Activation of AMPK under Hypoxia: Many Roads Leading to Rome


Abstract
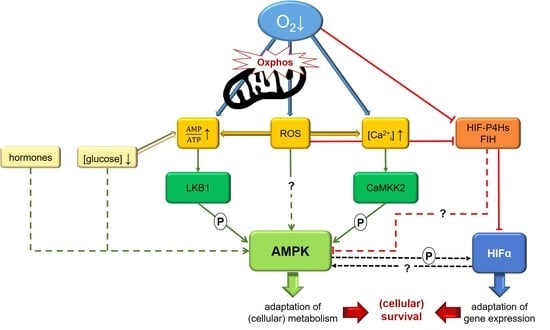
:
{kind=link}
{kind=link}
1. Introduction
2. General Characteristics of AMPK
3. Definition of Hypoxia
4. The Role of AMPK under Hypoxia
5. Activation of AMPK under Hypoxia
5.1. AMP
5.2. LKB1 versus CaMKK2
5.3. CaMKK2 and Intracellular [Ca2+]
5.4. Reactive Oxygen Species
5.5. HIF-hydroxylase Enzymes
5.6. Hormones and Other Mediators
5.7. Glucose and Coupling to mTORC1
6. Crosstalk between AMPK and HIF
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| CaMKK | Ca2+/calmodulin-dependent protein kinase |
| DMOG | Dimethyloxalylglycine |
| FIH | Factor inhibiting HIF |
| HIF | Hypoxia-inducible factor |
| HIF-P4H | HIF-prolyl-4-hydroxylase |
| LKB | Liver kinase B |
| MDPI | Multidisciplinary Digital Publishing Institute |
| mTORC | Mammalian target of rapamycin complex |
| PGC-1α | Peroxisome proliferator-activated receptor-γ coactivator-1α |
| ROS | Reactive oxygen species |
| SIRT | Sirtuin |
| TAK | Transforming growth factor β-activated kinase |
References
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996, 271, 27879–27887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Ashford, M.L.J. AMPK: Regulating energy balance at the cellular and whole body levels. Physiology 2014, 29, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faubert, B.; Vincent, E.E.; Poffenberger, M.C.; Jones, R.G. The AMP-activated protein kinase (AMPK) and cancer: Many faces of a metabolic regulator. Cancer Lett. 2015, 356, 165–170. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. AMPK and HIF signaling pathways regulate both longevity and cancer growth: The good news and the bad news about survival mechanisms. Biogerontology 2016, 17, 655–680. [Google Scholar] [CrossRef]
- Brown, E.; Taylor, C.T. Hypoxia-sensitive pathways in intestinal inflammation. J. Physiol. 2018, 596, 2985–2989. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Carling, D. The AMP-activated protein kinase--fuel gauge of the mammalian cell? Eur. J. Biochem. 1997, 246, 259–273. [Google Scholar] [CrossRef]
- Ross, F.A.; Jensen, T.E.; Hardie, D.G. Differential regulation by AMP and ADP of AMPK complexes containing different γ subunit isoforms. Biochem. J. 2016, 473, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.D.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.-P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, B.; Pulinilkunnil, T.; Murano, I.; Bence, K.K.; He, H.; Minokoshi, Y.; Asakura, K.; Lee, A.; Haj, F.; Furukawa, N.; et al. Neuronal protein tyrosine phosphatase 1B deficiency results in inhibition of hypothalamic AMPK and isoform-specific activation of AMPK in peripheral tissues. Mol. Cell. Biol. 2009, 29, 4563–4573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carling, D.; Sanders, M.J.; Woods, A. The regulation of AMP-activated protein kinase by upstream kinases. Int. J. Obes. 2008, 32 (Suppl. 4), S55–S59. [Google Scholar] [CrossRef] [Green Version]
- Antonia, R.J.; Baldwin, A.S. IKK promotes cytokine-induced and cancer-associated AMPK activity and attenuates phenformin-induced cell death in LKB1-deficient cells. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Neumann, D. Is TAK1 a Direct Upstream Kinase of AMPK? Int. J. Mol. Sci. 2018, 19, 2412. [Google Scholar] [CrossRef] [Green Version]
- Corton, J.M.; Gillespie, J.G.; Hardie, D.G. Role of the AMP-activated protein kinase in the cellular stress response. Curr. Biol. 1994, 4, 315–324. [Google Scholar] [CrossRef]
