Regenerative Potential of Carbon Monoxide in Adult Neural Circuits of the Central Nervous System



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Overview of Repairing the Damaged Brain and Heme Oxygenase (HO) Metabolites
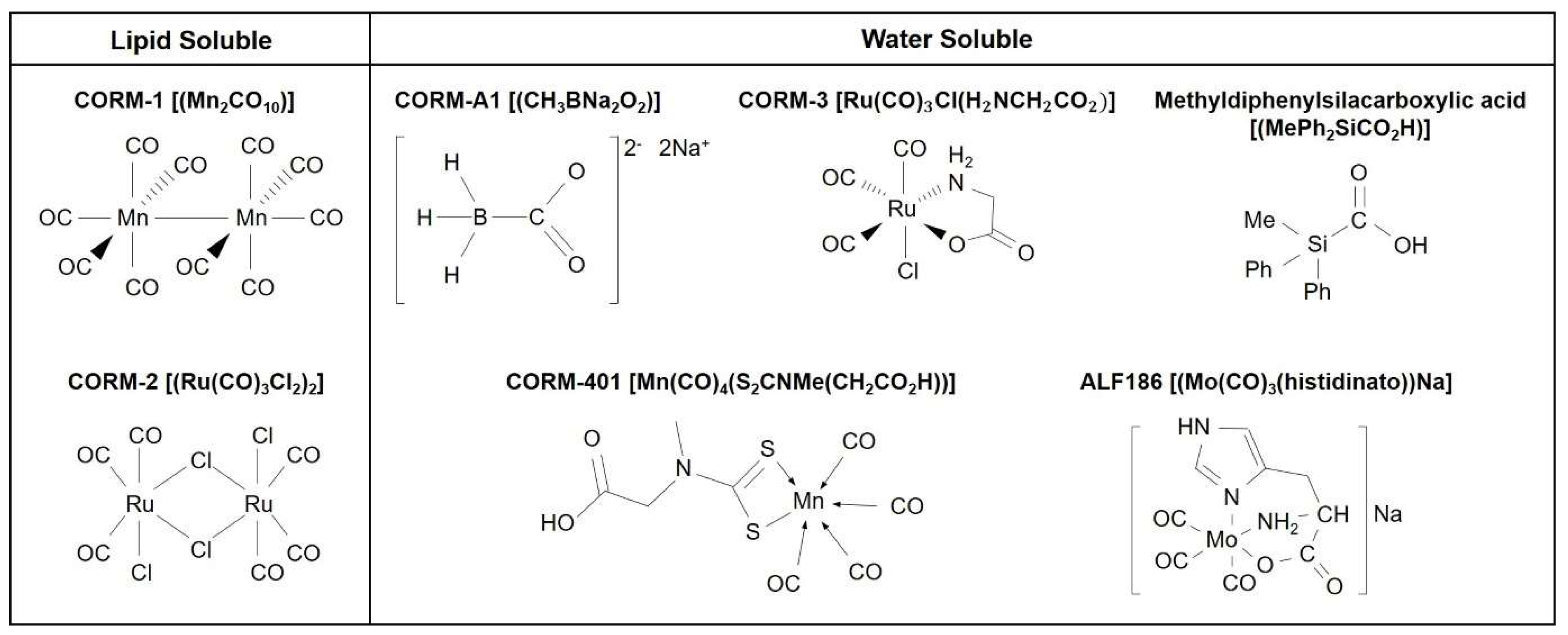
2. Biological Signaling of CO: Use of CO Gas and CORMs
3. Effects of CO on Neuronal Intrinsic Mechanisms
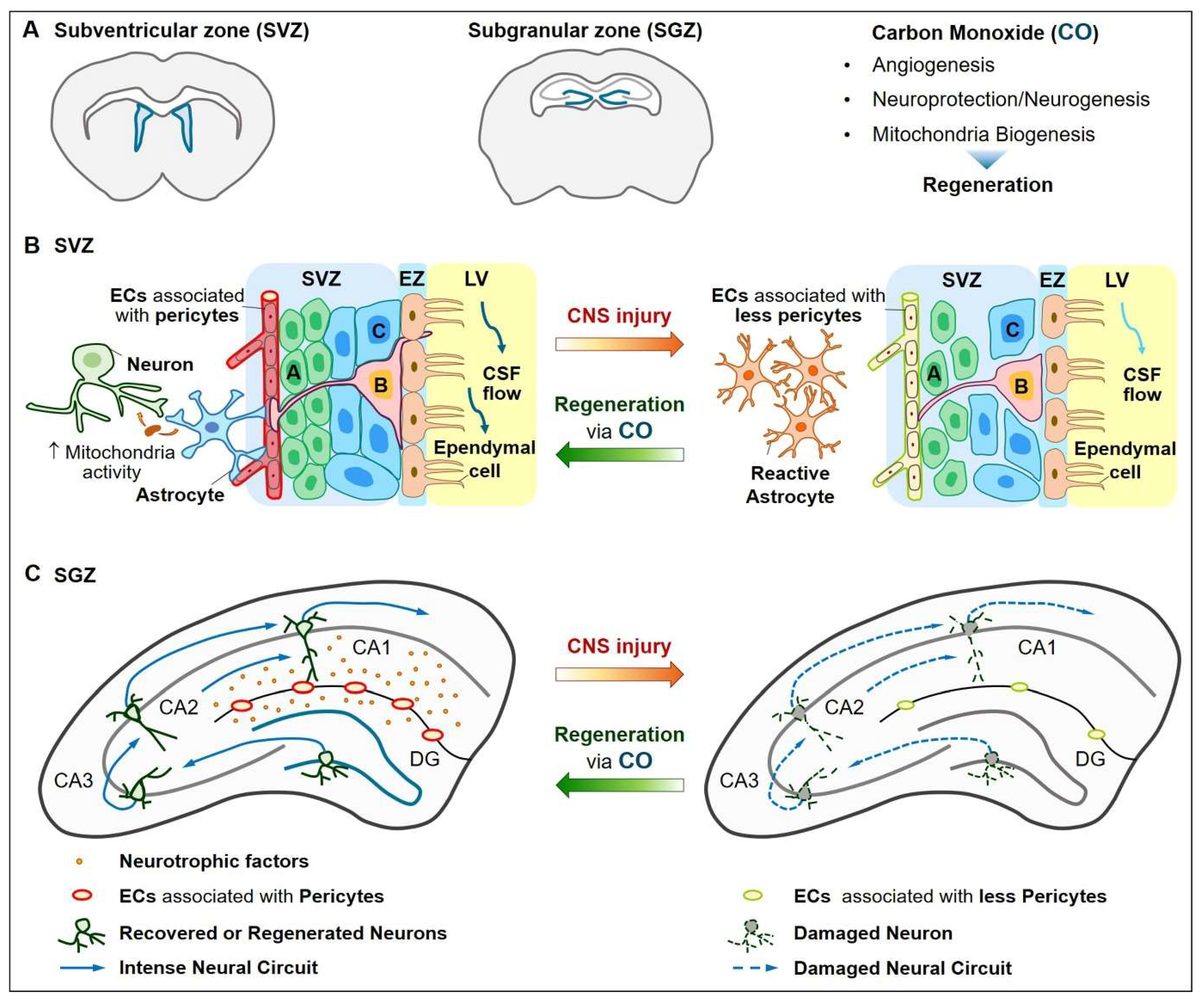
4. Neurovascular Regeneration
4.1. Vascular Functions
4.2. Neuroprotection and Neurogenesis
4.3. Mitochondria Function
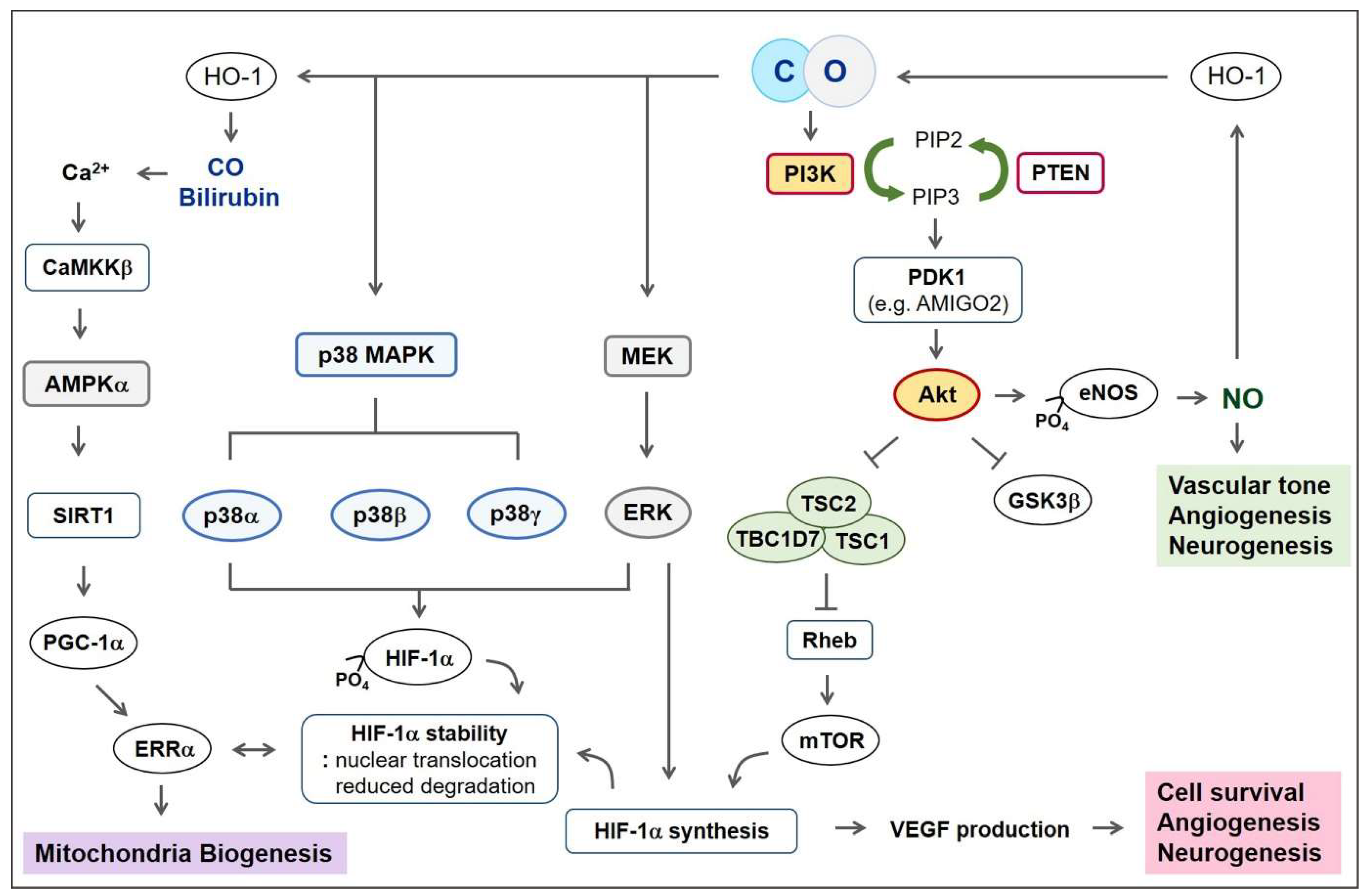
5. Signaling Pathways for Regeneration
5.1. PI3K-Akt Pathway
5.2. Mitogen-Activated Protein Kinase (MAPK)
6. Possible Regenerative Signaling Molecules through the Cell-Cell Network
6.1. Ciliary Neurotrophic Factors (CNTFs)
6.2. Brain-Derived Neurotrophic Factor (BDNF)
6.3. Glial Cell-Line Derived Neurotrophic Factor (GDNF)
6.4. Nitric Oxide (NO)
7. Functional Recovery by CO/HO-1 Pathway in CNS Injury
7.1. Stroke
7.2. Traumatic Brain Injury (TBI)
7.3. Multiple Sclerosis
7.4. Alzheimer’s Disease (AD)
8. Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | beta-amyloid |
| AD | Alzheimer’s disease |
| AMP | adenosine monophosphate |
| AMPK | AMP kinase |
| AMIGO2 | adhesion molecule with IgG-like domain 2 |
| Armcx1 | armadillo repeat-containing X-linked 1 |
| BDNF | brain-derived neurotrophic factor |
| CA | cornus ammonis |
| CaMKKβ | Ca2+-calmodulin kinase kinase β |
| CO | carbon monoxide |
| CORMs | CO-releasing molecules |
| CNS | central nervous system |
| CNTF | ciliary neurotrophic factor |
| cAMP | cyclic AMP |
| CREB | cAMP-responsive element-binding protein |
| DG | dentate gyrus |
| DLK | dual leucine zipper-bearing kinase |
| EAE | experimental autoimmune encephalomyelitis |
| ERK | extracellular-signal-related kinase |
| ERRα | estrogen-related receptor α |
| GAP43 | growth-associated protein 43 |
| GDNF | glial cell-line derived neurotrophic factor |
| GSK3β | glycogen synthase kinase-3β |
| gp130 | glycoprotein 130 |
| HDAC5 | histone deacetylase 5 |
| HIF-1α | Hypoxia-inducible factor-1α |
| Hsp90 | heat shock protein 90 |
| HO | heme oxygenase |
| IL-8 | Interleukin-8 |
| JAKs | Janus kinases |
| JNKs | c-Jun N-terminal kinases |
| JIP3 | JNK-interacting protein 3 |
| MAPKs | mitogen-activated protein kinases |
| MEKs | MAPK kinases |
| mTOR | mammalian target of rapamycin |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NOX | NADPH oxidase |
| NO | nitric oxide |
| eNOS | endothelial NO synthase |
| NF-κB | nuclear factor κ-light-chain-enhancer of activated B cells |
| Nrf2 | nuclear transcription factor NF-E2-related factor 2 |
| NSC | neural stem cell |
| OGD | oxygen-glucose deprivation |
| PKA | protein kinase A |
| PDK1 | 3-phosphoinositide-dependent kinase 1 |
| PGC-1α | peroxisome proliferator-activated receptor γ-coactivator-1α |
| PI3K | phosphatidylinositide 3-kinase |
| Prox1 | prospero homeobox 1 |
| PTEN | phosphatase and tensin homolog |
| RGCs | retinal ganglion cells |
| ROS | reactive oxygen species |
| SIRT1 | sirtuin 1 |
| SGZ | subgranular zone |
| SVZ | subventricular zone |
| SOCS3 | suppressor of cytokine signaling 3 |
| STAT3 | signal transducer and activator of transcription 3 |
| TAZ | transcriptional coactivator with PDZ-binding motif |
| TBI | traumatic brain injury |
| TNF-α | tumor necrosis factor-α |
| TSC | tuberous sclerosis complex |
| TSC1 | hamartin |
| TSC2 | tuberin |
| TrkB | tropomyosin-related kinase B |
| VEGF | vascular endothelial growth factor |
| VEGFR1 | VEGF receptor 1 |
| YAP | Yes-associated protein |
References
- Huebner, E.A.; Strittmatter, S.M. Axon regeneration in the peripheral and central nervous systems. Results Probl Cell Differ 2009, 48, 339–351. [Google Scholar] [PubMed] [Green Version]
- He, Z.; Jin, Y. Intrinsic control of axon regeneration. Neuron 2016, 90, 437–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.L.; Yu, W.M.; Strickland, S. Peripheral regeneration. Annu. Rev. Neurosci. 2007, 30, 209–233. [Google Scholar] [CrossRef]
- Schwab, M.E.; Strittmatter, S.M. Nogo limits neural plasticity and recovery from injury. Curr. Opin. Neurobiol. 2014, 27, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Tedeschi, A.; Bradke, F. Spatial and temporal arrangement of neuronal intrinsic and extrinsic mechanisms controlling axon regeneration. Curr. Opin. Neurobiol. 2017, 42, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Mahar, M.; Cavalli, V. Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 2018, 19, 323–337. [Google Scholar] [CrossRef]
- Hilton, B.J.; Bradke, F. Can injured adult CNS axons regenerate by recapitulating development? Development 2017, 144, 3417–3429. [Google Scholar] [CrossRef] [Green Version]
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743. [Google Scholar] [CrossRef]
- Choi, Y.K.; Maki, T.; Mandeville, E.T.; Koh, S.H.; Hayakawa, K.; Arai, K.; Kim, Y.M.; Whalen, M.J.; Xing, C.; Wang, X.; et al. Dual effects of carbon monoxide on pericytes and neurogenesis in traumatic brain injury. Nat. Med. 2016, 22, 1335–1341. [Google Scholar] [CrossRef]
