miR-1285-3p Controls Colorectal Cancer Proliferation and Escape from Apoptosis through DAPK2

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
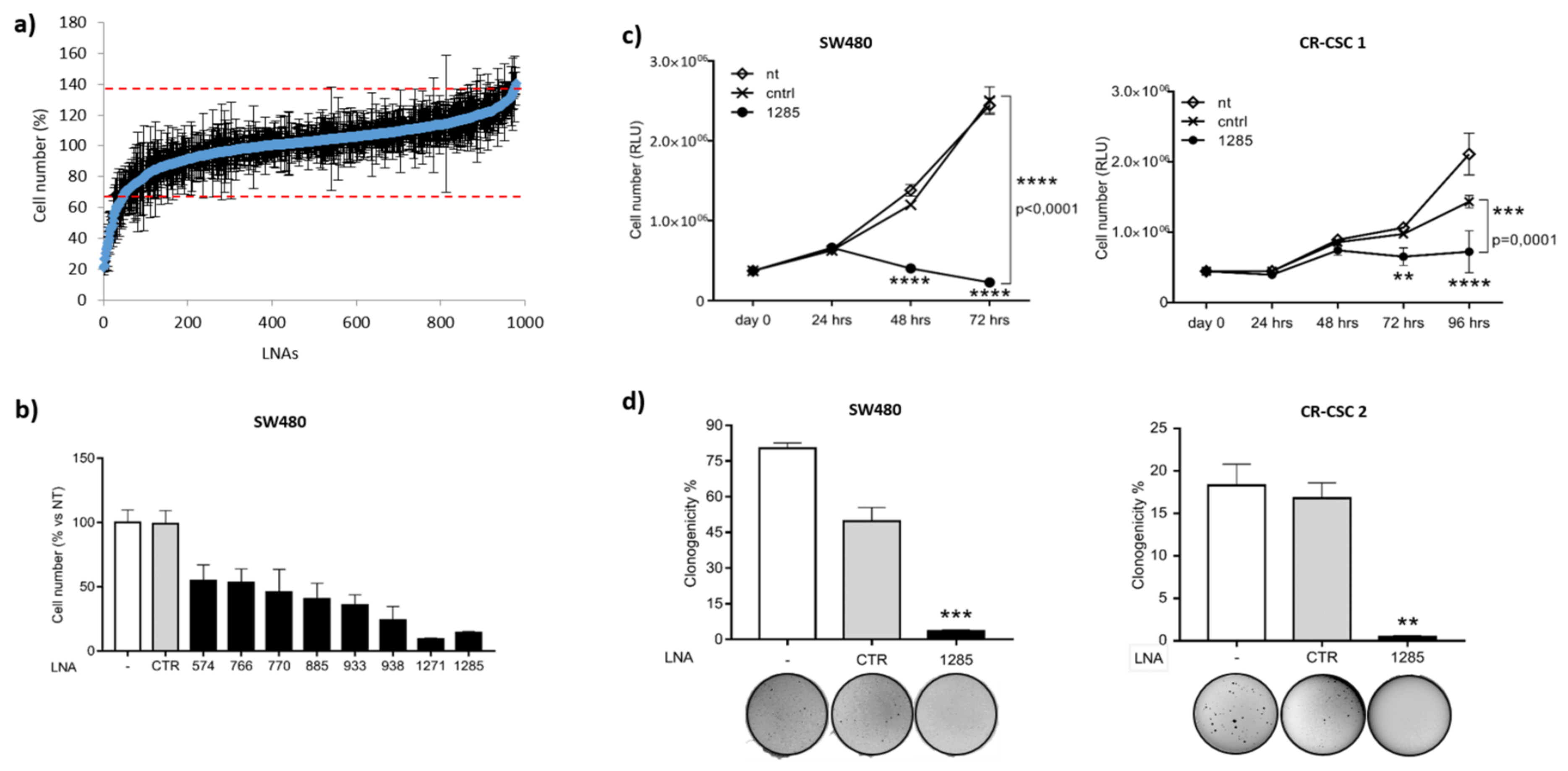
2.1. Genome-Wide Functional Screening with microRNA Inhibitors Identifies miR-1285-3p as a Potential Oncogene in Colorectal Cancer
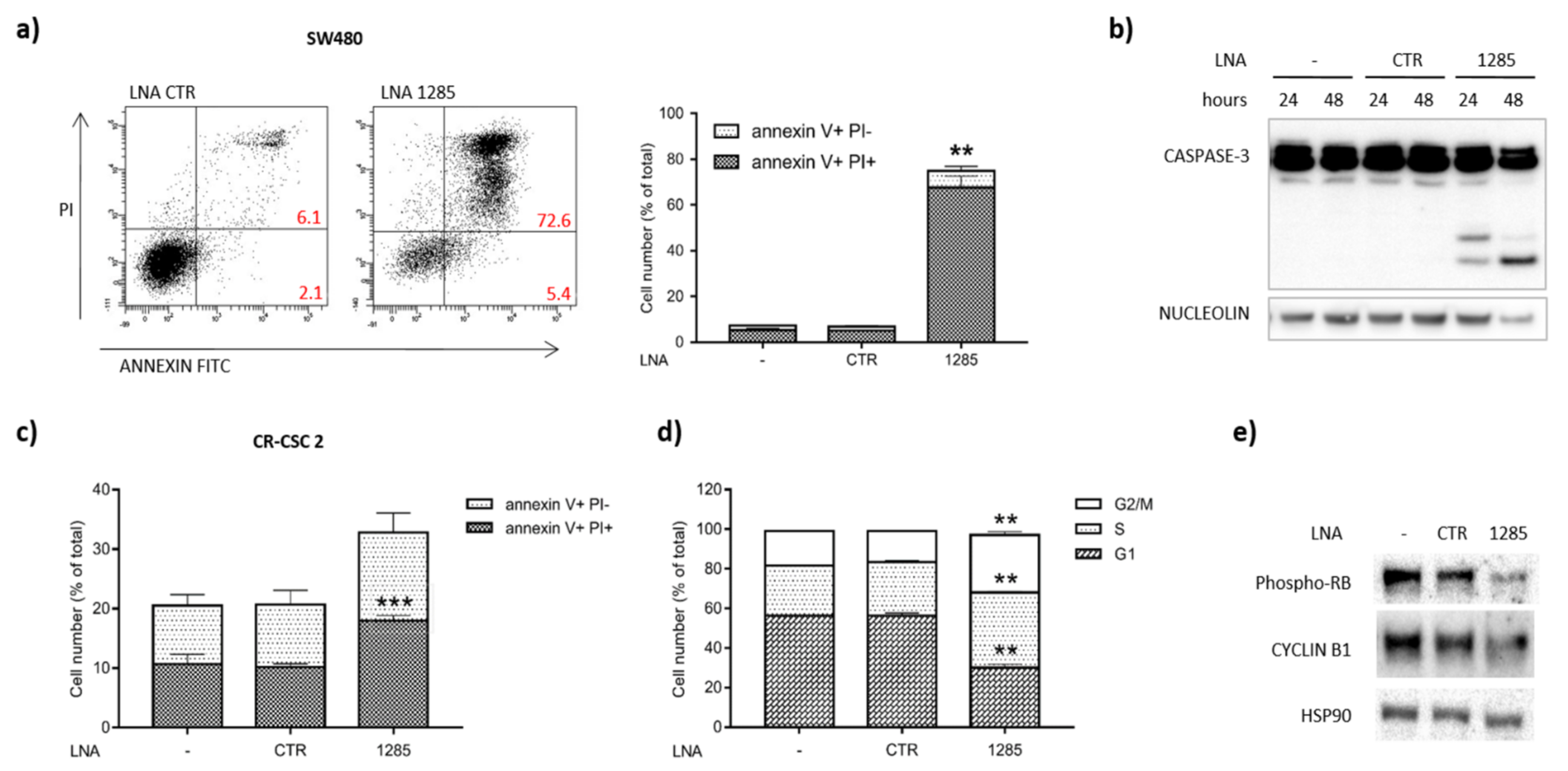
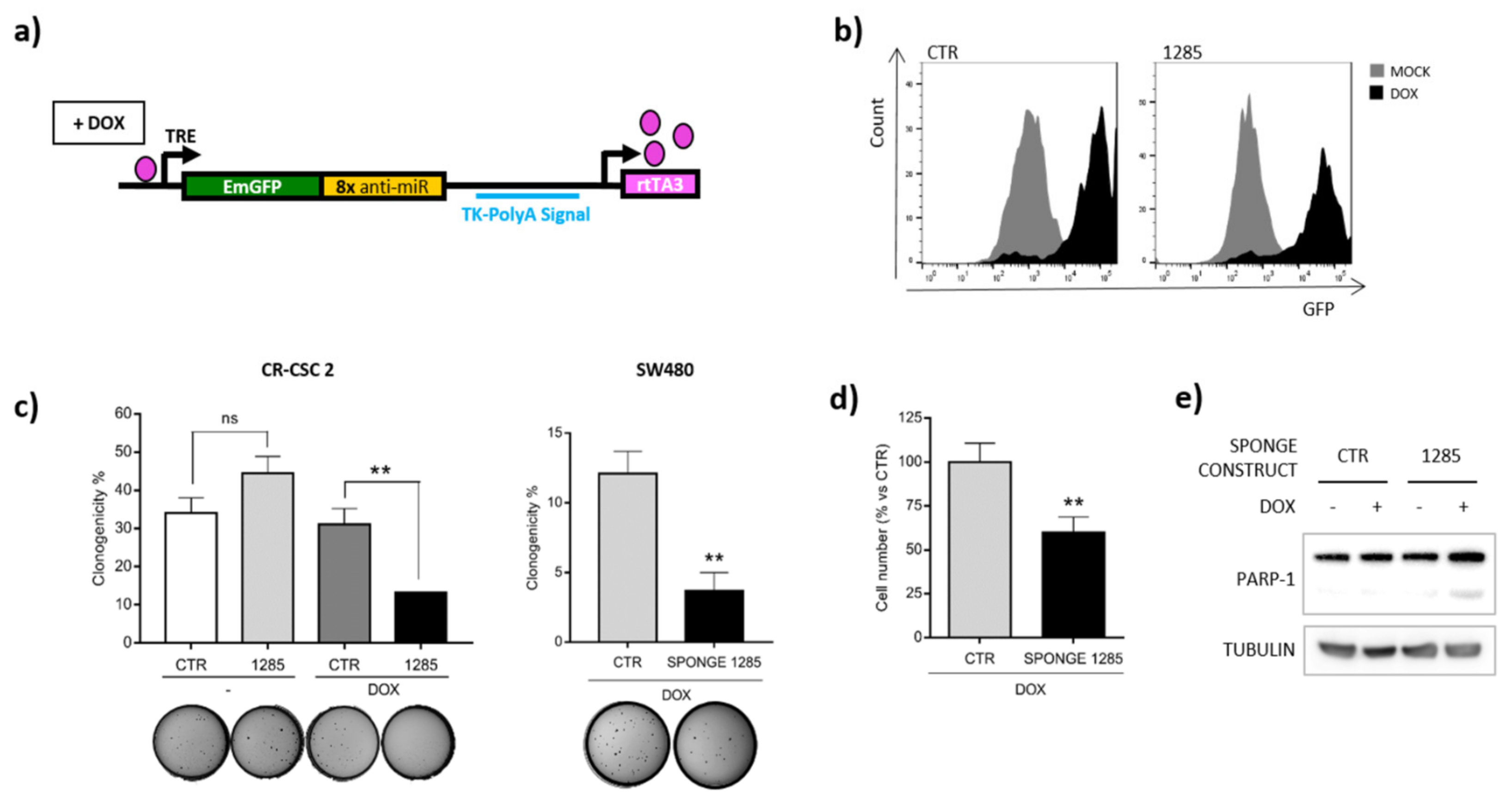
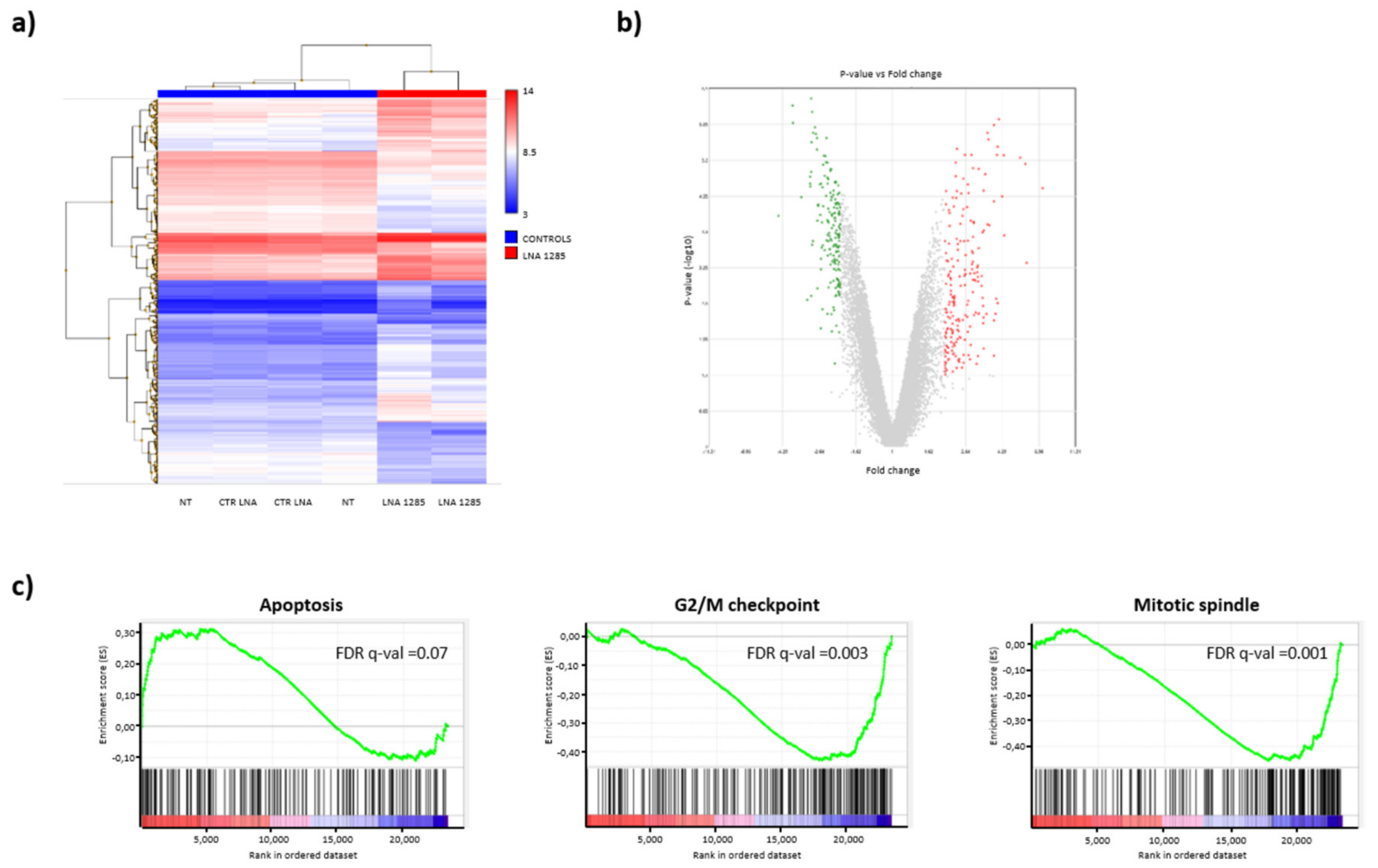
2.2. Inhibition of miR-1285 Induces Cell Cycle Arrest and Apoptosis in Colorectal Cancer
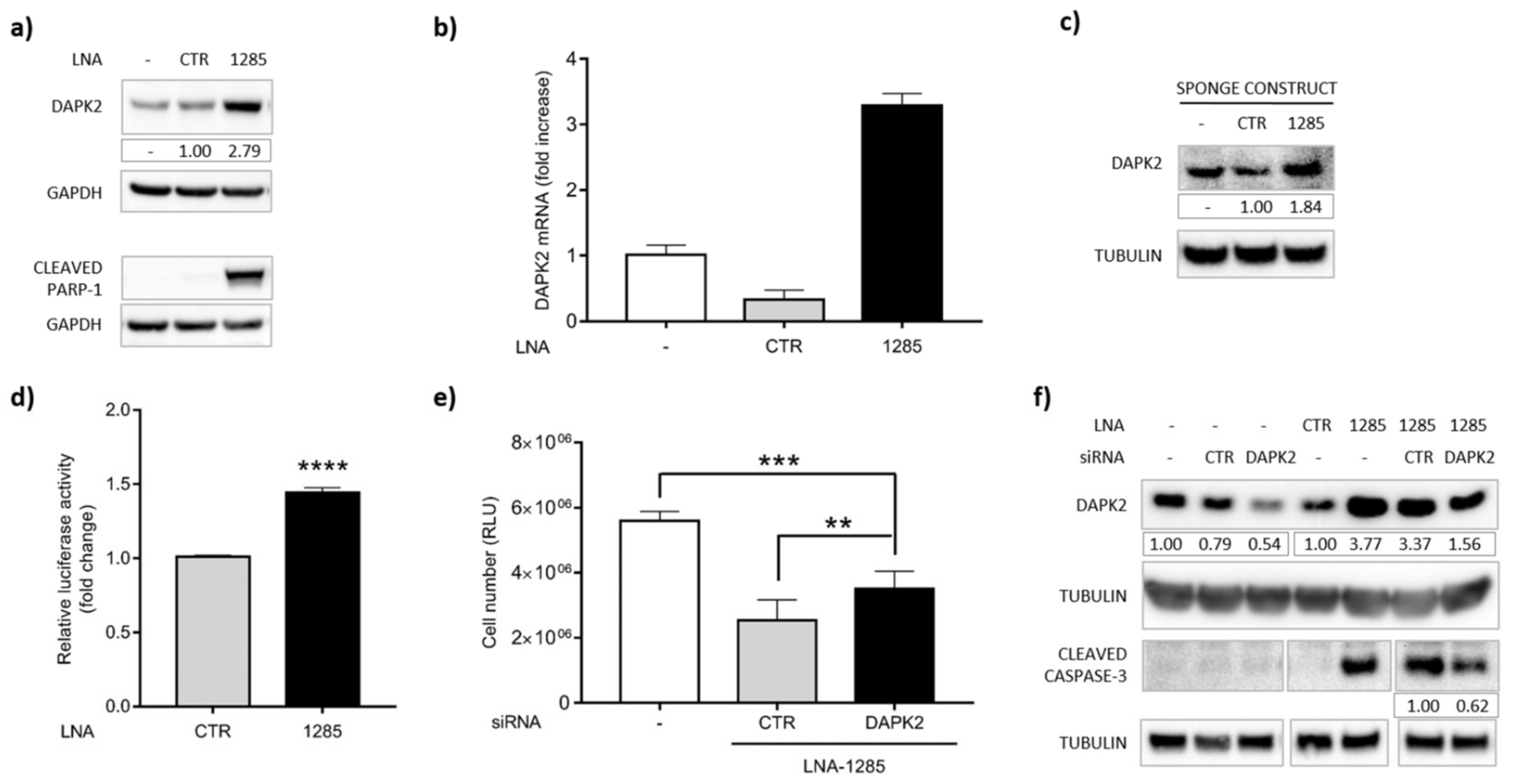
2.3. DAPK2 Is a Novel Direct Target of miR-1285 in Colorectal Cancer
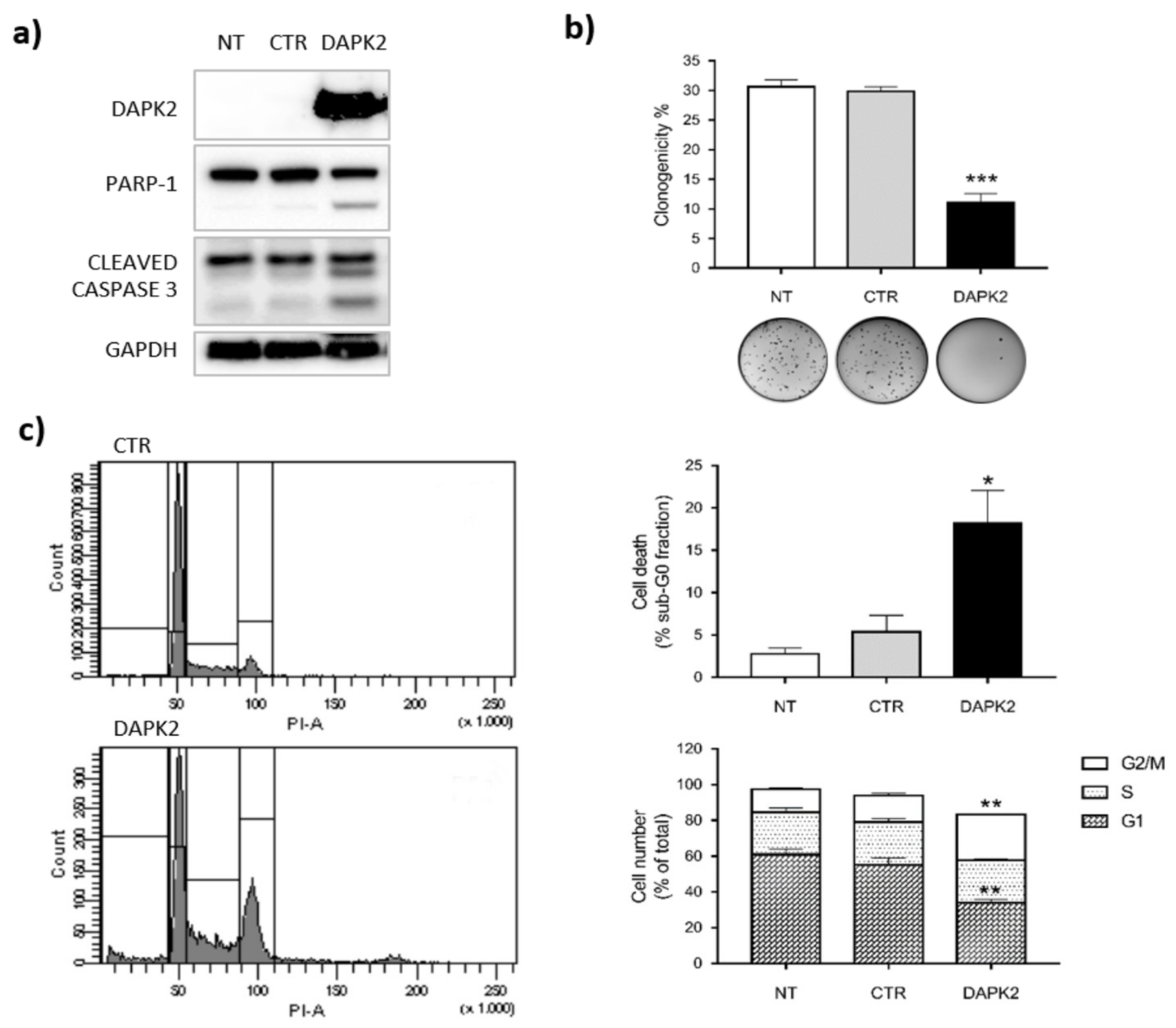
2.4. DAPK2 Mediates miR-1285 Oncogenic Role
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. DNA Constructs and Transduction/Transfection Procedures
4.3. LNA Library Screening
4.4. Anchorage-Independent Assay (Soft Agar Colony Formation Assay)
4.5. Cell Proliferation and Apoptosis Detection
4.6. Cell Cycle Analysis
4.7. Immunoblotting
4.8. Luciferase Reporter Assay
4.9. RT-qPCR Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McGuire, S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv. Nutr. 2016, 7, 418–419. [Google Scholar] [CrossRef] [Green Version]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Strubberg, A.M.; Madison, B.B. MicroRNAs in the etiology of