- Lang, F.; Föller, M. Regulation of ion channels and transporters by AMP-activated kinase (AMPK). Channels 2014, 8, 20–28. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef]
- Ross, F.A.; MacKintosh, C.; Hardie, D.G. AMP-activated protein kinase: A cellular energy sensor that comes in 12 flavours. FEBS J. 2016, 283, 2987–3001. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, J.B.J.; Keely, S.J.; Keely, S.J. Oxygen in the regulation of intestinal epithelial transport. J. Physiol. 2014, 592, 2473–2489. [Google Scholar] [CrossRef] [PubMed]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitouni, N.E.; Chotikatum, S.; von Köckritz-Blickwede, M.; Naim, H.Y. The impact of hypoxia on intestinal epithelial cell functions: Consequences for invasion by bacterial pathogens. Mol. Cell. Pediatr. 2016, 3, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.R.; Li, J.; Coven, D.L.; Pypaert, M.; Zechner, C.; Palmeri, M.; Giordano, F.J.; Mu, J.; Birnbaum, M.J.; Young, L.H. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Investig. 2004, 114, 495–503. [Google Scholar] [CrossRef]
- Seo-Mayer, P.W.; Thulin, G.; Zhang, L.; Alves, D.S.; Ardito, T.; Kashgarian, M.; Caplan, M.J. Preactivation of AMPK by metformin may ameliorate the epithelial cell damage caused by renal ischemia. Am. J. Physiol. Ren. Physiol. 2011, 301, F1346–F1357. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.-C.; Zhu, J.-H.; Ye, L.-X.; Dai, Y.-Y.; Fang, M.-C.; Hu, Y.-Y.; Pan, S.-L.; Chen, S.; Li, P.-J.; Fu, X.-Q.; et al. Glycine Protects against Hypoxic-Ischemic Brain Injury by Regulating Mitochondria-Mediated Autophagy via the AMPK Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 4248529. [Google Scholar] [CrossRef]
- Hu, H.; Li, X.; Ren, D.; Tan, Y.; Chen, J.; Yang, L.; Chen, R.; Li, J.; Zhu, P. The cardioprotective effects of carvedilol on ischemia and reperfusion injury by AMPK signaling pathway. Biomed. Pharmacother. 2019, 117, 109106. [Google Scholar] [CrossRef]
- Michiels, C. Physiological and pathological responses to hypoxia. Am. J. Pathol. 2004, 164, 1875–1882. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Sueyoshi, K.; Zhang, J.; Bao, Y.; Li, X.; Fakhari, M.; Slubowski, C.J.; Bahrami, S.; Ledderose, C.; Junger, W.G. Adenosine 5’-Monophosphate Protects From Hypoxia By Lowering Mitochondrial Metabolism and Oxygen Demand. Shock 2019. [Google Scholar] [CrossRef] [PubMed]
- Rousset, C.I.; Leiper, F.C.; Kichev, A.; Gressens, P.; Carling, D.; Hagberg, H.; Thornton, C. A dual role for AMP-activated protein kinase (AMPK) during neonatal hypoxic-ischaemic brain injury in mice. J. Neurochem. 2015, 133, 242–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.Y.; Choi, Y.-W.; Park, G. Nrf2-mediated neuroprotection against oxygen-glucose deprivation/reperfusion injury by emodin via AMPK-dependent inhibition of GSK-3β. J. Pharm. Pharmacol. 2018, 70, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Xu, B.; Sun, Y.; Lian, M.; Li, Y.; Lin, Y.; Chen, D.; Diao, Y.; Almoiliqy, M.; Wang, L. Paeoniflorin protects against intestinal ischemia/reperfusion by activating LKB1/AMPK and promoting autophagy. Pharmacol. Res. 2019, 146, 104308. [Google Scholar] [CrossRef]
- Mulligan, J.D.; Gonzalez, A.A.; Kumar, R.; Davis, A.J.; Saupe, K.W. Aging elevates basal adenosine monophosphate-activated protein kinase (AMPK) activity and eliminates hypoxic activation of AMPK in mouse liver. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Mu, J.; Brozinick, J.T.; Valladares, O.; Bucan, M.; Birnbaum, M.J. A Role for AMP-Activated Protein Kinase in Contraction- and Hypoxia-Regulated Glucose Transport in Skeletal Muscle. Mol. Cell 2001, 7, 1085–1094. [Google Scholar] [CrossRef]
