1. Introduction
Antibacterial resistance has intensified over the past several decades and threatens current standards of clinical care [
1,
2]. The majority of antibacterial drugs that are used today prevent or subvert just five essential cellular processes conserved across bacteria [
3]. Although we have multiple antibiotic classes at our disposal, the rapid acquisition of resistance remains an ongoing issue [
4]. Therefore, new antibacterial therapies are urgently needed. One of the pathogens where there is an unmet need for new antibacterial agents is
Pseudomonas aeruginosa, an opportunistic pathogen renowned for its metabolic versatility and high-level intrinsic antibiotic resistance. Although traditionally cited as being “ubiquitous”, recent work has indicated that
P. aeruginosa is primarily associated with human activity and the built environment [
5].
P. aeruginosa encounters several metabolic challenges in vivo, particularly during infection scenarios, where nutrients are limited. When the primary source of carbon is derived from C
2 molecules, the glyoxylate shunt is utilized to provide gluconeogenic precursors. The glyoxylate shunt is composed of two enzymes: Isocitrate lyase (ICL) and malate synthase (MS). ICL cleaves isocitrate to yield glyoxylate and succinate in a reversible reaction. This reaction is then followed by an irreversible condensation of glyoxylate and acetyl coenzyme A by MS, leading to formation of the gluconeogenic precursor, malate, and CoA [
6,
7,
8].
ICL from
P. aeruginosa (ICL
Pa) is a tetramer, with each protomer comprised of 17 α-helices and 14 β-strands arranged as a TIM barrel-like core. The active site is located between β4 and β5 [
9]. MS from
P. aeruginosa (MS
Pa) has also been characterized. The enzyme is a monomer comprised of an 8α/8β TIM barrel fold and an α-helical C-terminal domain, which borders the active site along with the TIM barrel [
10].
Beyond their role in carbon fixation, ICL
Pa and MS
Pa have been implicated in virulence, persistence, and antibiotic resistance [
11,
12]. The glyoxylate shunt is conditionally essential for survival in mammalian systems, and a double deletion mutant (ΔICL ΔMS) of
P. aeruginosa was found to be completely avirulent in a mouse pulmonary infection model [
13]. Given their importance in pathogenicity and the fact that there are no human orthologues of the glyoxylate shunt enzymes, ICL
Pa and MS
Pa have become attractive targets for drug discovery efforts. Indeed, nature has already targeted the glyoxylate shunt as an antibacterial strategy. The human enzyme, Irg1, synthesizes the ICL-inhibitory metabolite, itaconate, during macrophage activation [