colorectal cancer: Pathways and clinical implications. Dis. Model Mech. 2017, 10, 197–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.E.; Barbini, C.; Haas, T.L.; Marroncelli, N.; Patrizii, M.; Biffoni, M.; De Maria, R. Antitumor effect of miR-197 targeting in p53 wild-type lung cancer. Cell Death Differ. 2014, 21, 774–782. [Google Scholar] [CrossRef]
- Shiloh, R.; Bialik, S.; Kimchi, A. The DAPK family: A structure-function analysis. Apoptosis 2014, 19, 286–297. [Google Scholar] [CrossRef]
- Kawai, T.; Nomura, F.; Hoshino, K.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Akira, S. Death-associated protein kinase 2 is a new calcium/calmodulin-dependent protein kinase that signals apoptosis through its catalytic activity. Oncogene 1999, 18, 3471–3480. [Google Scholar] [CrossRef] [Green Version]
- Inbal, B.; Shani, G.; Cohen, O.; Kissil, J.L.; Kimchi, A. Death-associated protein kinase-related protein 1, a novel serine/threonine kinase involved in apoptosis. Mol. Cell. Biol. 2000, 20, 1044–1054. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ray, G.; Yoo, B.H.; Erdogan, M.; Rosen, K.V. Down-regulation of death-associated protein kinase-2 is required for beta-catenin-induced anoikis resistance of malignant epithelial cells. J. Biol. Chem. 2009, 284, 2012–2022. [Google Scholar] [CrossRef] [Green Version]
