Carnosine as a Possible Drug for Zinc-Induced Neurotoxicity and Vascular Dementia

 ,
,
Abstract
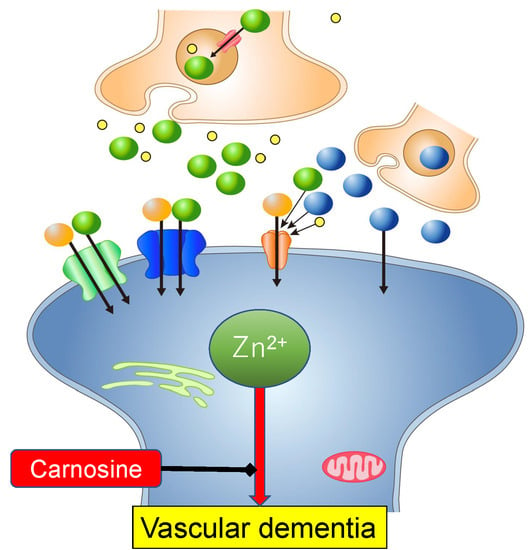
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Zinc and Vascular Dementia
3. Zn2+-Induced Neurotoxicity
3.1. GT1–7 Cells as a Model System for Investigating Zn2+-Induced Neurotoxicity
3.2. Molecular Pathways Underlying Zn2+-Induced Neurotoxicity
3.2.1. Energy Production Pathway
3.2.2. Ca2+ Homeostasis
3.2.3. Endoplasmic Reticulum (ER) Stress Pathway
3.2.4. SAPK/JNK Pathway
3.2.5. ROS Pathway
3.3. Hypothesis Regarding the Molecular Pathways Underlying Zn2+-Induced Neurotoxicity
4. Carnosine as a Protective Substance Against Zn2+-Induced Neurotoxicity
4.1. Screening System for Protective Substances Against Zn2+-Induced Neurotoxicity
4.2. Carnosine as an Endogenous Neuroprotector
4.3. Carnosine in the Brain
4.4. The roles of Carnosine in Protection from Zn2+-Induced Neurotoxicity
5. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AβP | Alzheimer’s β-amyloid protein |
| AMPA | α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid |
| Arc | activity-regulated cytoskeleton |
| Ca-EDTA | calcium ethylenediaminetetraacetic acid |
| CHOP | CCAAT-enhancer-binding protein homologous protein |
| D-APV | 2-amino-5-phosphonovalerate |
| ER | endoplasmic reticulum |
| GABA | γ-aminobutyric acid |
| GnRH | gonadotropin-releasing hormone |
| GADD34 | growth-arrest and DNA-damage-inducible gene 34 |
| HPLC | high performance liquid chromatography |
| LTP | long-term potentiation |
| [Ca2+]i | intracellular calcium levels |
| NMDA | N-methyl-D-aspartate, ROS; reactive oxygen species |
| SAPK/JNK | stress-activated protein kinases/c-Jun amino-terminal kinases |
| VD | vascular dementia |
| VGCC | voltage-dependent Ca2+ channel |
References
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Brás, I.C.; Dominguez-Meijide, A.; Gerhardt, E.; Koss, D.; Lázaro, D.F.; Santos, P.I.; Vasili, E.; Xylaki, M.; Outeiro, T.F. Synucleinopathies: Where we are and where we need to go. J. Neurochem. 2020. [Google Scholar] [CrossRef] [Green Version]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Dementia. Fact sheets of WHO. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 28 March 2020).