- Kim, Y.M.; Pae, H.O.; Park, J.E.; Lee, Y.C.; Woo, J.M.; Kim, N.H.; Choi, Y.K.; Lee, B.S.; Kim, S.R.; Chung, H.T. Heme oxygenase in the regulation of vascular biology: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 14, 137–167. [Google Scholar] [CrossRef] [Green Version]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maines, M.D.; Trakshel, G.M. Purification and characterization of human biliverdin reductase. Arch. Biochem. Biophys. 1993, 300, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Tenhunen, R.; Marver, H.S.; Schmid, R. Microsomal heme oxygenase. Characterization of the enzyme. J. Biol. Chem. 1969, 244, 6388–6394. [Google Scholar] [PubMed]
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar] [PubMed]
- Schipper, H.M.; Song, W.; Tavitian, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 2019, 172, 40–70. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Hayashi, S.; Omata, Y.; Sakamoto, H.; Higashimoto, Y.; Hara, T.; Sagara, Y.; Noguchi, M. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene 2004, 336, 241–250. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme oxygenase 1 in the nervous system: Does it favor neuronal cell survival or induce neurodegeneration? Int. J. Mol. Sci. 2018, 19, E2260. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Peng, Y.; Hui, Y.; Zhang, S.; Zhou, Y.; Li, D.; Li, J.; Si, Z.; Li, J.; Wang, D.; et al. Overexpression of heme oxygenase 1 impairs cognitive ability and changes the plasticity of the synapse. J. Alzheimers Dis. 2015, 47, 595–608. [Google Scholar] [CrossRef]
- Lin, C.C.; Yang, C.C.; Hsiao, L.D.; Chen, S.Y.; Yang, C.M. Heme oxygenase-1 induction by carbon Monoxide releasing molecule-3 suppresses interleukin-1beta-mediated neuroinflammation. Front. Mol. Neurosci. 2017, 10, 387. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Choi, Y.K. The Role of astrocytes in the central nervous system focused on BK channel and heme oxygenase metabolites: A review. Antioxidants 2019, 8, E121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stifter, J.; Ulbrich, F.; Goebel, U.; Bohringer, D.; Lagreze, W.A.; Biermann, J. Neuroprotection and neuroregeneration of retinal ganglion cells after intravitreal carbon monoxide release. PLoS ONE 2017, 12, e0188444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbin, T.J.; Benignus, V.A.; Muller, K.E.; Barton, C.N. The effects of low-level carbon monoxide exposure upon evoked cortical potentials in young and elderly men. Neurotoxicol. Teratol. 1988, 10, 93–100. [Google Scholar] [CrossRef]
- Verma, A.; Hirsch, D.J.; Glatt, C.E.; Ronnett, G.V.; Snyder, S.H. Carbon monoxide: A putative neural messenger. Science 1993, 259, 381–384. [Google Scholar] [CrossRef]
- Sammut, I.A.; Foresti, R.; Clark, J.E.; Exon, D.J.; Vesely, M.J.; Sarathchandra, P.; Green, C.J.; Motterlini, R. Carbon monoxide is a major contributor to the regulation of vascular tone in aortas expressing high levels of haeme oxygenase-1. Br. J. Pharmacol. 1998, 125, 1437–1444. [Google Scholar] [CrossRef] [Green Version]
- Schipper, H.M. Heme oxygenase expression in human central nervous system disorders. Free Radic. Biol. Med. 2004, 37, 1995–2011. [Google Scholar] [CrossRef]
- Motterlini, R.; Foresti, R. Biological signaling by carbon monoxide and carbon monoxide-releasing molecules. Am. J. Physiol. Cell Physiol. 2017, 312, C302–C313. [Google Scholar] [CrossRef] [Green Version]
- Fayad-Kobeissi, S.; Ratovonantenaina, J.; Dabire, H.; Wilson, J.L.; Rodriguez, A.M.; Berdeaux, A.; Dubois-Rande, J.L.; Mann, B.E.; Motterlini, R.; Foresti, R. Vascular and angiogenic activities of CORM-401, an oxidant-sensitive CO-releasing molecule. Biochem. Pharmacol. 2016, 102, 64–77. [Google Scholar] [CrossRef]
- Lo Iacono, L.; Boczkowski, J.; Zini, R.; Salouage, I.; Berdeaux, A.; Motterlini, R.; Morin, D. A carbon monoxide-releasing molecule (CORM-3) uncouples mitochondrial respiration and modulates the production of reactive oxygen species. Free Radic. Biol. Med. 2011, 50, 1556–1564. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.K. Role of carbon monoxide in neurovascular repair processing. Biomol. Ther. 2018, 26, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.S.; Figueiredo-Pereira, C.; Vieira, H.L. Carbon monoxide and mitochondria-modulation of cell metabolism, redox response and cell death. Front. Physiol. 2015, 6, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Karpus, J.; Zhao, B.S.; Luo, Z.; Chen, P.R.; He, C. A selective fluorescent probe for carbon monoxide imaging in living cells. Angew. Chem. Int. Ed. Engl. 2012, 51, 9652–9656. [Google Scholar] [CrossRef]
- Dreyer-Andersen, N.; Almeida, A.S.; Jensen, P.; Kamand, M.; Okarmus, J.; Rosenberg, T.; Friis, S.D.; Martinez Serrano, A.; Blaabjerg, M.; Kristensen, B.W.; et al. Intermittent, low dose carbon monoxide exposure enhances survival and dopaminergic differentiation of human neural stem cells. PLoS ONE 2018, 13, e0191207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Stein, A.B.; Wu, W.J.; Tan, W.; Zhu, X.; Li, Q.H.; Dawn, B.; Motterlini, R.; Bolli, R. Administration of a CO-releasing molecule at the time of reperfusion reduces infarct size in vivo. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1649–H1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katada, K.; Bihari, A.; Mizuguchi, S.; Yoshida, N.; Yoshikawa, T.; Fraser, D.D.; Potter, R.F.; Cepinskas, G. Carbon monoxide liberated from CO-releasing molecule (CORM-2) attenuates ischemia/reperfusion (I/R)-induced inflammation in the small intestine. Inflammation 2010, 33, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Yabluchanskiy, A.; Sawle, P.; Homer-Vanniasinkam, S.; Green, C.J.; Foresti, R.; Motterlini, R. CORM-3, a carbon monoxide-releasing molecule, alters the inflammatory response and reduces brain damage in a rat model of hemorrhagic stroke. Crit. Care Med. 2012, 40, 544–552. [Google Scholar] [CrossRef]