- Rizzi, M.; Tschan, M.P.; Britschgi, C.; Britschgi, A.; Hugli, B.; Grob, T.J.; Leupin, N.; Mueller, B.U.; Simon, H.U.; Ziemiecki, A.; et al. The death-associated protein kinase 2 is up-regulated during normal myeloid differentiation and enhances neutrophil maturation in myeloid leukemic cells. J. Leukoc. Biol. 2007, 81, 1599–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.M.; Wang, M.Y.; Hong, C.C.; Chen, H.A.; Su, Y.H.; Wu, C.H.; Huang, M.T.; Chang, Y.W.; Jiang, S.S.; Sung, S.Y.; et al. miR-520h is crucial for DAPK2 regulation and breast cancer progression. Oncogene 2016, 35, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Villanova, L.; Careccia, S.; De Maria, R.; Fiori, M.E. Micro-Economics of Apoptosis in Cancer: ncRNAs Modulation of BCL-2 Family Members. Int. J. Mol. Sci. 2018, 19, 958. [Google Scholar] [CrossRef] [Green Version]
- Fiori, M.E.; Villanova, L.; Barbini, C.; De Angelis, M.L.; De Maria, R. miR-663 sustains NSCLC by inhibiting mitochondrial outer membrane permeabilization (MOMP) through PUMA/BBC3 and BTG2. Cell Death Dis. 2018, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Zeuner, A.; Todaro, M.; Stassi, G.; De Maria, R. Colorectal cancer stem cells: From the crypt to the clinic. Cell Stem Cell 2014, 15, 692–705. [Google Scholar] [CrossRef] [Green Version]
