; Romih, R.; Baptiste, S.; Deng, F.M.; Wu, X.R.; Shapiro, E.; Sun, T.T. Cellular basis of urothelial squamous metaplasia: Roles of lineage heterogeneity and cell replacement. J. Cell Biol. 2005, 171, 835–844. [Google Scholar] [CrossRef] [Green Version]
- Elias, P.M.; Friend, D.S. Vitamin-A-induced mucous metaplasia. An in vitro system for modulating tight and gap junction differentiation. J. Cell Biol. 1976, 68, 173–188. [Google Scholar] [CrossRef]
- Ellis, C.N.; Gold, R.C.; Grekin, R.C.; Anderson, T.F.; Swanson, N.A.; Voorhees, J.J. Etretinate therapy stimulates deposition of mucus-like material in epidermis of patients with psoriasis. J. Am. Acad. Dermatol. 1982, 6, 699–704. [Google Scholar] [CrossRef]
- Hohl, D.; Lichti, U.; Breitkreutz, D.; Steinert, P.M.; Roop, D.R. Transcription of the human loricrin gene in vitro is induced by calcium and cell density and suppressed by retinoic acid. J. Investig. Dermatol. 1991, 96, 414–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, L.J.; Geesin, J.C.; Rothnagel, J.A.; Roop, D.R.; Gordon, J.S. Retinoic acid suppression of loricrin expression in reconstituted human skin cultured at the liquid-air interface. J. Investig. Dermatol. 1994, 102, 886–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnaldo, T.; Bernerd, F.; Asselineau, D.; Darmon, M. Expression of loricrin is negatively controlled by retinoic acid in human epidermis reconstructed in vitro. Differentiation 1992, 49, 39–46. [Google Scholar] [CrossRef]
- Iwata, M.; Hirakiyama, A.; Eshima, Y.; Kagechika, H.; Kato, C.; Song, S.Y. Retinoic acid imprints gut-homing specificity on T cells. Immunity 2004, 21, 527–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkelens, M.N.; Mebius, R.E. Retinoic Acid and Immune Homeostasis: A Balancing Act. Trends Immunol. 2017, 38, 168–180. [Google Scholar] [CrossRef]
- Agace, W.W.; Persson, E.K. How vitamin A metabolizing dendritic cells are generated in the gut mucosa. Trends Immunol. 2012, 33, 42–48. [Google Scholar] [CrossRef]
- Koenig, U.; Amatschek, S.; Mildner, M.; Eckhart, L.; Tschachler, E. Aldehyde dehydrogenase 1A3 is transcriptionally activated by all-trans-retinoic acid in human epidermal keratinocytes. Biochem. Biophys. Res. Commun. 2010, 400, 207–211. [Google Scholar] [CrossRef]
- Pavez Lorie, E.; Li, H.; Vahlquist, A.; Torma, H. The involvement of cytochrome p450 (CYP) 26 in the retinoic acid metabolism of human epidermal keratinocytes. Biochim. Biophys. Acta 2009, 1791, 220–228. [Google Scholar] [CrossRef]
- Hodam, J.R.; Creek, K.E. Comparison of the metabolism of retinol delivered to human keratinocytes either bound to serum retinol-binding protein or added directly to the culture medium. Exp. Cell Res. 1998, 238, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Randolph, R.K.; Simon, M. Characterization of retinol metabolism in cultured human epidermal keratinocytes. J. Biol. Chem. 1993, 268, 9198–9205. [Google Scholar] [PubMed]
- Sigmundsdottir, H.; Butcher, E.C. Environmental cues, dendritic cells and the programming of tissue-selective lymphocyte trafficking. Nat. Immunol. 2008, 9, 981–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigmundsdottir, H.; Pan, J.; Debes, G.F.; Alt, C.; Habtezion, A.; Soler, D.; Butcher, E.C. DCs metabolize sunlight-induced vitamin D3 to ‘program’ T cell attraction to the epidermal chemokine CCL27. Nat. Immunol. 2007, 8, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Indra, A.K.; Warot, X.; Brocard, J.; Messaddeq, N.; Kato, S.; Metzger, D.; Chambon, P. Skin abnormalities generated by temporally controlled RXRalpha mutations in mouse epidermis. Nature 2000, 407, 633–636. [Google Scholar] [CrossRef]