14]. Several other ICL inhibitors have been identified, such as 3-bromopyruvate, 3-nitropropionate, and 2-vinyl-D-isocitrate [
15,
16,
17]. However, these inhibitors display nonspecific hepatotoxicity, making them unsuitable as drug candidates [
18].
Phenyl-diketo acid (PDKA) inhibitors of MS from
Mycobacterium tuberculosis (MS
Mt) have been developed through structure-based drug design. Their mechanism of action appears to be via chelation of the active site-bound Mg
2+ by the 1-2-diol [
19]. Further analysis of the structure-activity relationships indicated that these PDKAs adopt an unusual conformational pose once bound, which allows close contact between the PDKA aromatic ring and the carboxylic acid of a nearby aspartate side chain [
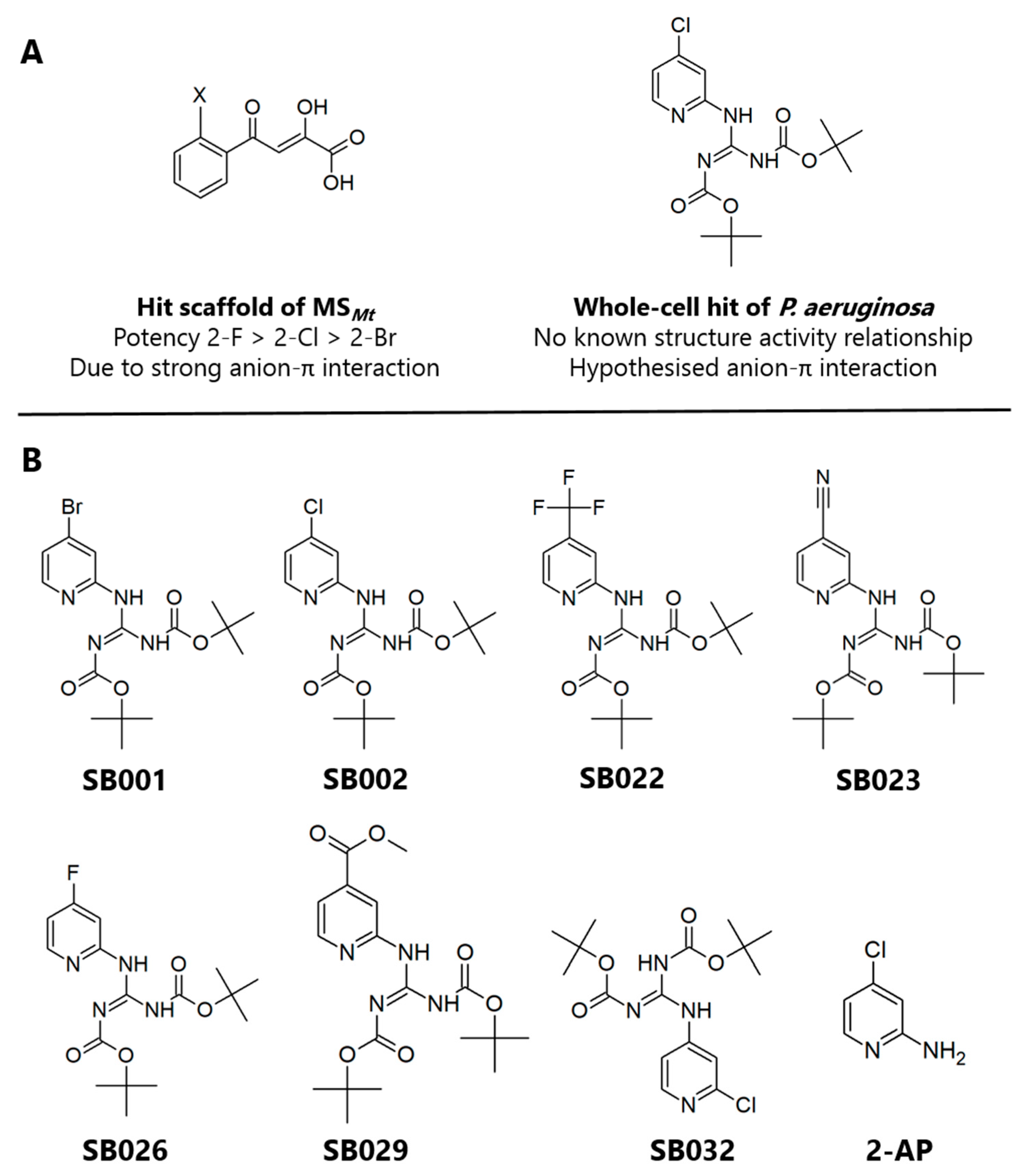
20]. This pose permits the formation of an unusual face-on anion-π interaction. Optimization of the PDKA hits revealed that the potency of the inhibitors improved with increasing electrophilicity of the substituent at the ortho-position on the aromatic ring (2-F > 2-Cl > 2-Br > 2-Me > 2-H), resulting in stronger anion-π interactions [
21].
Fahnoe and coworkers were the first to demonstrate the tractability of the glyoxylate shunt in
P. aeruginosa as an antibacterial target using a combined chemical and genetic approach [
13]. They identified a suite of eight compounds capable of preventing
P. aeruginosa growth on acetate as a sole carbon source. Remarkably, these compounds also inhibited purified ICL
Pa and MS
Pa with IC
50 values in the low micromolar range. However, the mechanism of inhibition, binding affinity, cytotoxicity profile, drug–drug interactions, metabolic clearance properties, and specificity of the compounds were not investigated further.
In the current study, we hypothesized that by combining the structural and electronic features of known inhibitors of the glyoxylate shunt enzymes, we might be able to improve their efficacy. Given its inhibitory activity against both ICL
Pa and MS
Pa, the 2-aminopyridine framework described by Fahnoe et al. [
13] was considered a good starting point, especially given the π-acidity of the scaffold (
Figure 1A). We, therefore, synthesized a suite of compounds, which all shared the core 2-aminopyridine structure but varied in the electronegativity of the substituents on the aromatic ring (
Figure 1B). (Synthetic routes are outlined in the
Supplementary Materials.) The targeted analogs were tested for their ability to inhibit purified ICL
Pa and MS
Pa, and the mechanism of inhibition. Crucially, we also further characterized the hits for cytotoxicity, in vitro drug clearance and cytochrome P450 inhibition, and protein binding.