- Fiori, M.E.; Villanova, L.; De Maria, R. Cancer stem cells: At the forefront of personalized medicine and immunotherapy. Curr. Opin. Pharmacol. 2017, 35, 1–11. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallavalle, C.; Albino, D.; Civenni, G.; Merulla, J.; Ostano, P.; Mello-Grand, M.; Rossi, S.; Losa, M.; D’Ambrosio, G.; Sessa, F.; et al. MicroRNA-424 impairs ubiquitination to activate STAT3 and promote prostate tumor progression. J. Clin. Investig. 2016, 126, 4585–4602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valeri, N.; Braconi, C.; Gasparini, P.; Murgia, C.; Lampis, A.; Paulus-Hock, V.; Hart, J.R.; Ueno, L.; Grivennikov, S.I.; Lovat, F.; et al. MicroRNA-135b promotes cancer progression by acting as a downstream effector of oncogenic pathways in colon cancer. Cancer Cell 2014, 25, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Leone, E.; Morelli, E.; Di Martino, M.T.; Amodio, N.; Foresta, U.; Gulla, A.; Rossi, M.; Neri, A.; Giordano, A.; Munshi, N.C.; et al. Targeting miR-21 inhibits in vitro and in vivo multiple myeloma cell growth. Clin. Cancer Res. 2013, 19, 2096–2106. [Google Scholar] [CrossRef] [Green Version]
- Huynh, C.; Segura, M.F.; Gaziel-Sovran, A.; Menendez, S.; Darvishian, F.; Chiriboga, L.; Levin, B.; Meruelo, D.; Osman, I.; Zavadil, J.; et al. Efficient in vivo microRNA targeting of liver metastasis. Oncogene 2011, 30, 1481–1488. [Google Scholar] [CrossRef] [Green