- Choi, Y.K.; Kim, C.K.; Lee, H.; Jeoung, D.; Ha, K.S.; Kwon, Y.G.; Kim, K.W.; Kim, Y.M. Carbon monoxide promotes VEGF expression by increasing HIF-1alpha protein level via two distinct mechanisms, translational activation and stabilization of HIF-1alpha protein. J. Biol. Chem. 2010, 285, 32116–32125. [Google Scholar]
- Choi, Y.K.; Kim, J.H.; Lee, D.K.; Lee, K.S.; Won, M.H.; Jeoung, D.; Lee, H.; Ha, K.S.; Kwon, Y.G.; Kim, Y.M. Carbon Monoxide Potentiation of L-Type Ca2+ Channel Activity Increases HIF-1alpha-Independent VEGF Expression via an AMPKalpha/SIRT1-Mediated PGC-1alpha/ERRalpha Axis. Antioxid. Redox Signal. 2017, 27, 21–36. [Google Scholar] [CrossRef]
- Choi, Y.K.; Park, J.H.; Baek, Y.Y.; Won, M.H.; Jeoung, D.; Lee, H.; Ha, K.S.; Kwon, Y.G.; Kim, Y.M. Carbon monoxide stimulates astrocytic mitochondrial biogenesis via L-type Ca2+ channel-mediated PGC-1alpha/ERRalpha activation. Biochem. Biophys. Res. Commun. 2016, 479, 297–304. [Google Scholar] [CrossRef]
- Choi, Y.K.; Park, J.H.; Yun, J.A.; Cha, J.H.; Kim, Y.; Won, M.H.; Kim, K.W.; Ha, K.S.; Kwon, Y.G.; Kim, Y.M. Heme oxygenase metabolites improve astrocytic mitochondrial function via a Ca2+-dependent HIF-1alpha/ERRalpha circuit. PLoS ONE 2018, 13, e0202039. [Google Scholar]
- Lancel, S.; Hassoun, S.M.; Favory, R.; Decoster, B.; Motterlini, R.; Neviere, R. Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. J. Pharmacol. Exp. Ther. 2009, 329, 641–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaarmann, A.; Mandel, M.; Zeb, A.; Wareski, P.; Liiv, J.; Kuum, M.; Antsov, E.; Liiv, M.; Cagalinec, M.; Choubey, V.; et al. Mitochondrial biogenesis is required for axonal growth. Development 2016, 143, 1981–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queiroga, C.S.; Vercelli, A.; Vieira, H.L. Carbon monoxide and the CNS: Challenges and achievements. Br. J. Pharmacol. 2015, 172, 1533–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egawa, N.; Lok, J.; Washida, K.; Arai, K. Mechanisms of axonal damage and repair after central nervous system injury. Transl. Stroke Res. 2017, 8, 14–21. [Google Scholar] [CrossRef]
- Rishal, I.; Fainzilber, M. Axon-soma communication in neuronal injury. Nat. Rev. Neurosci. 2014, 15, 32–42. [Google Scholar] [CrossRef]
- Kulbatski, I.; Cook, D.J.; Tator, C.H. Calcium entry through L-type calcium channels is essential for neurite regeneration in cultured sympathetic neurons. J. Neurotrauma 2004, 21, 357–374. [Google Scholar] [CrossRef]
- Cho, Y.; Sloutsky, R.; Naegle, K.M.; Cavalli, V. Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell 2013, 155, 894–908. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef]
- Wang, H.; Xu, J.; Lazarovici, P.; Quirion, R.; Zheng, W. cAMP Response Element-Binding Protein (CREB): A possible signaling molecule link in the pathophysiology of schizophrenia. Front. Mol. Neurosci. 2018, 11, 255. [Google Scholar] [CrossRef]
- Tedeschi, A.; Dupraz, S.; Laskowski, C.J.; Xue, J.; Ulas, T.; Beyer, M.; Schultze, J.L.; Bradke, F. The calcium channel subunit alpha2delta2 suppresses axon regeneration in the adult CNS. Neuron 2016, 92, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Zaccolo, M.; Pozzan, T. CAMP and Ca2+ interplay: A matter of oscillation patterns. Trends Neurosci. 2003, 26, 53–55. [Google Scholar] [CrossRef]
- Hao, Y.; Frey, E.; Yoon, C.; Wong, H.; Nestorovski, D.; Holzman, L.B.; Giger, R.J.; DiAntonio, A.; Collins, C. An evolutionarily conserved mechanism for cAMP elicited axonal regeneration involves direct activation of the dual leucine zipper kinase DLK. Elife 2016, 5, e14048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, S.M.; Collura, K.M.; Ketschek, A.; Noma, K.; Ferguson, T.A.; Jin, Y.; Gallo, G.; Thomas, G.M. Palmitoylation controls DLK localization, interactions and activity to ensure effective axonal injury signaling. Proc. Natl. Acad. Sci. USA 2016, 113, 763–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saura, C.A.; Cardinaux, J.R. Emerging roles of CREB-regulated transcription coactivators in brain physiology and pathology. Trends Neurosci. 2017, 40, 720–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canettieri, G.; Coni, S.; Della Guardia, M.; Nocerino, V.; Antonucci, L.; Di Magno, L.; Screaton, R.; Screpanti, I.; Giannini, G.; Gulino, A. The coactivator CRTC1 promotes cell proliferation and transformation via AP-1. Proc. Natl. Acad. Sci. USA 2009, 106, 1445–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raivich, G.; Bohatschek, M.; Da Costa, C.; Iwata, O.; Galiano, M.; Hristova, M.; Nateri, A.S.; Makwana, M.; Riera-Sans, L.; Wolfer, D.P.; et al. The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron 2004, 43, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Ruff, C.A.; Staak, N.; Patodia, S.; Kaswich, M.; Rocha-Ferreira, E.; Da Costa, C.; Brecht, S.; Makwana, M.; Fontana, X.; Hristova, M.; et al. Neuronal c-Jun is required for successful axonal regeneration, but the effects of phosphorylation of its N-terminus are moderate. J. Neurochem. 2012, 121, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.; Cavalli, V. HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J. 2012, 31, 3063–3078. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Yin, C.; Lao, T.; Liang, D.; He, D.; Wang, C.; Sang, N. AMPK-HDAC5 pathway facilitates nuclear accumulation of HIF-1alpha and functional activation of HIF-1 by deacetylating Hsp70 in the cytosol. Cell Cycle 2015, 14, 2520–2536. [Google Scholar] [CrossRef] [Green Version]