3. Discussion
In 2012, Fahnoe et al. reported that a pair of 2-aminopyridine (2-AP) derivatives, discovered following a high-throughput screen, could apparently inhibit both enzymes (ICL
Pa and MS
Pa) of the glyoxylate shunt [
13]. This was a remarkable observation since ICL
Pa and MS
Pa exhibit very different structures and activities [
9,
10]. However, no detailed SAR was carried out, and, even for the hits, the mode of action, enzyme binding properties, cytotoxicity profiles, drug–drug interactions, and drug clearance properties of the compounds were not investigated further by Fahnoe et al. [
13] In the current work, we rectified this and also carried out a limited SAR analysis of the 2-AP derivatives.
The most potent inhibitor that we discovered (SB002, 4-chloro-2-AP) was identical to one of the 2-AP hits (referred to as compound 3) obtained by Fahnoe et al. in their high-throughput screen. However, in our hands, SB002 was a more potent inhibitor of MSPa than it was of ICLPa (IC50 = 4.5 µM and 13.3 µM, respectively), whereas Fahnoe et al. reported the opposite (IC50 = 0.17 µM for ICLPa and 5.3 µM for MSPa). The fact that the MIC of SB002 was lower than the IC50 for ICLPa and MSPa may indicate additional off-target effects or possibly differential partitioning of the compound into cells. SB002 lost activity as an enzyme inhibitor when the Boc protecting groups were removed and showed diminished activity when the chlorine atom at the 4-position on the pyridine ring was substituted with either bromine (SB001) or fluorine (SB026). Interestingly, a close analogue (SB029) of the most potent inhibitor identified by Fahnoe et al. (their compound 4, which is identical to SB029 except with a propionic acid substituent on the 2-AP ring instead of a methyl ester moiety) showed only weak activity in our experiments, indicating that inhibition may be exquisitely sensitive to even small structural variations.
SB002 and SB023 could completely inhibit
P. aeruginosa growth in M9-acetate, indicating that they are cell-permeable. The MIC for SB002 of 1.6 µM is comparable with the MIC reported previously by Fahnoe et al. for compound 3 (8 μg/mL, or 21.6 µM) [
13]. Furthermore, the compounds had little or no impact on
P. aeruginosa growth in LB, indicating that they are not generically toxic to the organism. It is notable that SB002 and SB023 are structurally dissimilar to the endogenous substrates of the glyoxylate shunt enzymes or with any of the known inhibitors of ICL. Crucially, SB002 and SB023 did not exhibit profound cytotoxicity towards any of the human cell lines we tested; although for the most part, they did exhibit a relatively narrow potential therapeutic index (TI < 100). This is not necessarily a major problem; there are several approved antibiotics or classes of antibiotics on the market with narrow TIs, including the aminoglycosides, vancomycin and polymyxin B [
30]. SB002 and SB023 were metabolized by cytochrome P450 in human liver microsomes, with half-lives of 81 and 72 min, respectively (
Table 4). These data suggested that both compounds are direct substrates of the CYP450 isozymes. However, neither compound prevented the microsomal CYP450 isozymes from metabolizing other drugs.