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s disease: how far have we come in the clinic? J. Alzheimers Dis. 2018, 62, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.R. Metalloproteins and neuronal death. Metallomics 2010, 2, 186–194. [Google Scholar] [CrossRef]
- Jiang, H.; Song, N.; Jiao, Q.; Shi, L.; Du, X. Iron pathophysiology in parkinson diseases. Adv. Exp. Med. Biol. 2019, 1173, 45–66. [Google Scholar]
- Weiss, J.H.; Sensi, S.L.; Koh, J.Y. Zn(2+): a novel ionic mediator of neural injury in brain disease. Trends Pharmacol. Sci. 2000, 21, 395–401. [Google Scholar] [CrossRef]
- Kawahara, M.; Tanaka, K.I.; Kato-Negishi, M. Zinc, Carnosine, and Neurodegenerative Diseases. Nutrients 2018, 10, 147. [Google Scholar]
- Tanaka, K.I.; Shimoda, M.; Kasai, M.; Ikeda, M.; Ishima, Y.; Kawahara, M. Involvement of SAPK/JNK Signaling Pathway in Copper Enhanced Zinc-Induced Neuronal Cell Death. Toxicol. Sci. 2019, 169, 293–302. [Google Scholar] [CrossRef]
- Kawahara, M.; Konoha, K.; Nagata, T.; Sadakane, Y. Protective substances against zinc-induced neuronal death after ischemia: carnosine a target for drug of vascular type of dementia. Recent Pat. CNS Drug Discov. 2007, 2, 145–149. [Google Scholar]
- Sadakane, Y.; Konoha, K.; Nagata, T.; Kawahara, M. Protective activity of the extracts from Japanese eel (Anguilla japonica) against zinc-induced neuronal cell death: Carnosine and an unknown substance. Trace Nutrient Res. 2007, 24, 98–105. [Google Scholar]
- Hipkiss, A.R. On the relationship between energy metabolism, proteostasis, aging and Parkinson’s disease: possible causative role of methylglyoxal and alleviative potential of carnosine. Aging Dis. 2017, 8, 334–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldyrev, A.A.; Aldini, G.; Derave, W. Physiology and pathophysiology of carnosine. Physiol Rev. 2013, 93, 1803–1845. [Google Scholar] [CrossRef] [PubMed]
- Berezhnoy, D.S.; Stvolinsky, S.L.; Lopachev, A.V.; Devyatov, A.A.; Lopacheva, O.M.; Kulikova, O.I.; Abaimov, D.A.; Fedorova, T.N. Carnosine as an effective neuroprotector in brain pathology and potential neuromodulator in normal conditions. Amino Acids. 2019, 51, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Baye, E.; Menon, K.; de Courten, M.P.; Earnest, A.; Cameron, J.; de Courten, B. Does supplementation with carnosine improve cardiometabolic health and cognitive function in patients with pre-diabetes and type 2 diabetes? study protocol for a randomised, double-blind, placebo-controlled trial. BMJ Open 2017, 7, e017691. [Google Scholar] [CrossRef]
- Dubois, V.D.; Bastawrous, A. N-acetylcarnosine (NAC) drops for age-related cataract. Cochrane Database Syst. Rev. 2017, 2, CD009493. [Google Scholar] [CrossRef]
- Araminia, B.; Shalbafan, M.; Mortezaei, A.; Shirazi, E.; Ghaffari, S.; Sahebolzamani, E.; Mortazavi, S.H.; Shariati, B.; Ardebili, M.E.; Aqamolaei, A.; et al. L-Carnosine combination therapy for major depressive disorder: A randomized, double-blind, placebo-controlled trial. J. Affect. Disord. 2020, 267, 131–136. [Google Scholar] [CrossRef]
- Chester, J.E.; Rowneki, M.; Van Doren, W.; Helmer, D.A. Progression of intervention-focused research for Gulf War illness. Mil. Med. Res. 2019, 6, 31. [Google Scholar] [CrossRef]
- Mori, M.; Mizuno, D.; Konoha-Mizuno, K.; Sadakane, Y.; Kawahara, M. Quantitative analysis of carnosine and anserine in foods by performing high performance liquid chromatography. Biomed. Res. Trace Elem. 2015, 26, 147–152. [Google Scholar]