Version]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Orum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef] [Green Version]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Yan, J.; Zhou, C.; Ma, Q.; Jin, Q.; Yang, Z. miR-1285-3p acts as a potential tumor suppressor miRNA via downregulating JUN expression in hepatocellular carcinoma. Tumor Biol. 2015, 36, 219–225. [Google Scholar] [CrossRef]
- Katoch, A.; George, B.; Iyyappan, A.; Khan, D.; Das, S. Interplay between PTB and miR-1285 at the p53 3’UTR modulates the levels of p53 and its isoform Delta40p53alpha. Nucleic Acids Res. 2017, 45, 10206–10217. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.H.; Dai, J.; Shang, H.L.; Zhao, Z.X.; Hao, Y.D. miR-1285-3p is a potential prognostic marker in human osteosarcoma and functions as a tumor suppressor by targeting YAP1. Cancer Biomark. 2019, 25, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gilad, Y.; Shiloh, R.; Ber, Y.; Bialik, S.; Kimchi, A. Discovering protein-protein interactions within the programmed cell death network using a protein-fragment complementation screen. Cell Rep. 2014, 8, 909–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isshiki, K.; Hirase, T.; Matsuda, S.; Miyamoto, K.; Tsuji, A.; Yuasa, K. Death-associated protein kinase 2 mediates nocodazole-induced apoptosis through interaction with tubulin. Biochem. Biophys. Res. Commun. 