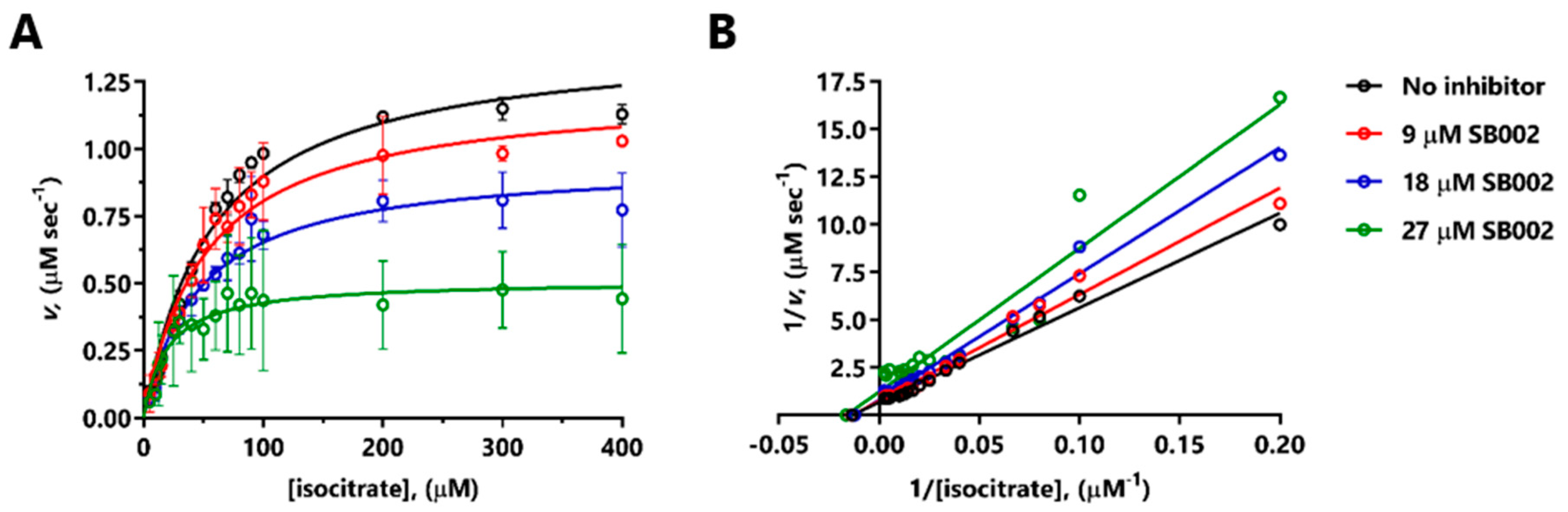
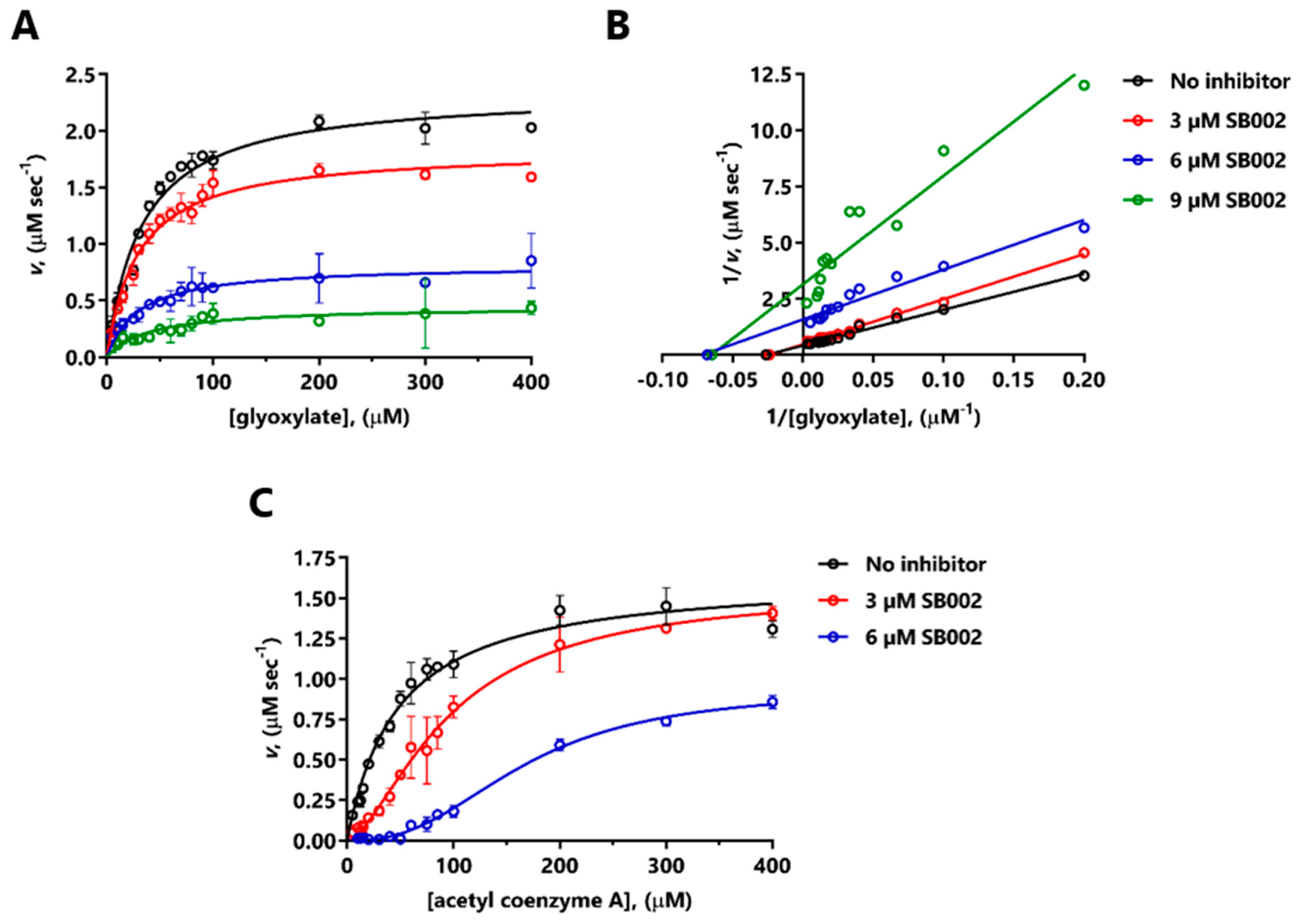
Given that SB002 was (in our hands) the most potent inhibitor of both ICL
Pa and MS
Pa, we used enzyme kinetics to investigate its mode of action on each enzyme. SB002 displayed complex, concentration-dependent inhibition kinetics with MS
Pa. At lower concentrations, SB002 elicited uncompetitive inhibition of MS
Pa (indicative of inhibitor binding to the MS
Pa-glyoxylate complex) but this changed to noncompetitive inhibition (inhibitor binding to the free MS
Pa enzyme) at higher SB002 concentrations. The effects of SB002 on MS
Pa kinetics with respect to acetyl coenzyme A were equally complex, although, overall, they were consistent with competitive inhibition. This was in line with our previous demonstration that the MS
Pa acetyl coenzyme A binding site is a highly druggable binding pocket [
10]. We speculate that SB002 could potentially interact with this pocket, thereby blocking the binding of acetyl coenzyme A at the protein’s surface. However, attempts to co-crystallize MS
Pa with SB002 (± glyoxylate) or to obtain structures of the bound inhibitor-MS
Pa complex through crystal soaking all failed.
SB002 inhibition of ICL
Pa was best described using an uncompetitive model, although the double-reciprocal plot data indicated more complex, “mixed” uncompetitive and noncompetitive inhibition (
Figure 2B). Crousilles et al. reported that ICL
Pa has a flexible loop between β4 and β5, which points away from the active site [
9]. This loop contains the general catalytic base, Cys222. However, in the crystal structure of ICL from
M. tuberculosis, the loop is positioned inwards. This closes off the active site and presumably brings Cys222 in close proximity to the substrate (isocitrate) for catalysis [