- Kalaria, R.N. The pathology and pathophysiology of vascular dementia. Neuropharmacology 2018, 134, 226–239. [Google Scholar] [CrossRef]
- Pendlebury, S.T.; Rothwell, P.M. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: A systematic review and meta-analysis. Lancet Neurol. 2009, 8, 1006–1018. [Google Scholar] [CrossRef]
- Shuttleworth, C.W.; Weiss, J.H. Zinc: New clues to diverse roles in brain ischemia. Trends Pharmacol. Sci. 2011, 32, 480–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, M.; Mizuno, D.; Koyama, H.; Konoha, K.; Ohkawara, S.; Sadakane, Y. Disruption of zinc homeostasis and the pathogenesis of senile dementia. Metallomics. 2014, 6, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Stork, C.J.; Li, Y.V. Elevated cytoplasmic free zinc and increased reactive oxygen species generation in the context of brain injury. Acta Neurochir. Suppl. 2016, 121, 347–353. [Google Scholar] [PubMed]
- Becker, J.S.S.; Matusch, A.; Palm, C.; Salber, D.; Morton, K.; Becker, S. Bioimaging of metals in brain tissue by laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) and metallomics. Metallomics 2010, 2, 104–111. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Suh, S.W.; Silva, D.; Frederickson, C.J.; Thompson, R.B. Importance of zinc in the central nervous system: the zinc-containing neuron. J. Nutr. 2000, 130, 1471S–1483S. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Bush, A.I. Synaptically released zinc: physiological functions and pathological effects. Biometals. 2001, 14, 353–366. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat Rev Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef]
- Ueno, S.; Tsukamoto, M.; Hirano, T.; Kikuchi, K.; Yamada, M.K.; Nishiyama, N.; Nagano, T.; Matsuki, N.; Ikegaya, Y. Mossy fiber Zn2+ spillover modulates heterosynaptic N-methyl-D-aspartate receptor activity in hippocampal CA3 circuits. J. Cell Biol. 2002, 158, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Takeda, A.; Nakamura, M.; Fujii, H.; Tamano, H. Synaptic Zn(2+) homeostasis and its significance. Metallomics 2013, 5, 417–423. [Google Scholar] [CrossRef]
- Takeda, A.; Tamano, H. The impact of synaptic Zn2+ dynamics on cognition and its decline. Int. J. Mol. Sci. 2017, 18, 2411. [Google Scholar] [CrossRef] [Green Version]
- McAllister, B.B.; Dyck, R.H. Zinc transporter 3 (ZnT3) and vesicular zinc in central nervous system function. Neurosci. Biobehav. Rev. 2017, 80, 329–350. [Google Scholar] [CrossRef] [PubMed]
- Sandstead, H.H. Subclinical zinc deficiency impairs human brain function. J. Trace Elem. Med. Biol. 2012, 26, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Reid, C.A.; Hildebrand, M.S.; Mullen, S.A.; Hildebrand, J.M.; Berkovic, S.F.; Petrou, S. Synaptic Zn2+ and febrile seizure susceptibility. Br. J. Pharmacol. 2017, 174, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ambrosi, N.; Rossi, L. Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem. Int. 2015, 90, 36–45. [Google Scholar] [CrossRef]
- Kawahara, M.; Kato-Negishi, M.; Tanaka, K.I. Amyloids: Regulators of Metal Homeostasis in the Synapse. Molecules 2020, 25, 1441. [Google Scholar] [CrossRef] [Green Version]
- Frederickson, C.J.; Klitenick, M.A.; Manton, W.I.; Kirkpatrick, J.B. Cytoarchitectonic distribution of zinc in the hippocampus of man and the rat. Brain Res. 1983, 27, 335–339. [Google Scholar] [CrossRef]