- Sun, S.; Gu, Z.; Fu, H.; Zhu, J.; Ge, X.; Wu, X. Hypoxia Induces Changes in AMP-Activated Protein Kinase Activity and Energy Metabolism in Muscle Tissue of the Oriental River Prawn Macrobrachium nipponense. Front. Physiol. 2018, 9, 751. [Google Scholar] [CrossRef] [Green Version]
- Siques, P.; Brito, J.; Flores, K.; Ordenes, S.; Arriaza, K.; Pena, E.; León-Velarde, F.; López de Pablo, Á.L.; Gonzalez, M.C.; Arribas, S. Long-Term Chronic Intermittent Hypobaric Hypoxia Induces Glucose Transporter (GLUT4) Translocation Through AMP-Activated Protein Kinase (AMPK) in the Soleus Muscle in Lean Rats. Front. Physiol. 2018, 9, 799. [Google Scholar] [CrossRef]
- Marsin, A.-S.; Bertrand, L.; Rider, M.H.; Deprez, J.; Beauloye, C.; Vincent, M.F.; van den Berghe, G.; Carling, D.; Hue, L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr. Biol. 2000, 10, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.-W.; Zhu, M.; et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef]
- Gusarova, G.A.; Trejo, H.E.; Dada, L.A.; Briva, A.; Welch, L.C.; Hamanaka, R.B.; Mutlu, G.M.; Chandel, N.S.; Prakriya, M.; Sznajder, J.I. Hypoxia leads to Na,K-ATPase downregulation via Ca(2+) release-activated Ca(2+) channels and AMPK activation. Mol. Cell. Biol. 2011, 31, 3546–3556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.D.; Smolenski, R.T.; Harhun, M.I.; Patel, H.K.; Ahmed, S.G.; Wanisch, K.; Yáñez-Muñoz, R.J.; Baines, D.L. AMP-activated protein kinase (AMPK)-dependent and -independent pathways regulate hypoxic inhibition of transepithelial Na+ transport across human airway epithelial cells. Br. J. Pharmacol. 2012, 167, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Dengler, F.; Rackwitz, R.; Pfannkuche, H.; Gäbel, G. Glucose transport across lagomorph jejunum epithelium is modulated by AMP-activated protein kinase under hypoxia. J. Appl. Physiol. 2017, 123, 1487–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dengler, F.; Gäbel, G. The Fast Lane of Hypoxic Adaptation: Glucose Transport Is Modulated via A HIF-Hydroxylase-AMPK-Axis in Jejunum Epithelium. Int. J. Mol. Sci. 2019, 20, 4993. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Murata, Y.; Urushihara, Y.; Shiga, S.; Takeda, K.; Hosoi, Y. Severe hypoxia increases expression of ATM and DNA-PKcs and it increases their activities through Src and AMPK signaling pathways. Biochem. Biophys. Res. Commun. 2018, 505, 13–19. [Google Scholar] [CrossRef]
- De Theije, C.C.; Schols, A.M.W.J.; Lamers, W.H.; Neumann, D.; Köhler, S.E.; Langen, R.C.J. Hypoxia impairs adaptation of skeletal muscle protein turnover- and AMPK signaling during fasting-induced muscle atrophy. PLoS ONE 2018, 13, e0203630. [Google Scholar] [CrossRef]
- Wilson, D.F.; Matschinsky, F.M. Hyperbaric oxygen toxicity in brain: A case of hyperoxia induced hypoglycemic brain syndrome. Med. Hypotheses 2019, 132, 109375. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Chi, Y.; Shi, C.; Zhao, Y.; Guo, C. Forkhead box O (FOXO) 3 modulates hypoxia-induced autophagy through AMPK signalling pathway in cardiomyocytes. Biosci. Rep. 2016, 36, e00345. [Google Scholar] [CrossRef] [Green Version]