9]. We speculate that when the substrate binds to ICL
Pa, this leads to closure of the active cleft, and it seems plausible that SB002 binding to a nearby site somehow prevents this. Unfortunately, attempts to co-crystallize ICL
Pa with SB002 failed to yield diffracting crystals.
ITC revealed that SB002 binds to MS
Pa with a greater affinity than to ICL
Pa, which correlates well with the kinetic data. However, and as outlined above, Fahnoe et al. found that SB002 is a better inhibitor of ICL
Pa [
13]. The reason for this apparent discrepancy is not yet clear, although one possibility is that batch-to-batch variation in the enzyme preparations (i.e., the amount of integrally active enzyme present) might be responsible. We note that our preparations were “crystallization grade” enzymes, although we cannot exclude the possibility that some of the enzyme in each preparation could have been inactive.
In summary, we have shown that the 2-aminopyridines display the remarkable property of being able to inhibit two consecutive enzymes in a conditionally essential metabolic pathway, the glyoxylate shunt. This is all the more surprising given the very different structures and mode of action of each enzyme. Our top hit compounds, SB002 and SB023, displayed better potency than itaconate (a naturally occurring ICL inhibitor) and low cytotoxicity and drug–drug interactions. Current efforts are aimed at exploring the SAR of these compounds further and in establishing where they bind on the enzyme targets (through X-ray crystallography) with a view to employing structure-guided improvements in inhibitor design.
4. Materials and Methods
4.1. Protein Purification of P. aeruginosa ICL and MS
ICL
Pa and MS
Pa were purified essentially as previously described [
9,
10].