- Koh, J.Y.; Suh, S.W.; Gwag, B.J.; He, Y.Y.; Hsu, C.Y.; Choi, D.W. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 1996, 272, 1013–1016. [Google Scholar] [CrossRef]
- Calderone, A.; Jover, T.; Mashiko, T.; Noh, K.M.; Tanaka, H.; Bennett, M.V.; Zukin, R.S. Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J. Neurosci. 2004, 24, 9903–9913. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, Y.; Iida, Y.; Abe, J.; Mifune, M.; Kasuya, F.; Ohta, M.; Igarashi, K.; Saito, Y.; Saji, H. Release of vesicular Zn2+ in a rat transient middle cerebral artery occlusion model. Brain Res. Bull. 2006, 69, 622–625. [Google Scholar] [CrossRef]
- Qi, Z.; Shi, W.; Zhao, Y.; Ji, X.; Liu, K.J. Zinc accumulation in mitochondria promotes ischemia-induced BBB disruption through Drp1-dependent mitochondria fission. Toxicol. Appl. Pharmacol. 2019, 377, 114601. [Google Scholar] [CrossRef]
- Vogt, K.; Mellor, J.; Tong, G.; Nicoll, R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron 2000, 26, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Hopt, A.; Korte, S.; Fink, H.; Panne, U.; Niessner, R.; Jahn, R.; Herms, J. Methods for studying synaptosomal copper release. J. Neurosci. Methods 2003, 128, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Schikorski, T.; Stevens, C.F. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J. Neurosci. 1997, 17, 5858–5867. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Liao, S.L. Zinc toxicity on neonatal cortical neurons: involvement of glutathione chelation. J. Neurochem. 2003, 85, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Ukiana, J.; Uson-Lopez, R.; Sikder, M.T.; Saito, T.; Kurasaki, M. Cytotoxic effects of cadmium and zinc co-exposure in PC12 cells and the underlying mechanism. Chem. Biol. Interact. 2017, 269, 41–49. [Google Scholar] [CrossRef]
- Kawahara, M.; Kato-Negishi, M.; Kuroda, Y. Pyruvate blocks zinc-induced neurotoxicity in immortalized hypothalamic neurons. Cell. Mol. Neurobiol. 2002, 22, 87–93. [Google Scholar] [CrossRef]
- Koyama, H.; Konoha, K.; Sadakane, Y.; Ohkawara, S.; Kawahara, M. Zinc neurotoxicity and the pathogenesis of vascular-type dementia: Involvement of calcium dyshomeostasis and carnosine. J. Clin. Toxicol. 2011, S3, 2161. [Google Scholar]
- Mellon, P.L.; Windle, J.J.; Goldsmith, P.C.; Padula, C.A.; Roberts, J.L.; Weiner, R.I. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 1990, 5, 1–10. [Google Scholar] [CrossRef]
- Mahesh, V.B.; Zamorano, P.; De Sevilla, L.; Lewis, D.; Brann, D.W. Characterization of ionotropic glutamate receptors in rat hypothalamus, pituitary and immortalized gonadotropin-releasing hormone (GnRH) neurons (GT1–7 cells). Neuroendocrinology 1999, 69, 397–407. [Google Scholar] [CrossRef]
- Nissen, E.; Pauli, G.; Vollenbroich, D. WST-1 assay--a simple colorimetric method for virus titration. In Vitro Cell Dev. Biol. Anim. 1997, 33, 28–29. [Google Scholar] [CrossRef]
- Kawahara, M.; Kato-Negishi, M.; Hosoda, R.; Kuroda, Y. Characterization of zinc-induced apoptosis of GT1–7 cells. Biomed. Res. Trace Elements 2002, 13, 280–281. [Google Scholar]
- Kawahara, M.; Konoha, K.; Sadakane, Y. Neurotoxicity of zinc: the involvement of calcium homeostasis and carnosine. Biomed. Res. Trace Elements. 2007, 18, 26–34. [Google Scholar]