- Kietzmann, T.; Mennerich, D.; Dimova, E.Y. Hypoxia-Inducible Factors (HIFs) and phosphorylation: Impact on stability, localization, and transactivity. Front. Cell Dev. Biol. 2016, 4, 11. [Google Scholar] [CrossRef]
- Tanabe, N.; Kuboyama, T.; Tohda, C. Matrine directly activates extracellular heat shock protein 90, resulting in axonal growth and functional recovery in spinal cord injured-mice. Front. Pharmacol. 2018, 9, 446. [Google Scholar] [CrossRef] [Green Version]
- Ben-Yaakov, K.; Dagan, S.Y.; Segal-Ruder, Y.; Shalem, O.; Vuppalanchi, D.; Willis, D.E.; Yudin, D.; Rishal, I.; Rother, F.; Bader, M.; et al. Axonal transcription factors signal retrogradely in lesioned peripheral nerve. EMBO J. 2012, 31, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Ribeiro, M.; Bray, E.R.; Lee, D.H.; Yungher, B.J.; Mehta, S.T.; Thakor, K.A.; Diaz, F.; Lee, J.K.; Moraes, C.T.; et al. Enhanced transcriptional activity and mitochondrial localization of STAT3 co-induce axon regrowth in the adult central nervous system. Cell Rep. 2016, 15, 398–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Shan, P.; Jiang, G.; Zhang, S.S.; Otterbein, L.E.; Fu, X.Y.; Lee, P.J. Endothelial STAT3 is essential for the protective effects of HO-1 in oxidant-induced lung injury. FASEB J. 2006, 20, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Snipes, G.J.; Chan, S.Y.; McGuire, C.B.; Costello, B.R.; Norden, J.J.; Freeman, J.A.; Routtenberg, A. Evidence for the coidentification of GAP-43, a growth-associated protein, and F1, a plasticity-associated protein. J. Neurosci. 1987, 7, 4066–4075. [Google Scholar] [CrossRef] [Green Version]
- De la Monte, S.M.; Federoff, H.J.; Ng, S.C.; Grabczyk, E.; Fishman, M.C. GAP-43 gene expression during development: Persistence in a distinctive set of neurons in the mature central nervous system. Brain Res. Dev. Brain Res. 1989, 46, 161–168. [Google Scholar] [CrossRef]
- Dijk, F.; Bergen, A.A.; Kamphuis, W. GAP-43 expression is upregulated in retinal ganglion cells after ischemia/reperfusion-induced damage. Exp. Eye Res. 2007, 84, 858–867. [Google Scholar] [CrossRef]
- Cui, J.; Cui, C.; Cui, Y.; Li, R.; Sheng, H.; Jiang, X.; Tian, Y.; Wang, K.; Gao, J. Bone marrow mesenchymal stem cell transplantation increases GAP-43 expression via ERK1/2 and PI3K/Akt pathways in intracerebral hemorrhage. Cell. Physiol. Biochem. 2017, 42, 137–144. [Google Scholar] [CrossRef]
- Ulbrich, F.; Hagmann, C.; Buerkle, H.; Romao, C.C.; Schallner, N.; Goebel, U.; Biermann, J. The Carbon monoxide releasing molecule ALF-186 mediates anti-inflammatory and neuroprotective effects via the soluble guanylate cyclase ss1 in rats’ retinal ganglion cells after ischemia and reperfusion injury. J. Neuroinflammation 2017, 14, 130. [Google Scholar] [CrossRef] [Green Version]
- Friedlander, M. Fibrosis and diseases of the eye. J. Clin. Investig. 2007, 117, 576–586. [Google Scholar] [CrossRef]
- Ming, G.L.; Song, H. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J. Angiogenesis. Annu. Rev. Med. 2006, 57, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.E.; Zurakowski, D.; Italiano, J.E., Jr.; Michel, L.V.; Connors, S.; Oenick, M.; D’Amato, R.J.; Klement, G.L.; Folkman, J. VEGF, PF4 and PDGF are elevated in platelets of colorectal cancer patients. Angiogenesis 2012, 15, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Choi, J.; Yang, M.J.; Hong, S.P.; Lee, C.K.; Kubota, Y.; Lim, D.S.; Koh, G.Y. A MST1-FOXO1 cascade establishes endothelial tip cell polarity and facilitates sprouting angiogenesis. Nat. Commun. 2019, 10, 838. [Google Scholar] [CrossRef] [Green Version]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Ahn, J.H.; Cho, H.; Kim, J.H.; Kim, S.H.; Ham, J.S.; Park, I.; Suh, S.H.; Hong, S.P.; Song, J.H.; Hong, Y.K.; et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature 2019, 572, 62–66. [Google Scholar] [CrossRef]
- Da Mesquita, S.; Fu, Z.; Kipnis, J. The meningeal lymphatic system: A new player in neurophysiology. Neuron 2018, 100, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Song, E.; Mao, T.; Dong, H.; Boisserand, L.S.B.; Antila, S.; Bosenberg, M.; Alitalo, K.; Thomas, J.L.; Iwasaki, A. VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature 2020, 577, 689–694. [Google Scholar] [CrossRef]
- Preston, J.E. Ageing choroid plexus-cerebrospinal fluid system. Microsc. Res. Tech. 2001, 52, 31–37. [Google Scholar] [CrossRef]