- Li, M.; Messaddeq, N.; Teletin, M.; Pasquali, J.L.; Metzger, D.; Chambon, P. Retinoid X receptor ablation in adult mouse keratinocytes generates an atopic dermatitis triggered by thymic stromal lymphopoietin. Proc. Natl. Acad. Sci. USA 2005, 102, 14795–14800. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Hener, P.; Zhang, Z.; Kato, S.; Metzger, D.; Chambon, P. Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc. Natl. Acad. Sci. USA 2006, 103, 11736–11741. [Google Scholar] [CrossRef] [Green Version]
- Ruzicka, T.; Larsen, F.G.; Galewicz, D.; Horvath, A.; Coenraads, P.J.; Thestrup-Pedersen, K.; Ortonne, J.P.; Zouboulis, C.C.; Harsch, M.; Brown, T.C.; et al. Oral alitretinoin (9-cis-retinoic acid) therapy for chronic hand dermatitis in patients refractory to standard therapy: Results of a randomized, double-blind, placebo-controlled, multicenter trial. Arch. Dermatol. 2004, 140, 1453–1459. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Komuves, L.; Yu, Q.C.; Elalieh, H.; Ng, D.C.; Leary, C.; Chang, S.; Crumrine, D.; Yoshizawa, T.; Kato, S.; et al. Lack of the vitamin D receptor is associated with reduced epidermal differentiation and hair follicle growth. J. Investig. Dermatol. 2002, 118, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, T.; Ishitsuka, Y.; Roop, D.; Fujimoto, M. 314 Loricrin protects against chemical carcinogenesis but affects cancer immunoediting. J. Investig. Dermatol. 2019, 139, S54. [Google Scholar] [CrossRef] [Green Version]
- Kehren, J.; Desvignes, C.; Krasteva, M.; Ducluzeau, M.T.; Assossou, O.; Horand, F.; Hahne, M.; Kagi, D.; Kaiserlian, D.; Nicolas, J.F. Cytotoxicity is mandatory for CD8(+) T cell-mediated contact hypersensitivity. J. Exp. Med. 1999, 189, 779–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez-Farinas, M.; Tintle, S.J.; Shemer, A.; Chiricozzi, A.; Nograles, K.; Cardinale, I.; Duan, S.; Bowcock, A.M.; Krueger, J.G.; Guttman-Yassky, E. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J. Allergy Clin. Immunol. 2011, 127, 954–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, P.Y.; Leung, D.Y. Bacterial and Viral Infections in Atopic Dermatitis: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2016, 51, 329–337. [Google Scholar] [CrossRef]
- Griffith, T.S.; Brunner, T.; Fletcher, S.M.; Green, D.R.; Ferguson, T.A. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science 1995, 270, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Bellgrau, D.; Gold, D.; Selawry, H.; Moore, J.; Franzusoff, A.; Duke, R.C. A role for CD95 ligand in preventing graft rejection. Nature 1995, 377, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Hill, L.L.; Ouhtit, A.; Loughlin, S.M.; Kripke, M.L.; Ananthaswamy, H.N.; Owen-Schaub, L.B. Fas ligand: A sensor for DNA damage critical in skin cancer etiology. Science 1999, 285, 898–900. [Google Scholar] [CrossRef]