- Gui, D.; Cui, Z.; Zhang, L.; Yu, C.; Yao, D.; Xu, M.; Chen, M.; Wu, P.; Li, G.; Wang, L.; et al. Salidroside attenuates hypoxia-induced pulmonary arterial smooth muscle cell proliferation and apoptosis resistance by upregulating autophagy through the AMPK-mTOR-ULK1 pathway. BMC Pulm. Med. 2017, 17, 191. [Google Scholar] [CrossRef] [Green Version]
- Seok, J.-Y.; Jeong, Y.-J.; Hwang, S.-K.; Kim, C.-H.; Magae, J.; Chang, Y.-C. Upregulation of AMPK by 4-O-methylascochlorin promotes autophagy via the HIF-1α expression. J. Cell. Mol. Med. 2018, 22, 6345–6356. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.; Merceron, C.; Mangiavini, L.; Seifert, E.L.; Schipani, E.; Shapiro, I.M.; Risbud, M.V. Hypoxia promotes noncanonical autophagy in nucleus pulposus cells independent of MTOR and HIF1A signaling. Autophagy 2016, 12, 1631–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaachouay, H.; Fehrenbacher, B.; Toulany, M.; Schaller, M.; Multhoff, G.; Rodemann, H.P. AMPK-independent autophagy promotes radioresistance of human tumor cells under clinical relevant hypoxia in vitro. Radiother. Oncol. 2015, 116, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Mungai, P.T.; Waypa, G.B.; Jairaman, A.; Prakriya, M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol. Cell. Biol. 2011, 31, 3531–3545. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Guan, M.; Townsend, K.L.; Huang, T.L.; An, D.; Yan, X.; Xue, R.; Schulz, T.J.; Winnay, J.; Mori, M.; et al. MicroRNA-455 regulates brown adipogenesis via a novel HIF1an-AMPK-PGC1α signaling network. EMBO Rep. 2015, 16, 1378–1393. [Google Scholar] [CrossRef] [Green Version]
- Moral-Sanz, J.; Lewis, S.A.; MacMillan, S.; Ross, F.A.; Thomson, A.; Viollet, B.; Foretz, M.; Moran, C.; Hardie, D.G.; Evans, A.M. The LKB1-AMPK-α1 signaling pathway triggers hypoxic pulmonary vasoconstriction downstream of mitochondria. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Stahmann, N.; Woods, A.; Carling, D.; Heller, R. Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase beta. Mol. Cell. Biol. 2006, 26, 5933–5945. [Google Scholar] [CrossRef] [Green Version]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [Green Version]
- Evans, A.M.; Mustard, K.J.W.; Wyatt, C.N.; Dipp, M.; Kinnear, N.P.; Hardie, D.G. Does AMP-activated protein kinase couple inhibition of mitochondrial oxidative phosphorylation by hypoxia to pulmonary artery constriction? Adv. Exp. Med. Biol. 2006, 580, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Zarrinpashneh, E.; Budas, G.R.; Pouleur, A.-C.; Dutta, A.; Prescott, A.R.; Vanoverschelde, J.-L.; Ashworth, A.; Jovanović, A.; Alessi, D.R.; et al. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPKalpha2 but not AMPKalpha1. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E780–E788. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, C.N.; Mustard, K.J.; Pearson, S.A.; Dallas, M.L.; Atkinson, L.; Kumar, P.; Peers, C.; Hardie, D.G.; Evans, A.M. AMP-activated protein kinase mediates carotid body excitation by hypoxia. J. Biol. Chem. 2007, 282, 8092–8098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.; Belaidi, E.; Moulin, S.; Horman, S.; van der Zon, G.C.; Viollet, B.; Levy, P.; Bertrand, L.; Pepin, J.-L.; Godin-Ribuot, D.; et al. Chronic Intermittent Hypoxia Impairs Insulin Sensitivity but Improves Whole-Body Glucose Tolerance by Activating Skeletal Muscle AMPK. Diabetes 2017, 66, 2942–2951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aley, P.K.; Porter, K.E.; Boyle, J.P.; Kemp, P.J.; Peers, C. Hypoxic modulation of Ca2+ signaling in human venous endothelial cells. Multiple roles for reactive oxygen species. J. Biol. Chem. 2005, 280, 13349–13354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.K.; Park, J.H.; Yun, J.-A.; Cha, J.-H.; Kim, Y.; Won, M.-H.; Kim, K.-W.; Ha, K.-S.; Kwon, Y.-G.; Kim, Y.-M. Heme oxygenase metabolites improve astrocytic mitochondrial function via a Ca2+-dependent HIF-1α/ERRα circuit. PLoS ONE 2018, 13, e0202039. [Google Scholar] [CrossRef] [Green Version]