- Toni, N.; Schinder, A.F. Maturation and functional integration of new granule cells into the adult hippocampus. Cold Spring Harb. Perspect. Biol. 2015, 8, a018903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, M. VEGFR and type-V RTK activation and signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a009092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Claesson-Welsh, L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp. Cell Res. 2006, 312, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Foo, S.Y.; Ma, Y.; Ruas, J.L.; Bommi-Reddy, A.; Girnun, G.; Cooper, M.; Laznik, D.; Chinsomboon, J.; Rangwala, S.M.; et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature 2008, 451, 1008–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [Green Version]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.; Powner, M.B.; Gandhi, P.; Clarkin, C.; Gutmann, D.H.; Johnson, R.S.; Ferrara, N.; Fruttiger, M. Astrocyte-derived vascular endothelial growth factor stabilizes vessels in the developing retinal vasculature. PLoS ONE 2010, 5, e11863. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Kim, J.; Ahn, J.H.; Hong, Y.K.; Makinen, T.; Lim, D.S.; Koh, G.Y. YAP and TAZ negatively regulate prox1 during developmental and pathologic lymphangiogenesis. Circ. Res. 2019, 124, 225–242. [Google Scholar] [CrossRef]
- Johnson, N.C.; Dillard, M.E.; Baluk, P.; McDonald, D.M.; Harvey, N.L.; Frase, S.L.; Oliver, G. Lymphatic endothelial cell identity is reversible and its maintenance requires Prox1 activity. Genes Dev. 2008, 22, 3282–3291. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kim, Y.H.; Kim, J.; Park, D.Y.; Bae, H.; Lee, D.H.; Kim, K.H.; Hong, S.P.; Jang, S.P.; Kubota, Y.; et al. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J. Clin. Investig. 2017, 127, 3441–3461. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Freire Valls, A.; Schermann, G.; Shen, Y.; Moya, I.M.; Castro, L.; Urban, S.; Solecki, G.M.; Winkler, F.; Riedemann, L.; et al. YAP/TAZ orchestrate VEGF signaling during developmental angiogenesis. Dev. Cell 2017, 42, 462–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Bao, Q.; Zhang, Y.; Liu, M.; Lv, H.; Liu, Y.; Yao, L.; Li, B.; Zhang, C.; He, S.; et al. Yes-associated protein promotes angiogenesis via signal transducer and activator of transcription 3 in endothelial cells. Circ. Res. 2018, 122, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Charest-Marcotte, A.; Dufour, C.R.; Wilson, B.J.; Tremblay, A.M.; Eichner, L.J.; Arlow, D.H.; Mootha, V.K.; Giguere, V. The homeobox protein Prox1 is a negative modulator of ERR{alpha}/PGC-1{alpha} bioenergetic functions. Genes Dev. 2010, 24, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.J.; Zhou, Y.; Stadel, R.P.; Moss, J.; Yong, J.H.; Ito, S.; Kawasaki, N.K.; Phan, A.T.; Oh, J.H.; Modak, N.; et al. Tangential migration of neuronal precursors of glutamatergic neurons in the adult mammalian brain. Proc. Natl. Acad. Sci. USA 2015, 112, 9484–9489. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Lee, M.; Choi, Y.K. The role of a neurovascular signaling pathway involving hypoxia-inducible factor and notch in the function of the central nervous system. Biomol. Ther. 2020, 28, 45–57. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, D.; Fu, X.; Yu, L.; Lu, Z.; Gao, Y.; Liu, X.; Man, J.; Li, S.; Li, N.; et al. Carbon monoxide-releasing molecule-3 protects against ischemic stroke by suppressing neuroinflammation and alleviating blood-brain barrier disruption. J. Neuroinflammation 2018, 15, 188. [Google Scholar] [CrossRef]
- Obernier, K.; Alvarez-Buylla, A. Neural stem cells: Origin, heterogeneity and regulation in the adult mammalian brain. Development 2019, 146, dev156059. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, P.S.; Perfilieva, E.; Bjork-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Asselin, C.; et al. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science 2018, 361, eaan8821. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Han, P.; Cui, Z.; Wang, B.; Zhong, Z.; Sun, Y.; Yang, G.; Sun, Q.; Bian, L. Pretreatment of mouse neural stem cells with carbon monoxide-releasing molecule-2 interferes with NF-kappaB p65 signaling and suppresses iron overload-induced apoptosis. Cell. Mol. Neurobiol. 2016, 36, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Gunter, K.; Maines, M.D. Neurons overexpressing heme oxygenase-1 resist oxidative stress-mediated cell death. J. Neurochem. 2000, 75, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Scheiblich, H.; Bicker, G. Regulation of microglial migration, phagocytosis, and neurite outgrowth by HO-1/CO signaling. Dev. Neurobiol. 2015, 75, 854–876. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.S.; Soares, N.L.; Vieira, M.; Gramsbergen, J.B.; Vieira, H.L. Carbon monoxide releasing molecule-A1 (CORM-A1) improves neurogenesis: Increase of neuronal differentiation yield by preventing cell death. PLoS ONE 2016, 11, e0154781. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.M.; Gallo, G. The role of mitochondria in axon development and regeneration. Dev. Neurobiol. 2018, 78, 221–237. [Google Scholar] [CrossRef]
- Cartoni, R.; Norsworthy, M.W.; Bei, F.; Wang, C.; Li, S.; Zhang, Y.; Gabel, C.V.; Schwarz, T.L.; He, Z. The mammalian-specific protein armcx1 regulates mitochondrial transport during axon regeneration. Neuron 2016, 92, 1294–1307. [Google Scholar] [CrossRef] [Green Version]
- Park, H.H.; Han, M.H.; Choi, H.; Lee, Y.J.; Kim, J.M.; Cheong, J.H.; Ryu, J.I.; Lee, K.Y.; Koh, S.H. Mitochondria damaged by oxygen glucose deprivation can be restored through activation of the PI3K/Akt pathway and inhibition of calcium influx by amlodipine camsylate. Sci. Rep. 2019, 9, 15717. [Google Scholar] [CrossRef]