2015, 468, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Geering, B.; Zokouri, Z.; Hurlemann, S.; Gerrits, B.; Auslander, D.; Britschgi, A.; Tschan, M.P.; Simon, H.U.; Fussenegger, M. Identification of Novel Death-Associated Protein Kinase 2 Interaction Partners by Proteomic Screening Coupled with Bimolecular Fluorescence Complementation. Mol. Cell. Biol. 2016, 36, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Ber, Y.; Shiloh, R.; Gilad, Y.; Degani, N.; Bialik, S.; Kimchi, A. DAPK2 is a novel regulator of mTORC1 activity and autophagy. Cell Death Differ. 2015, 22, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Chi, H.C.; Chen, S.L.; Tsai, C.Y.; Chuang, W.Y.; Huang, Y.H.; Tsai, M.M.; Wu, S.M.; Sun, C.P.; Yeh, C.T.; Lin, K.H. Thyroid hormone suppresses hepatocarcinogenesis via DAPK2 and SQSTM1-dependent selective autophagy. Autophagy 2016, 12, 2271–2285. [Google Scholar] [CrossRef]
- Shiloh, R.; Gilad, Y.; Ber, Y.; Eisenstein, M.; Aweida, D.; Bialik, S.; Cohen, S.; Kimchi, A. Non-canonical activation of DAPK2 by AMPK constitutes a new pathway linking metabolic stress to autophagy. Nat. Commun. 2018, 9, 1759. [Google Scholar] [CrossRef] [Green Version]
- Shani, G.; Henis-Korenblit, S.; Jona, G.; Gileadi, O.; Eisenstein, M.; Ziv, T.; Admon, A.; Kimchi, A. Autophosphorylation restrains the apoptotic activity of DRP-1 kinase by controlling dimerization and calmodulin binding. EMBO J. 2001, 20, 1099–1113. [Google Scholar] [CrossRef] [Green Version]