- Konoha, K.; Sadakane, Y.; Kawahara, M. Zinc neurotoxicity and its role in neurodegenerative diseases. J. Health Sci. 2006, 52, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, M.; Sadakane, Y.; Koyama, H.; Konoha, K.; Ohkawara, S. D-histidine and L-histidine attenuate zinc-induced neuronal death in GT1–7 cells. Metallomics 2013, 5, 453–460. [Google Scholar] [CrossRef]
- Mizuno, D.; Konoha-Mizuno, D.; Mori, M.; Sadakane, Y.; Koyama, H.; Ohkawara, S.; Kawahara, M. Protective activity of carnosine and anserine against zinc-induced neurotoxicity: A possible treatment for vascular dementia. Metallomics 2015, 7, 1233–1239. [Google Scholar] [CrossRef]
- Konoha, K.; Sadakane, Y.; Kawahara, M. Effects of gadolinium and other metal on the neurotoxicity of immortalized hypothalamic neurons induced by zinc. Biomed. Res. Trace Elements. 2004, 15, 275–277. [Google Scholar]
- Tanaka, K.; Kawahara, M. Copper enhances zinc-induced neurotoxicity and the endoplasmic reticulum stress response in a neuronal model of vascular dementia. Front Neurosci. 2017, 11, 58. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.I.; Kasai, M.; Shimoda, M.; Shimizu, A.; Kubota, M.; Kawahara, M. Nickel enhances zinc-induced neuronal cell death by priming the endoplasmic reticulum stress response. Oxid Med Cell Longev. 2019, 9693726. [Google Scholar] [CrossRef]
- Sheline, C.T.; Behrens, M.M.; Choi, D.W. Zinc-induced cortical neuronal death: contribution of energy failure attributable to loss of NAD(+) and inhibition of glycolysis. J. Neurosci. 2000, 20, 3139–3146. [Google Scholar] [CrossRef] [Green Version]
- Cai, A.L.; Zipfel, G.J.; Sheline, C.T. Zinc neurotoxicity is dependent on intracellular NAD levels and the sirtuin pathway. Eur. J. Neurosci. 2006, 24, 2169–2176. [Google Scholar] [CrossRef]
- Kelland, E.E.; Kelly, M.D.; Toms, N.J. Pyruvate limits zinc-induced rat oligodendrocyte progenitor cell death. Eur. J. Neurosci. 2004, 19, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.H.; Lee, J.Y.; Lee, S.E.; Koh, J.Y.; Yoon, Y.H. Protection by pyruvate of rat retinal cells against zinc toxicity in vitro, and pressure-induced ischemia in vivo. Invest Ophthalmol Vis Sci. 2004, 45, 1523–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Kim, Y.H.; Koh, J.Y. Protection by pyruvate against transient forebrain ischemia in rats. J. Neurosci. 2001, 21, RC171. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.I.; Shimoda, M.; Kawahara, M. Pyruvic acid prevents Cu2+/Zn2+-induced neurotoxicity by suppressing mitochondrial injury. Biochem Biophys Res Commun. 2018, 495, 1335–1341. [Google Scholar] [CrossRef]
- Ji, S.G.; Medvedeva, Y.V.; Weiss, J.H. Zn2+ entry through the mitochondrial calcium uniporter is a critical contributor to mitochondrial dysfunction and neurodegeneration. Exp. Neurol. 2020, 325, 113161. [Google Scholar] [CrossRef]
- Olgar, Y.; Tuncay, E.; Turan, B. Mitochondria-targeting antioxidant provides cardioprotection through regulation of cytosolic and mitochondrial Zn2+ levels with re-distribution of Zn2+-transporters in aged rat cardiomyocytes. Int. J. Mol. Sci. 2019, 20, 3783. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.H.; Sheline, C.T.; Tian, M.; Higashi, T.; McMahon, R.J.; Cousins, R.J.; Choi, D.W. L-type Ca(2+) channel-mediated Zn(2+) toxicity and modulation by ZnT-1 in PC12 cells. Brain Res. 2000, 886, 99–107. [Google Scholar] [CrossRef]