- Davis, C.H.; Kim, K.Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Queiroga, C.S.; Alves, R.M.; Conde, S.V.; Alves, P.M.; Vieira, H.L. Paracrine effect of carbon monoxide—Astrocytes promote neuroprotection through purinergic signaling in mice. J Cell Sci 2016, 129, 3178–3188. [Google Scholar] [CrossRef] [Green Version]
- Queiroga, C.S.; Almeida, A.S.; Martel, C.; Brenner, C.; Alves, P.M.; Vieira, H.L. Glutathionylation of adenine nucleotide translocase induced by carbon monoxide prevents mitochondrial membrane permeabilization and apoptosis. J. Biol. Chem. 2010, 285, 17077–17088. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Choi, Y.K. Regenerative effects of heme oxygenase metabolites on neuroinflammatory diseases. Int. J. Mol. Sci. 2018, 20, 78. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.M.; Huang, Y.T.; Zhang, Y.Q.; Hsieh, C.W.; Wung, B.S. Carbon monoxide releasing molecule induces endothelial nitric oxide synthase activation through a calcium and phosphatidylinositol 3-kinase/Akt mechanism. Vasc. Pharmacol. 2016, 87, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.H.; Lo, E.H. The role of the PI3K pathway in the regeneration of the damaged brain by neural stem cells after cerebral infarction. J. Clin. Neurol. 2015, 11, 297–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.Y.; Luciano, A.K.; Ackah, E.; Rodriguez-Vita, J.; Bancroft, T.A.; Eichmann, A.; Simons, M.; Kyriakides, T.R.; Morales-Ruiz, M.; Sessa, W.C. Endothelial Akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proc. Natl. Acad. Sci. USA 2014, 111, 12865–12870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltier, J.; O’Neill, A.; Schaffer, D.V. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev. Neurobiol. 2007, 67, 1348–1361. [Google Scholar] [CrossRef]
- Park, W.; Baek, Y.Y.; Kim, J.; Jo, D.H.; Choi, S.; Kim, J.H.; Kim, T.; Kim, S.; Park, M.; Kim, J.Y.; et al. Arg-Leu-Tyr-Glu suppresses retinal endothelial permeability and choroidal neovascularization by inhibiting the VEGF receptor 2 signaling pathway. Biomol. Ther. 2019, 27, 474–483. [Google Scholar] [CrossRef]
- Park, K.K.; Liu, K.; Hu, Y.; Smith, P.D.; Wang, C.; Cai, B.; Xu, B.; Connolly, L.; Kramvis, I.; Sahin, M.; et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008, 322, 963–966. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Lu, Y.; Lee, J.K.; Samara, R.; Willenberg, R.; Sears-Kraxberger, I.; Tedeschi, A.; Park, K.K.; Jin, D.; Cai, B.; et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat. Neurosci. 2010, 13, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Lee, S.; Shrestha, P.; Kim, J.; Park, J.A.; Ko, Y.; Ban, Y.H.; Park, D.Y.; Ha, S.J.; Koh, G.Y.; et al. AMIGO2, a novel membrane anchor of PDK1, controls cell survival and angiogenesis via Akt activation. J. Cell Biol. 2015, 211, 619–637. [Google Scholar] [CrossRef] [Green Version]
- Kuja-Panula, J.; Kiiltomaki, M.; Yamashiro, T.; Rouhiainen, A.; Rauvala, H. AMIGO, a transmembrane protein implicated in axon tract development, defines a novel protein family with leucine-rich repeats. J. Cell Biol. 2003, 160, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Kuja-Panula, J.; Sundvik, M.; Chen, Y.C.; Aho, V.; Peltola, M.A.; Porkka-Heiskanen, T.; Panula, P.; Rauvala, H. Amigo adhesion protein regulates development of neural circuits in zebrafish brain. J. Biol. Chem. 2014, 289, 19958–19975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Meng, Z.; Cui, M.; Zhang, X.; Chen, F.; Xiao, J.; Shen, L.; Zhang, Y. An Ang1-Tie2-PI3K axis in neural progenitor cells initiates survival responses against oxygen and glucose deprivation. Neuroscience 2009, 160, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Son, J.W.; Choi, H.; Yoo, A.; Park, H.H.; Kim, Y.S.; Lee, K.Y.; Lee, Y.J.; Koh, S.H. Activation of the phosphatidylinositol 3-kinase pathway plays important roles in reduction of cerebral infarction by cilnidipine. J. Neurochem. 2015, 135, 186–193. [Google Scholar] [CrossRef] [Green Version]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [Green Version]
- Nagasawa-Masuda, A.; Terai, K. ERK activation in endothelial cells is a novel marker during neovasculogenesis. Genes Cells 2016, 21, 1164–1175. [Google Scholar] [CrossRef] [Green Version]
- Chierzi, S.; Ratto, G.M.; Verma, P.; Fawcett, J.W. The ability of axons to regenerate their growth cones depends on axonal type and age, and is regulated by calcium, cAMP and ERK. Eur. J. Neurosci. 2005, 21, 2051–2062. [Google Scholar] [CrossRef]
- Perlson, E.; Hanz, S.; Ben-Yaakov, K.; Segal-Ruder, Y.; Seger, R.; Fainzilber, M. Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron 2005, 45, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.E.; Cho, Y.; Beirowski, B.; Milbrandt, J.; Cavalli, V.; DiAntonio, A. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron 2012, 74, 1015–1022. [Google Scholar] [CrossRef] [Green Version]
- Hammarlund, M.; Nix, P.; Hauth, L.; Jorgensen, E.M.; Bastiani, M. Axon regeneration requires a conserved MAP kinase pathway. Science 2009, 323, 802–806. [Google Scholar] [CrossRef] [Green Version]
- Nix, P.; Hisamoto, N.; Matsumoto, K.; Bastiani, M. Axon regeneration requires coordinate activation of p38 and JNK MAPK pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 10738–10743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M.P. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 2000, 192, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poyhonen, S.; Er, S.; Domanskyi, A.; Airavaara, M. Effects of neurotrophic factors in glial cells in the central nervous system: Expression and properties in neurodegeneration and injury. Front. Physiol. 2019, 10, 486. [Google Scholar] [CrossRef] [PubMed]