- Pinkus, H.; Mehregan, A.H. The primary histologic lesion of seborrheic dermatitis and psoriasis. J. Investig. Dermatol. 1966, 46, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Ishida-Yamamoto, A.; Eady, R.A.; Watt, F.M.; Roop, D.R.; Hohl, D.; Iizuka, H. Immunoelectron microscopic analysis of cornified cell envelope formation in normal and psoriatic epidermis. J. Histochem. Cytochem. 1996, 44, 167–175. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, B.S.; Kastelein, R.A.; Cua, D.J. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 2006, 27, 17–23. [Google Scholar] [CrossRef]
- Geissmann, F.; Dieu-Nosjean, M.C.; Dezutter, C.; Valladeau, J.; Kayal, S.; Leborgne, M.; Brousse, N.; Saeland, S.; Davoust, J. Accumulation of immature Langerhans cells in human lymph nodes draining chronically inflamed skin. J. Exp. Med. 2002, 196, 417–430. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Gomez, A.; Perretti, M.; Soehnlein, O. Resolution of inflammation: An integrated view. EMBO Mol. Med. 2013, 5, 661–674. [Google Scholar] [CrossRef]
- Verhasselt, V.; Vanden Berghe, W.; Vanderheyde, N.; Willems, F.; Haegeman, G.; Goldman, M. N-acetyl-L-cysteine inhibits primary human T cell responses at the dendritic cell level: Association with NF-kappaB inhibition. J. Immunol. 1999, 162, 2569–2574. [Google Scholar] [PubMed]
- Kim, H.J.; Barajas, B.; Chan, R.C.; Nel, A.E. Glutathione depletion inhibits dendritic cell maturation and delayed-type hypersensitivity: Implications for systemic disease and immunosenescence. J. Allergy Clin. Immunol. 2007, 119, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Al-Huseini, L.M.; Aw Yeang, H.X.; Sethu, S.; Alhumeed, N.; Hamdam, J.M.; Tingle, Y.; Djouhri, L.; Kitteringham, N.; Park, B.K.; Goldring, C.E.; et al. Nuclear factor-erythroid 2 (NF-E2) p45-related factor-2 (Nrf2) modulates dendritic cell immune function through regulation of p38 MAPK-cAMP-responsive element binding protein/activating transcription factor 1 signaling. J. Biol. Chem. 2013, 288, 22281–22288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, T.; Maeda, S.; Yamane, S.; Ogino, K.; Kasai, M.; Kajiura, F.; Matsumoto, M.; Inoue, J. Dependence of self-tolerance on TRAF6-directed development of thymic stroma. Science 2005, 308, 248–251. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Origin | Involucrin | Loricrin | |
|---|---|---|---|
| Hair follicle | Interfollicular | + | + |
| Infundibulm | + | + | |
| Sebaceous gland | + | − | |
| Inner root sheath | + | − | |
| Hair shaft | + | − | |
| Oral/throat | Hard plate | + | + |
| Buccal | + | − | |
| Phalynx | + | − | |
| Upper digestive tract | Esophagus | + | − |
| Stomach | − | − | |
| Duodenum | − | − | |
| Lower digestive tract | Colon | − | − |
| Anus | + | − | |
| Genital | Foreskin | + | + |
| Cervix | + | − | |
| Vagina | + | ± | |
| Eye | Conjunctiva | + | − |
| Lymphoid organ | Thyms (mTEC *) | + | − |
| Lymph nodes | − | − | |
| Spleen | − | − | |
| Solid organ | Liver | − | − |
| Pancreas | − | − | |
| Kidney | − | − | |
| Urothelium | Urinary bladder | + | − |
| Endocrine organ | Thyroid | − | − |
| Adrenal gland | − | − | |
| Target | Primary Defect | Remarkable Feature | Reference PMID | ||
|---|---|---|---|---|---|
| Permeability Barrier | CEs | ||||
| Gene Knockout | |||||
| Cornified cell envelope (CE) components | Envoplakin (EPL) | − | − | Proportionally increased immature CEs | 11564887 |
| Periplakin (PPL) | − | − | N/A *1 | 15226441 | |
| Involucrin (IVL) | − | − | N/A | 11038184 | |
| EPL/PPL/IVL | + | + | Atopy, Resistant against chemically induced cutaneous carcinogenesis | 18166659 24843010 | |