- Sallé-Lefort, S.; Miard, S.; Nolin, M.-A.; Boivin, L.; Paré, M.-È.; Debigaré, R.; Picard, F. Hypoxia upregulates Malat1 expression through a CaMKK/AMPK/HIF-1α axis. Int. J. Oncol. 2016, 49, 1731–1736. [Google Scholar] [CrossRef] [Green Version]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Brüne, B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef]
- Smith, K.A.; Waypa, G.B.; Schumacker, P.T. Redox signaling during hypoxia in mammalian cells. Redox Biol. 2017, 13, 228–234. [Google Scholar] [CrossRef]
- Emerling, B.M.; Weinberg, F.; Snyder, C.; Burgess, Z.; Mutlu, G.M.; Viollet, B.; Budinger, G.R.S.; Chandel, N.S. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic. Biol. Med. 2009, 46, 1386–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, S.-M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [PubMed]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.L.; Kim, S.J.; Lee, K.T.; Kim, J.; Mu, J.; Birnbaum, M.J.; Soo Kim, S.; Ha, J. The regulation of AMP-activated protein kinase by H(2)O(2). Biochem. Biophys. Res. Commun. 2001, 287, 92–97. [Google Scholar] [CrossRef]
- Gusarova, G.A.; Dada, L.A.; Kelly, A.M.; Brodie, C.; Witters, L.A.; Chandel, N.S.; Sznajder, J.I. Alpha1-AMP-activated protein kinase regulates hypoxia-induced Na,K-ATPase endocytosis via direct phosphorylation of protein kinase C zeta. Mol. Cell. Biol. 2009, 29, 3455–3464. [Google Scholar] [CrossRef] [Green Version]
- Vadász, I.; Dada, L.A.; Briva, A.; Trejo, H.E.; Welch, L.C.; Chen, J.; Tóth, P.T.; Lecuona, E.; Witters, L.A.; Schumacker, P.T.; et al. AMP-activated protein kinase regulates CO2-induced alveolar epithelial dysfunction in rats and human cells by promoting Na,K-ATPase endocytosis. J. Clin. Investig. 2008, 118, 752–762. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.-H.; Wen, J.; Asara, J.; McGraw, T.E.; et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-inducible factor 1: Master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 1998, 8, 588–594. [Google Scholar] [CrossRef]
- Semenza, G.L. Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiology 2004, 19, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Fandrey, J.; Schödel, J.; Eckardt, K.-U.; Katschinski, D.M.; Wenger, R.H. Now a Nobel gas: Oxygen. Pflugers Arch. 2019, 471, 1343–1358. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.J.; Ratcliffe, P.J. Signalling hypoxia by HIF hydroxylases. Biochem. Biophys. Res. Commun. 2005, 338, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhang, D.-X.; Shi, X.; Zhang, Q.; Huang, Y.-S. Activation of the prolyl-hydroxylase oxygen-sensing signal cascade leads to AMPK activation in cardiomyocytes. J. Cell. Mol. Med. 2012, 16, 2049–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cockman, M.E.; Lippl, K.; Tian, Y.-M.; Pegg, H.B.; Figg, W.D.; Abboud, M.I.; Heilig, R.; Fischer, R.; Myllyharju, J.; Schofield, C.J.; et al. Lack of activity of recombinant HIF prolyl hydroxylases (PHDs) on reported non-HIF substrates. Elife 2019, 8. [Google Scholar] [CrossRef]