- Zigmond, R.E. gp130 cytokines are positive signals triggering changes in gene expression and axon outgrowth in peripheral neurons following injury. Front. Mol. Neurosci. 2011, 4, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, F.; Park, K.K.; Belin, S.; Wang, D.; Lu, T.; Chen, G.; Zhang, K.; Yeung, C.; Feng, G.; Yankner, B.A.; et al. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature 2011, 480, 372–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yungher, B.J.; Ribeiro, M.; Park, K.K. Regenerative responses and axon pathfinding of retinal ganglion cells in chronically injured mice. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1743–1750. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; Zhang, Q.; Sheng, Z.; Li, Y.; Lu, H.H. Ciliary Neurotrophic Factor (CNTF) protects myocardial cells from Oxygen Glucose Deprivation (OGD)/re-oxygenation via activation of Akt-Nrf2 signaling. Cell. Physiol. Biochem. 2018, 51, 1852–1862. [Google Scholar] [CrossRef]
- Tron, K.; Samoylenko, A.; Musikowski, G.; Kobe, F.; Immenschuh, S.; Schaper, F.; Ramadori, G.; Kietzmann, T. Regulation of rat heme oxygenase-1 expression by interleukin-6 via the Jak/STAT pathway in hepatocytes. J. Hepatol. 2006, 45, 72–80. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Muller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Kim, E.; Ratan, R.; Lee, F.S.; Cho, S. Genetic variant of BDNF (Val66Met) polymorphism attenuates stroke-induced angiogenic responses by enhancing anti-angiogenic mediator CD36 expression. J. Neurosci. 2011, 31, 775–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougherty, K.D.; Dreyfus, C.F.; Black, I.B. Brain-derived neurotrophic factor in astrocytes, oligodendrocytes, and microglia/macrophages after spinal cord injury. Neurobiol. Dis. 2000, 7, 574–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Ma, X.L.; Geng, Z.; Huang, S.H.; Zhai, L.K.; Guo, Y.Y.; Chen, Z.Y. Up-regulation of c-Jun NH2-terminal kinase-interacting protein 3 (JIP3) contributes to BDNF-enhanced neurotransmitter release. J. Neurochem. 2015, 135, 453–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.H.; Duan, S.; Sun, T.; Wang, J.; Zhao, L.; Geng, Z.; Yan, J.; Sun, H.J.; Chen, Z.Y. JIP3 mediates TrkB axonal anterograde transport and enhances BDNF signaling by directly bridging TrkB with kinesin-1. J. Neurosci. 2011, 31, 10602–10614. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Galvez, G.; Zambrano, J.M.; Diaz Soto, J.C.; Zhan, W.Z.; Gransee, H.M.; Sieck, G.C.; Mantilla, C.B. TrkB gene therapy by adeno-associated virus enhances recovery after cervical spinal cord injury. Exp. Neurol. 2016, 276, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Puyang, Z.; Chen, H.; Liang, P.; Troy, J.B.; Liu, X. Overexpression of brain-derived neurotrophic factor protects large retinal ganglion cells after optic nerve crush in mice. eNeuro 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Bonafina, A.; Trinchero, M.F.; Rios, A.S.; Bekinschtein, P.; Schinder, A.F.; Paratcha, G.; Ledda, F. GDNF and GFRalpha1 are required for proper integration of adult-born hippocampal neurons. Cell Rep. 2019, 29, 4308–4319. [Google Scholar] [CrossRef] [Green Version]
- Irala, D.; Bonafina, A.; Fontanet, P.A.; Alsina, F.C.; Paratcha, G.; Ledda, F. The GDNF-GFRalpha1 complex promotes the development of hippocampal dendritic arbors and spines via NCAM. Development 2016, 143, 4224–4235. [Google Scholar] [CrossRef] [Green Version]
- Hung, S.Y.; Liou, H.C.; Kang, K.H.; Wu, R.M.; Wen, C.C.; Fu, W.M. Overexpression of heme oxygenase-1 protects dopaminergic neurons against 1-methyl-4-phenylpyridinium-induced neurotoxicity. Mol. Pharmacol. 2008, 74, 1564–1575. [Google Scholar] [CrossRef] [Green Version]
- Hung, S.Y.; Liou, H.C.; Fu, W.M. The mechanism of heme oxygenase-1 action involved in the enhancement of neurotrophic factor expression. Neuropharmacology 2010, 58, 321–329. [Google Scholar] [CrossRef]
- Quintino, L.; Avallone, M.; Brannstrom, E.; Kavanagh, P.; Lockowandt, M.; Garcia Jareno, P.; Breger, L.S.; Lundberg, C. GDNF-mediated rescue of the nigrostriatal system depends on the degree of degeneration. Gene Ther. 2019, 26, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.; Kim, J.; Kim, J.H.; Lee, D.K.; Park, W.; Park, M.; Kim, S.; Hwang, J.Y.; Won, M.H.; Choi, Y.K.; et al. Carbon monoxide prevents TNF-alpha-induced eNOS downregulation by inhibiting NF-kappaB-responsive miR-155–5p biogenesis. Exp. Mol. Med. 2017, 49, e403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Wang, S.; Cai, J.; Rao, M.S.; Mattson, M.P. Nitric oxide acts in a positive feedback loop with BDNF to regulate neural progenitor cell proliferation and differentiation in the mammalian brain. Dev. Biol. 2003, 258, 319–333. [Google Scholar] [CrossRef] [Green Version]
- Silver, J.; Miller, J.H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004, 5, 146–156. [Google Scholar] [CrossRef]
- Choi, Y.K.; Por, E.D.; Kwon, Y.G.; Kim, Y.M. Regulation of ROS production and vascular function by carbon monoxide. Oxid. Med. Cell. Longev. 2012, 2012, 794237. [Google Scholar] [CrossRef] [Green Version]