4.2. Antibacterial Assays
Growth assays were carried out using clear, U-bottom, sterile 96-well plates (Thermo Fisher Scientific, Bishop’s Stortford, UK). P. aeruginosa PAO1 overnight cultures were diluted to a starting OD600 of 0.05 in LB medium or M9 medium supplemented with 0.5% acetate. The test compounds were added to the indicated final concentrations, and the plates were covered with gas-permeable adhesive seals (Scientific Laboratory Supplies, Nottingham, UK). Assay controls included 1% DMSO, a positive antibiotic control (ciprofloxacin, 1 µg/mL), and medium-only wells as a control for background absorbance. Plates were incubated in a FLUOstar® Omega microplate reader (BMG Labtech, Aylesbury, UK) for 18 h at 37 °C with shaking at 200 rpm, and automatic OD600 readings were recorded every 60 min. MICs were defined as the lowest concentration of test compound, which inhibited >75% of bacterial growth.
4.3. Inhibitor Screening and Hit Confirmation
The enzymatic activity of purified ICL
Pa and MS
Pa was assayed essentially as previously described [
9,
10]. Compounds were added to the reaction mixtures (minus substrates) and incubated for 5 min at 37 °C. The enzyme concentration in the reaction mixtures was 170 nM for ICL and 25 nM for MS (based on the concentration of the monomer in each case). The DMSO solvent concentration did not exceed 1%
v/v. The reactions were initiated by the addition of pH-adjusted substrates, and the initial rates were recorded as A
324 nm (for ICL
Pa) and A
412 nm (for MS
Pa) using a Biospectrometer (Eppendorf, Stevenage, UK). Substrate turnover was calculated from the molar extinction coefficient of the assay products: Glyoxylate-phenylhydrazone (16,800/M/cm) for ICL
Pa and 2-nitro-5-thiobenzoic acid (14,150/M/cm) for MS
Pa.
4.4. Kinetic Mode of Action
The mode of action of SB002 on MS
Pa and ICL
Pa was determined by titrating the respective substrates of each enzyme in the presence of three fixed concentrations of SB002 (selected to lie around the measured IC
50 value) and measuring the reaction rates as outlined in
Section 4.3. Double-reciprocal plots and best-fit inhibition models (GraphPad Prism v. 7.04) were used to assess the mode of inhibition. Inhibition constants were calculated using GraphPad Prism v. 7.04 (GraphPad Software, Inc., La Jolla, CA, USA). Each data point represents the mean ± SD for three independent replicates.
4.5. Isothermal Titration Calorimetry
Protein samples were dialyzed (2 × 1 L) in a D-tube dialyzer (Merck, Watford, UK) for 3 h in 25 mM Tris-HCl, 100 mM NaCl, and 0.1 mM TCEP (pH 7.5). The protein samples were degassed and reverse-titrated into a calorimeter cell (VP-ITC, Malvern Panalytical, Malvern, UK) containing 11 µM of SB002 at 25 °C with 200 rpm stirring. The final protein concentration was 110 µM (MS
Pa) or 202 µM (ICL
Pa). The syringe and the cell both contained a final concentration of 0.1% DMSO. The injection parameters for MS
Pa were: Reference power 15 µcal/sec, initial injection of 3 µL (3.6 sec per injection) with subsequent injections of 10 µL (12 sec per injection). Injections were spaced by 150 sec with a filter period of 2 sec. The injection parameters for ICL
Pa were: Reference power 15 µcal/sec, initial injection of 3 µL (3.6 sec per injection) with subsequent injections of 8 µL (10 sec per injection). Injections were spaced by 170 sec with a filter period of 2 sec. Data were analyzed in NITPIC (NITPIC, Bethesda, MD, USA) [
31] for baseline calculations and then fit for thermodynamic parameters using the OneSite model in Origin 7.0 software (Origin Software Solutions, Bristol, UK). Each titration was carried out in duplicate.
4.6. Cell Viability Assays
The number of viable cells based on ATP quantification was determined using the CellTiter-Glo® luminescent cell viability assay (Promega, Madison, WI, USA, lot no. 0000200446). Briefly, cells were seeded into white, flat-bottomed, sterile 96-well plates (PerkinElmer, Beaconsfield, UK) and incubated for 24 h. For A-375 cells, we used 10,000 cells/well, and for H27 cells, we used 7500 cells/well. Test compounds were diluted in assay medium (Glutamax DMEM with 5% FBS (Gibco)) and added to the plates in two-fold dilutions starting at 100 µM. Negative controls (0.5% DMSO) and positive controls (camptothecin, 1.6 µM for the A-375 cells and 50 µM for the H27 cells) were also included, and the plates were incubated for an additional 48 h (95% humidity, 5% CO2 at 37 °C). After incubation, the cells were washed with PBS. To each well, we then added 50 µL of assay medium plus 50 µL of CellTiter-Glo® reagent. The samples were then shaken for 2 min at 100 rpm at 25 °C, followed by a further 10 min at 25 °C to stabilize the luminescence signal. Luminescence was measured using a VarioSkan LUX multimode microplate reader (Thermo Fisher Scientific, Vantaa, Finland).