- Scholz, C.C.; Rodriguez, J.; Pickel, C.; Burr, S.; Fabrizio, J.-A.; Nolan, K.A.; Spielmann, P.; Cavadas, M.A.S.; Crifo, B.; Halligan, D.N.; et al. FIH Regulates Cellular Metabolism through Hydroxylation of the Deubiquitinase OTUB1. PLoS Biol. 2016, 14, e1002347. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Liu, W.; Doycheva, D.M.; Gamdzyk, M.; Lu, W.; Tang, J.; Zhang, J.H. Ghrelin attenuates oxidative stress and neuronal apoptosis via GHSR-1α/AMPK/Sirt1/PGC-1α/UCP2 pathway in a rat model of neonatal HIE. Free Radic. Biol. Med. 2019, 141, 322–337. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, H.; Zhang, Y.; Wang, Z.; Liu, S.; Cui, L. Pretreatment of ghrelin protects H9c2 cells against hypoxia/reoxygenation-induced cell death via PI3K/AKT and AMPK pathways. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2179–2187. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Cheng, K.K.-Y. Hypothalamic AMPK as a Mediator of Hormonal Regulation of Energy Balance. Int. J. Mol. Sci. 2018, 19, 3552. [Google Scholar] [CrossRef] [Green Version]
- Birnbaum, M.J. Activating AMP-activated protein kinase without AMP. Mol. Cell 2005, 19, 289–290. [Google Scholar] [CrossRef]
- Lim, C.T.; Kola, B.; Korbonits, M. AMPK as a mediator of hormonal signalling. J. Mol. Endocrinol. 2010, 44, 87–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minokoshi, Y.; Kim, Y.-B.; Peroni, O.D.; Fryer, L.G.D.; Müller, C.; Carling, D.; Kahn, B.B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, J.; Gao, J.; Yang, H.; Wang, Y.; Manithody, C.; Li, J.; Rezaie, A.R. Antithrombin up-regulates AMP-activated protein kinase signalling during myocardial ischaemia/reperfusion injury. Thromb. Haemost. 2015, 113, 338–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-L.; Guo, H.; Zhang, C.-S.; Lin, S.-Y.; Yin, Z.; Peng, Y.; Luo, H.; Shi, Y.; Lian, G.; Zhang, C.; et al. AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab. 2013, 18, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Kles, K.A.; Tappenden, K.A. Hypoxia differentially regulates nutrient transport in rat jejunum regardless of luminal nutrient present. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G1336–G1342. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [Green Version]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Chandel, N.S. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am. J. Physiol. Cell Physiol. 2011, 300, C385–C393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel Malik, R.; Zippel, N.; Frömel, T.; Heidler, J.; Zukunft, S.; Walzog, B.; Ansari, N.; Pampaloni, F.; Wingert, S.; Rieger, M.A.; et al. AMP-Activated Protein Kinase α2 in Neutrophils Regulates Vascular Repair via Hypoxia-Inducible Factor-1α and a Network of Proteins Affecting Metabolism and Apoptosis. Circ. Res. 2017, 120, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-N.; Yang, W.K.; Kim, J.; Kim, H.S.; Kim, E.J.; Yun, H.; Park, H.; Kim, S.S.; Choe, W.; Kang, I.; et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis 2008, 29, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Minet, E.; Michel, G.; Mottet, D.; Raes, M.; Michiels, C. Transduction pathways involved in Hypoxia-Inducible Factor-1 phosphorylation and activation. Free Radic. Biol. Med. 2001, 31, 847–855. [Google Scholar] [CrossRef]
- Kietzmann, T.; Mennerich, D.; Dimova, E.Y. Hypoxia-Inducible Factors (HIFs) and Phosphorylation: Impact on Stability, Localization, and Transactivity. Front. Cell Dev. Biol. 2016, 4, 11. [Google Scholar] [CrossRef]