- Zyśk, M.; Gapys, B.; Ronowska, A.; Gul-Hinc, S.; Erlandsson, A.; Iwanicki, A.; Sakowicz-Burkiewicz, M.; Szutowicz, A.; Bielarczyk, H. Protective effects of voltage-gated calcium channel antagonists against zinc toxicity in SN56 neuroblastoma cholinergic cells. PLoS ONE 2018, 13, e0209363. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Chang, S.Y.; Chung, J.M.; Ryu, B.R.; Joo, C.K.; Moon, H.S.; Kang, K.; Yoon, S.H.; Han, P.L.; Gwag, B.J. Attenuation of Zn2+ neurotoxicity by aspirin: role of N-type Ca2+ channel and the carboxyl acid group. Neurobiol. Dis. 2001, 8, 774–783. [Google Scholar]
- Kawahara, M.; Kato-Negishi, M.; Hosoda, R.; Imamura, L.; Tsuda, M.; Kuroda, Y. Brain-derived neurotrophic factor protects cultured rat hippocampal neurons from aluminum maltolate neurotoxicity. J. Inorg. Biochem. 2003, 97, 124–131. [Google Scholar] [CrossRef]
- Büsselberg, D.; Platt, B.; Haas, H.L.; Carpenter, D.O. Voltage gated calcium channel currents of rat dorsal root ganglion (DRG) cells are blocked by Al3+. Brain Res. 1993, 622, 163–168. [Google Scholar] [CrossRef]
- Kaneko, M.; Imaizumi, K.; Saito, A.; Kanemoto, S.; Asada, R.; Matsuhisa, K.; Ohtake, Y. ER stress and disease: toward prevention and treatment. Biol. Pharm. Bull. 2017, 40, 1337–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Liu, L.; Naik, I.; Braunstein, Z.; Zhong, J.; Ren, B. Transcription factor C/EBP homologous protein in health and diseases. Front Immunol. 2017, 8, 1612. [Google Scholar] [CrossRef] [PubMed]
- Paschen, W.; Hayashi, T.; Saito, A.; Chan, P.H. GADD34 protein levels increase after transient ischemia in the cortex but not in the CA1 subfield: implications for post-ischemic recovery of protein synthesis in ischemia-resistant cells. J. Neurochem. 2004, 90, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Nishina, H.; Wada, T.; Katada, T. Physiological roles of SAPK/JNK signaling pathway. J. Biochem. 2004, 136, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Dandekar, A.; Mendez, R.; Zhang, K. Cross talk between ER stress, oxidative stress, and inflammation in health and disease. Methods Mol. Biol. 2015, 1292, 205–214. [Google Scholar]
- Papaconstantinou, J. The role of signaling pathways of inflammation and oxidative stress in development of senescence and aging phenotypes in cardiovascular disease. Cells 2019, 8, 1383. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.I.; Shimoda, M.; Chuang, V.T.G.; Nishida, K.; Kawahara, M.; Ishida, T.; Otagiri, M.; Maruyama, T.; Ishima, Y. Thioredoxin-albumin fusion protein prevents copper enhanced zinc-induced neurotoxicity via its antioxidative activity. Int. J. Pharm. 2018, 535, 140–147. [Google Scholar] [CrossRef]
- Sadakane, Y.; Konoha, K.; Kawahara, M. Protective activity of mango (Mangifera indica L.) fruit against a zinc-induced neuronal cell death is independent of its antioxidant activity. Trace Nutr. Res. 2005, 22, 73–79. [Google Scholar]
- Harris, R.C.; Wise, J.A.; Price, K.A.; Kim, H.J.; Kim, C.K.; Sale, C. Determinants of muscle carnosine content. Amino Acids. 2012, 43, 5–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, H. Role of histidine-related compounds as intracellular proton buffering constituents in vertebrate muscle. Biochemistry (Mosc) 2000, 65, 757–765. [Google Scholar] [PubMed]
- Sale, C.; Artioli, G.G.; Gualano, B.; Saunders, B.; Hobson, R.M.; Harris, R.C. Carnosine: From exercise performance to health. Amino Acids. 2013, 44, 1477–1479. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Mizuno, D.; Konoha-Mizuno, K.; Sadakane, Y.; Kawahara, M. Carnosine concentration in the muscle of thoroughbred horses and its implications in exercise performance. Trace Nutrients Research 2015, 32, 49–53. [Google Scholar]