The CytoTox 96® nonradioactive cytotoxicity assay (Promega, Madison, WI, USA) was performed to determine the number of cells affected by necrosis measuring LDH release. A U937 cell suspension (5 × 105 cells/mL) was prepared, and 99.5 µL of this suspension were added to the wells of clear, sterile, 96-well, U-bottomed plates (Greiner, Dungannon, UK). The test compounds (0.5 µL of a 200 × concentrated stock) or 0.5% DMSO (control) were added to each well, and the plates were shaken for 1 min at 100 rpm, followed by 3.5 h at 37 °C with 5% CO2. At this point, CytoTox 96® 10× lysis solution was added to the wells reserved for total lysis. These samples represented the LDH released following complete cell lysis. After 30 min, 50 µL of supernatant from each test well and total lysis well was transferred to new 96-well plates (Thermo Fisher Scientific, Bishop’s Stortford, UK). Prethawed CytoTox 96® buffer was mixed with the CytoTox 96® substrate, and 50 µL were added into each well. The plates were incubated for 10 min in darkness at 25 °C. CytoTox 96® stop buffer (50 µL) was added, and the plates were incubated for a further 15 min. LDH release was quantified by measuring A492 nm in a CLARIOstar® multimode microplate reader (BMG Labtech, Aylesbury, UK).
4.7. In Vitro Drug Metabolism
The metabolic stability of the test compounds was measured following incubation with liver microsomes from humans (BioIVT, lot no. IHG; West Sussex, UK) and rats (BD Gentest, lot no. 60614; BD Biosciences, Milan, Italy). Verapamil and dextromethorphan were used as positive controls. An automated incubation procedure was performed using the RSP Freedom Evo liquid dispensing and incubation system (TECAN, Milan, Italy). Five microliters of 50 µM test compound or positive control were added to 445 µL of microsomal suspension in 96 deep-well plates. The microsomal preparation comprised 0.56 mg/mL of pooled and homogenized liver microsomes suspended in 50 mM potassium phosphate buffer, pH 7.4. After 5 min at 37 °C, the reactions were initiated by adding 50 µL of prewarmed NADPH regenerating buffer to each sample. The regenerating buffer comprised a 2% NaHCO
3 solution containing 1.7 mg NADP, 7.8 mg glucose-6-phosphate, and 6 units of glucose-6-phosphate dehydrogenase per mL. The samples were incubated at 37 °C, and 50 µL aliquots were withdrawn after 0, 3, 10, 15, 30, and 45 min and quenched in 150 µL acetonitrile. Aliquots (100 µL) of the quenched sample were mixed with 200 µL of internal standard (100 ng/mL Rolipram in water) prior to analysis on an API4000 Qtrap mass spectrometer (Applied Biosystems/MDS SCIEX, Monza, Italy) coupled to an HP1100 series HPLC system (Agilent Technologies, Milan, Italy) and a CTC-PAL auto-injector (CTC Analytics AG, Zwingen, Switzerland). The analytical column was a Synergi “Max-RP” reverse phase C12 with TMS endcapping (30 mm × 2 mm, 4 µm particle size) (Phenomenex, Inc., Bologna, Italy). The column temperature was maintained at 60 °C. The injected sample volume was 10 µL, and analytes were eluted at a flow rate of 800 µL/min in a gradient of eluent A (water with 0.1% formic acid) and eluent B (acetonitrile with 0.1% formic acid). The gradient conditions for elution were 5% B (0.00–0.20 min), 5–95% B (0.20–1.00 min), 95% B (1.00–1.30 min), 95–5% B (1.30–1.31 min), and 5% B (1.31–1.50 min). Analytes were quantified in multiple reaction-monitoring mode. The mass transitions used for positive controls and hit compounds are listed in
Table S2. Peak areas were integrated using Integrator Software from Agilent (Agilent Technologies, Milan, Italy) and were divided by the peak areas of the internal standard. The percentage of parent compound remaining was calculated by normalizing the peak area ratio of the parent compound to that of the internal standard at time 0. The half-lives of compound metabolism were derived from the linear portion of the slope of ln percent parent compound vs time. Intrinsic clearance in liver microsomes was estimated from:
Here,
k is the rate of depletion of the parent compound (mL/min),
V is the volume of the reaction mixture in mL, and
M is the concentration of microsomal protein (in mg/mL). The amounts of protein in the incubations were scaled relative to the average liver- and body-mass of each species to obtain intrinsic hepatic clearance (
Clh, int, in mg/min/kg) (
Table S3). These scaling factors were used along with the average hepatic blood flow (
Q, in mL/min) of each species to obtain in vivo correlations (
Clh, in mL/min/kg) using the well-stirred model [
32].