- Laderoute, K.R.; Amin, K.; Calaoagan, J.M.; Knapp, M.; Le, T.; Orduna, J.; Foretz, M.; Viollet, B. 5’-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol. Cell. Biol. 2006, 26, 5336–5347. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, Y.; Ohta, K.; Okoshi, R.; Suehara, M.; Kizaki, H.; Nakagawa, K. Hypoxia induces expression and activation of AMPK in rat dental pulp cells. J. Dent. Res. 2007, 86, 903–907. [Google Scholar] [CrossRef]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013, 17, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Lyu, X.; Wang, J.; Guo, X.; Wu, G.; Jiao, Y.; Faleti, O.D.; Liu, P.; Liu, T.; Long, Y.; Chong, T.; et al. EBV-miR-BART1-5P activates AMPK/mTOR/HIF1 pathway via a PTEN independent manner to promote glycolysis and angiogenesis in nasopharyngeal carcinoma. PLoS Pathog. 2018, 14, e1007484. [Google Scholar] [CrossRef] [Green Version]
- Zadra, G.; Batista, J.L.; Loda, M. Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies. Mol. Cancer Res. 2015, 13, 1059–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, K.; Seo, S.; Ki, S.H.; Shin, S.M. Sestrin2 inhibits hypoxia-inducible factor-1α accumulation via AMPK-mediated prolyl hydroxylase regulation. Free Radic. Biol. Med. 2016, 101, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Majd, S.; Power, J.H.T.; Chataway, T.K.; Grantham, H.J.M. A comparison of LKB1/AMPK/mTOR metabolic axis response to global ischaemia in brain, heart, liver and kidney in a rat model of cardiac arrest. BMC Cell Biol. 2018, 19, 7. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Puppala, D.; Feng, X.; Monetti, M.; Lapworth, A.L.; Geoghegan, K.F. Chemoproteomic analysis of intertissue and interspecies isoform diversity of AMP-activated protein kinase (AMPK). J. Biol. Chem. 2013, 288, 35904–35912. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Ruderman, N.B.; Xu, X.J.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: A long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef]
- Salminen, A.; Hyttinen, J.M.T.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, X.; Zhang, W.; He, J.; Xu, B.; Lei, B.; Wang, Z.; Cates, C.; Rousselle, T.; Li, J. Activation of AMPK inhibits inflammatory response during hypoxia and reoxygenation through modulating JNK-mediated NF-κB pathway. Metab. Clin. Exp. 2018, 83, 256–270. [Google Scholar] [CrossRef]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Oliver, K.M.; Taylor, C.T.; Cummins, E.P. Hypoxia. Regulation of NFkappaB signalling during inflammation: The role of hydroxylases. Arthritis Res. Ther. 2009, 11, 215. [Google Scholar] [CrossRef] [Green Version]
- D’Ignazio, L.; Batie, M.; Rocha, S. Hypoxia and Inflammation in Cancer, Focus on HIF and NF-κB. Biomedicines 2017, 5, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dengler, F. Activation of AMPK under Hypoxia: Many Roads Leading to Rome. Int. J. Mol. Sci. 2020, 21, 2428. https://doi.org/10.3390/ijms21072428
Dengler F. Activation of AMPK under Hypoxia: Many Roads Leading to Rome. International Journal of Molecular Sciences. 2020; 21(7):2428. https://doi.org/10.3390/ijms21072428
Chicago/Turabian StyleDengler, Franziska. 2020. "Activation of AMPK under Hypoxia: Many Roads Leading to Rome" International Journal of Molecular Sciences 21, no. 7: 2428. https://doi.org/10.3390/ijms21072428
APA StyleDengler, F. (2020). Activation of AMPK under Hypoxia: Many Roads Leading to Rome. International Journal of Molecular Sciences, 21(7), 2428. https://doi.org/10.3390/ijms21072428