- Ramamoorthy, P.; Xu, G.; Shi, H. Expression of hypoxia inducible factor 1alpha is protein kinase a-dependent in primary cortical astrocytes exposed to severe hypoxia. Neurochem. Res. 2019, 44, 258–268. [Google Scholar] [CrossRef]
- Panahian, N.; Yoshiura, M.; Maines, M.D. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J. Neurochem. 1999, 72, 1187–1203. [Google Scholar] [CrossRef]
- Shi, H. Hypoxia inducible factor 1 as a therapeutic target in ischemic stroke. Curr. Med. Chem. 2009, 16, 4593–4600. [Google Scholar] [CrossRef] [Green Version]
- Esposito, E.; Hayakawa, K.; Ahn, B.J.; Chan, S.J.; Xing, C.; Liang, A.C.; Kim, K.W.; Arai, K.; Lo, E.H. Effects of ischemic post-conditioning on neuronal VEGF regulation and microglial polarization in a rat model of focal cerebral ischemia. J. Neurochem. 2018, 146, 160–172. [Google Scholar] [CrossRef] [Green Version]
- Che, X.; Fang, Y.; Si, X.; Wang, J.; Hu, X.; Reis, C.; Chen, S. The role of gaseous molecules in traumatic brain injury: An updated review. Front. Neurosci. 2018, 12, 392. [Google Scholar] [CrossRef] [Green Version]
- Fagone, P.; Patti, F.; Mangano, K.; Mammana, S.; Coco, M.; Touil-Boukoffa, C.; Chikovani, T.; Di Marco, R.; Nicoletti, F. Heme oxygenase-1 expression in peripheral blood mononuclear cells correlates with disease activity in multiple sclerosis. J. Neuroimmunol. 2013, 261, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Picard-Riera, N.; Decker, L.; Delarasse, C.; Goude, K.; Nait-Oumesmar, B.; Liblau, R.; Pham-Dinh, D.; Baron-Van Evercooren, A. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc. Natl. Acad. Sci. USA 2002, 99, 13211–13216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chora, A.A.; Fontoura, P.; Cunha, A.; Pais, T.F.; Cardoso, S.; Ho, P.P.; Lee, L.Y.; Sobel, R.A.; Steinman, L.; Soares, M.P. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J. Clin. Investig. 2007, 117, 438–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagone, P.; Mangano, K.; Quattrocchi, C.; Motterlini, R.; Di Marco, R.; Magro, G.; Penacho, N.; Romao, C.C.; Nicoletti, F. Prevention of clinical and histological signs of proteolipid protein (PLP)-induced experimental allergic encephalomyelitis (EAE) in mice by the water-soluble carbon monoxide-releasing molecule (CORM)-A1. Clin. Exp. Immunol. 2011, 163, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Agundez, J.A.; Garcia-Martin, E.; Martinez, C.; Benito-Leon, J.; Millan-Pascual, J.; Diaz-Sanchez, M.; Calleja, P.; Pisa, D.; Turpin-Fenoll, L.; Alonso-Navarro, H.; et al. Heme oxygenase-1 and 2 common genetic variants and risk for multiple sclerosis. Sci. Rep. 2016, 6, 20830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Hu, X.; Zhu, C.; Wang, D.; Zheng, X.; Liu, Q. Overexpression of CNTF in mesenchymal stem cells reduces demyelination and induces clinical recovery in experimental autoimmune encephalomyelitis mice. J. Neuroimmunol. 2009, 206, 58–69. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Premkumar, D.R.; Smith, M.A.; Richey, P.L.; Petersen, R.B.; Castellani, R.; Kutty, R.K.; Wiggert, B.; Perry, G.; Kalaria, R.N. Induction of heme oxygenase-1 mRNA and protein in neocortex and cerebral vessels in Alzheimer’s disease. J. Neurochem. 1995, 65, 1399–1402. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Sultana, R.; Coccia, R.; Mancuso, C.; Perluigi, M.; Butterfield, D.A. Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic. Biol. Med. 2012, 52, 2292–2301. [Google Scholar] [CrossRef] [Green Version]
- Hui, Y.; Wang, D.; Li, W.; Zhang, L.; Jin, J.; Ma, N.; Zhou, L.; Nakajima, O.; Zhao, W.; Gao, X. Long-term overexpression of heme oxygenase 1 promotes tau aggregation in mouse brain by inducing tau phosphorylation. J. Alzheimers Dis. 2011, 26, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Hettiarachchi, N.T.; Boyle, J.P.; Dallas, M.L.; Al-Owais, M.M.; Scragg, J.L.; Peers, C. Heme oxygenase-1 derived carbon monoxide suppresses Abeta1–42 toxicity in astrocytes. Cell Death Dis. 2017, 8, e2884. [Google Scholar] [CrossRef]
- Hettiarachchi, N.; Dallas, M.; Al-Owais, M.; Griffiths, H.; Hooper, N.; Scragg, J.; Boyle, J.; Peers, C. Heme oxygenase-1 protects against Alzheimer’s amyloid-beta(1–42)-induced toxicity via carbon monoxide production. Cell Death Dis. 2014, 5, e1569. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, E.; Koh, S.-H.; Yoo, M.; Choi, Y.K. Regenerative Potential of Carbon Monoxide in Adult Neural Circuits of the Central Nervous System. Int. J. Mol. Sci. 2020, 21, 2273. https://doi.org/10.3390/ijms21072273
Jung E, Koh S-H, Yoo M, Choi YK. Regenerative Potential of Carbon Monoxide in Adult Neural Circuits of the Central Nervous System. International Journal of Molecular Sciences. 2020; 21(7):2273. https://doi.org/10.3390/ijms21072273
Chicago/Turabian StyleJung, Eunyoung, Seong-Ho Koh, Myeongjong Yoo, and Yoon Kyung Choi. 2020. "Regenerative Potential of Carbon Monoxide in Adult Neural Circuits of the Central Nervous System" International Journal of Molecular Sciences 21, no. 7: 2273. https://doi.org/10.3390/ijms21072273
APA StyleJung, E., Koh, S. -H., Yoo, M., & Choi, Y. K. (2020). Regenerative Potential of Carbon Monoxide in Adult Neural Circuits of the Central Nervous System. International Journal of Molecular Sciences, 21(7), 2273. https://doi.org/10.3390/ijms21072273