4.8. Cytochrome P450 Inhibition
The CYP450 inhibition was determined by measuring the reaction products of known CYP450 substrates (probes). Probe solutions were prepared by diluting stock solutions with buffer (100 mM Tris-HCl, pH 7.5, and 0.5 mM EDTA) to the final indicated concentrations (
Figure 5), and adding 180 µL of pooled human liver microsomes (20 mg/mL) (BioIVT, West Sussex, UK). These probe solutions were dispensed (195 µL) into clear 96-well plates with low evaporation lids (Corning, Turin, Italy) and the plates were prewarmed to 37 °C in a thermomixer (Hamilton, Agrate Brianza, Italy). Next, 5 µL of the test compound or appropriate positive control (
Table S4) were added to the wells and the plates were incubated for a further 5 min. Following this, the reactions were started by adding NADPH regenerating solution (50 µL, see previous
Section 4.7) to each well. CYP450 mixtures containing phenacetin, rosiglitazone, mephenytoin, atorvastatin, and nifedipine probes were incubated for 10 min at 37 °C, whereas those with midazolam and diclofenac were incubated for 5 min at 37 °C. Aliquots (100 µL) from each reaction mixture were quenched in 200 µL acetonitrile, and the mixtures were sedimented at 3000 ×g for 10 min at 4 °C. The supernatants were diluted in water containing 5 ng/mL Rolipram as an internal standard for LC-MS/MS analysis. All incubations were carried out in triplicate.
Probe products were analyzed on an API4000 Qtrap mass spectrometer (Applied Biosystems/MDS SCIEX, Monza, Italy) coupled with an Acquity UPLC system (Waters, Milan, Italy) and a CTC-PAL auto-injector (CTC Analytics AG, Zwingen, Switzerland). For the CYP2C8 and CYP3A4 probe products, the analytical column was an Acquity BEH reverse phase C18 (50 × 2.1 mm, 1.7-µm particle size) (Waters, Milan, Italy). The column temperature was maintained at 60 °C. The injected sample volume was 10 µL, and analytes were eluted at a flow rate of 800 µL/min in a gradient of eluent A (water with 0.1% formic acid) and eluent B (acetonitrile with 0.1% formic acid). The gradient conditions for elution were 5% B (0.00–0.20 min), 5–95% B (0.20–1.50 min), 95% B (1.50–1.75 min), 95–5% B (1.75–1.80 min), and 5% B (1.80–2.00 min). For the CYP1A2, CYP2C9, CYP2C19, and CYP2D6 probe products, the analytical column was an Acquity BEH reverse-phase phenyl (50 × 2.1 mm, 1.7-µm particle size) (Waters, Milan, Italy). The column temperature was maintained at 45 °C. The injected sample volume was 10 µL, and analytes were eluted at a flow rate of 700 µL/min in a gradient of eluent A (water with 0.1% formic acid) and eluent B (acetonitrile with 0.1% formic acid). The gradient conditions for elution were 100% B (0.00–0.10 min), 100–20% B (0.10–2.30 min), 20% B (2.30–2.90 min), 20–100% B (2.90–3.00 min), and 100% B (3.00–3.80 min). The analytes were quantified in multiple reaction-monitoring mode. The mass transitions used for positive controls and hit compounds are listed in
Table S5. Data were processed, and the percentage control activity was calculated based on a comparison between the ratio of the sample peak area and the peak area.
4.9. Data Analysis
All data fitting was performed with GraphPad Prism version 7.04 (GraphPad Software, Inc., La Jolla, CA, USA). The solid lines are the result of fitting the data to the equations denoted in relevant experimental sections. IC50 values were determined by a nonlinear fit using the equation for sigmoidal dose-response with variable slope. In all graphs, unless otherwise stated, the data points are the mean of experimental triplicates, and all error bars correspond to ± standard deviation.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}