- Britschgi, A.; Trinh, E.; Rizzi, M.; Jenal, M.; Ress, A.; Tobler, A.; Fey, M.F.; Helin, K.; Tschan, M.P. DAPK2 is a novel E2F1/KLF6 target gene involved in their proapoptotic function. Oncogene 2008, 27, 5706–5716. [Google Scholar] [CrossRef] [Green Version]
- Humbert, M.; Federzoni, E.A.; Britschgi, A.; Schlafli, A.M.; Valk, P.J.; Kaufmann, T.; Haferlach, T.; Behre, G.; Simon, H.U.; Torbett, B.E.; et al. The tumor suppressor gene DAPK2 is induced by the myeloid transcription factors PU.1 and C/EBPalpha during granulocytic differentiation but repressed by PML-RARalpha in APL. J. Leukoc. Biol. 2014, 95, 83–93. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, W.; Jin, M.; Chen, J.; Xu, W.; Kong, X. lncRNA MIAT functions as a competing endogenous RNA to upregulate DAPK2 by sponging miR-22-3p in diabetic cardiomyopathy. Cell Death Dis. 2017, 8, e2929. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, H.; Liu, Y.; Zhang, J.; Li, H.; Liu, W.; Cao, G.; Xv, P.; Zhang, J.; Lv, C.; et al. miR-30a as Potential Therapeutics by Targeting TET1 through Regulation of Drp-1 Promoter Hydroxymethylation in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2017, 18, 633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, L.; Sun, Y.; Xiang, J.; Zhou, D.; Wang, L.; Xu, H.; Yang, X.; Du, N.; Zhang, M.; et al. MicroRNA-520g promotes epithelial ovarian cancer progression and chemoresistance via DAPK2 repression. Oncotarget 2016, 7, 26516–26534. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villanova, L.; Barbini, C.; Piccolo, C.; Boe, A.; De Maria, R.; Fiori, M.E. miR-1285-3p Controls Colorectal Cancer Proliferation and Escape from Apoptosis through DAPK2. Int. J. Mol. Sci. 2020, 21, 2423. https://doi.org/10.3390/ijms21072423
Villanova L, Barbini C, Piccolo C, Boe A, De Maria R, Fiori ME. miR-1285-3p Controls Colorectal Cancer Proliferation and Escape from Apoptosis through DAPK2. International Journal of Molecular Sciences. 2020; 21(7):2423. https://doi.org/10.3390/ijms21072423
Chicago/Turabian StyleVillanova, Lidia, Chiara Barbini, Cristina Piccolo, Alessandra Boe, Ruggero De Maria, and Micol Eleonora Fiori. 2020. "miR-1285-3p Controls Colorectal Cancer Proliferation and Escape from Apoptosis through DAPK2" International Journal of Molecular Sciences 21, no. 7: 2423. https://doi.org/10.3390/ijms21072423
APA StyleVillanova, L., Barbini, C., Piccolo, C., Boe, A., De Maria, R., & Fiori, M. E. (2020). miR-1285-3p Controls Colorectal Cancer Proliferation and Escape from Apoptosis through DAPK2. International Journal of Molecular Sciences, 21(7), 2423. https://doi.org/10.3390/ijms21072423