| Loricrin | − | + | Delayed acquisition of permeability barrier, Compensatory response, ultraviolet B susceptible | 11038185 23237955 27167730 29932941 | |
| Filaggrin | − | − | Ichthyotic phenotype | 22409988 | |
| Caspase 14 | + | − *2 | Ultraviolet B susceptible, decreased urocanic acids, and free amino acids | 17515931 21654840 | |
| Transglutaminase (TG) | TG1 | + | + | Neonatal lethality | 9448282 |
| TG3 | − | − | Impaired hair development | 22496784 | |
| Lipid transport | ABCA12 | + | − | Neonatal lethality | 18957418 18802465 27551807 |
| TMEM79 | − | N/A | Mast cell-mediated histamigenic itch | 30463955 | |
| Transgenic overexpression | |||||
| CE components | Involucrin | N/A | N/A | Alopecia, scaly epidermis | 8405770 |
| Mutant loricrin | + | N/A | Mimicking Vohwinkel syndrome | 11038186 | |
| Human loricrin | N/A | N/A | Normal phenotype | 8248167 | |
| Filaggrin | N/A | N/A | Enhanced TEWL *3 recovery | 15304104 | |
| Target | Susceptibility/Challenge | Remarkable Feature | Reference PMID |
|---|---|---|---|
| Gene Knockout | |||
| Nuclear factor erythroid 2-related factor 2 (Nrf2) | Ultraviolet B | Impaired cytoprotection | 18200051 |
| Chemically induced cutaneous carcinogenesis | 11248092 | ||
| Topical imiquimod | Dysregulated innate immunity | 31953037 | |
| Contact hypersensitivity | Outcomes may depend on contexts. | 18325578 | |
| Kelch-like erythroid cell-derived protein with cap´n´collar homology-associated protein 1 (Keap1) | N/A * | Hyperorthokeratosis with loricrin overexpression and postnatal lethality | 14517554 |
| Transgenic Overexpression: Dominant-Negative NRF2 | |||
| Basal keratinocytes (K14 promoter-driven) | Chemically induced cutaneous carcinogenesis | Wound healing is not affected | 16648473 |
| Stratum granulosum keratinocytes (Loricrin promoter-driven) | Epidermal barrier recovery | Loricrin-knockout background can lead to postnatal lethality | 23237955 |
| Transgenic Overexpression: Constitutively Active NRF2 | |||
| Basal keratinocytes (K5 (or K10) promoter-driven) | ultraviolet B (resistant) | Phenotypes resemble autosomal recessive congenital ichthyosis or chloacne | 20478997 22383093 24503019 |
| Target/Gene Targeting Strategy | Remarkable Feature | Reference PMID |
|---|---|---|
| Retinoic acid receptor α (RARα) (Dominant-negative, K1 promoter driven) | Defective permeability barrier and premature cornification that resembles vitamin A deficiency | 7867929 |
| 9-cis retinoic acid receptor α (RXRα) (K10 promoter driven) | Attenuated response to topical all-trans retinoic acid (ATRA) | 9000050 |
| RXRα-conditional knockout (Tamoxifen-inducible K14-Cre-driven) | Vitamin D receptor-dependent atopic dermatitis | 16199515 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishitsuka, Y.; Roop, D.R. Loricrin: Past, Present, and Future. Int. J. Mol. Sci. 2020, 21, 2271. https://doi.org/10.3390/ijms21072271
Ishitsuka Y, Roop DR. Loricrin: Past, Present, and Future. International Journal of Molecular Sciences. 2020; 21(7):2271. https://doi.org/10.3390/ijms21072271
Chicago/Turabian StyleIshitsuka, Yosuke, and Dennis R. Roop. 2020. "Loricrin: Past, Present, and Future" International Journal of Molecular Sciences 21, no. 7: 2271. https://doi.org/10.3390/ijms21072271
APA StyleIshitsuka, Y., & Roop, D. R. (2020). Loricrin: Past, Present, and Future. International Journal of Molecular Sciences, 21(7), 2271. https://doi.org/10.3390/ijms21072271