- Quesnele, J.J.; Laframboise, M.A.; Wong, J.J.; Kim, P.; Wells, G.D. The effects of beta-alanine supplementation on performance: A systematic review of the literature. Int. J. Sport Nutr. Exerc. Metab. 2014, 24, 14–27. [Google Scholar] [CrossRef]
- Rezzani, R.; Favero, G.; Ferroni, M.; Lonati, C.; Moghadasian, M.H. A carnosine analog with therapeutic potentials in the treatment of disorders related to oxidative stress. PLoS ONE 2019, 14, e0215170. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, M.; Kato-Negishi, M.; Tanaka, K. Cross talk between neurometals and amyloidogenic proteins at the synapse and the pathogenesis of neurodegenerative diseases. Metallomics 2017, 9, 619–633. [Google Scholar] [CrossRef]
- Corona, C.; Frazzini, V.; Silvestri, E.; Lattanzio, R.; La Sorda, R.; Piantelli, M.; Canzoniero, L.M.; Ciavardelli, D.; Rizzarelli, E.; Sensi, S.L. Effects of dietary supplementation of carnosine on mitochondrial dysfunction, amyloid pathology, and cognitive deficits in 3xTg-AD mice. PLoS ONE 2011, 6, e17971. [Google Scholar] [CrossRef] [Green Version]
- Caruso, G.; Fresta, C.G.; Musso, N.; Giambirtone, M.; Grasso, M.; Spampinato, S.F.; Merlo, S.; Drago, F.; Lazzarino, G.; Sortinom, M.A.; et al. Carnosine prevents Aβ-induced oxidative stress and inflammation in microglial cells: A key role of TGF-β. Cells 2019, 8, 64. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, M.; Koyama, H.; Nagata, T.; Sadakane, Y. Zinc, copper, and carnosine attenuate neurotoxicity of prion fragment PrP106–126. Metallomics 2011, 3, 726–734. [Google Scholar] [CrossRef]
- Matsukura, T.; Tanaka, H. Applicability of zinc complex of L-carnosine for medical use. Biochemistry (Mosc) 2000, 65, 817–823. [Google Scholar] [PubMed]
- Kimura, K.; Nakano, Y.; Sugizaki, T.; Shimoda, M.; Kobayashi, N.; Kawahara, M.; Tanaka, K.I. Protective effect of polaprezinc on cadmium-induced injury of lung epithelium. Metallomics 2019, 11, 1310–1320. [Google Scholar] [CrossRef] [PubMed]
- Ommati, M.M.; Heidari, R.; Ghanbarinejad, V.; Aminian, A.; Abdoli, N.; Niknahad, H. The neuroprotective properties of carnosine in a mouse model of manganism is mediated via mitochondria regulating and antioxidative mechanisms. Nutr. Neurosci. 2019, 11, 1–13. [Google Scholar]
- Baslow, M.H.; Suckow, R.F.; Berg, M.J.; Marks, N.; Saito, M.; Bhakoo, K.K. Differential expression of carnosine, homocarnosine and N-acetyl-L-histidine hydrolytic activities in cultured rat macroglial cells. J. Mol. Neurosci. 2001, 17, 351–359. [Google Scholar] [CrossRef]
- Bakardjiev, A. Carnosine and beta-alanine release is stimulated by glutamatergic receptors in cultured rat oligodendrocytes. Glia 1998, 24, 346–351. [Google Scholar] [CrossRef]
- Kawahara, M.; Konoha, K. Drugs for prevention or treatment of vascular dementia [Translated from Japanese]. JP5382633, 11 October 2013. [Google Scholar]
- Kawahara, M.; Konoha, K. Drugs for prevention or treatment of vascular dementia [Translated from Japanese]. JP5294194, 21 June 2013. [Google Scholar]
- Zhang, X.; Song, L.; Cheng, X.; Yang, Y.; Luan, B.; Jia, L.; Xu, F.; Zhang, Z. Carnosine pretreatment protects against hypoxia-ischemia brain damage in the neonatal rat model. Eur. J. Pharmacol. 2011, 667, 202–207. [Google Scholar] [CrossRef]
- Park, H.S.; Han, K.H.; Shin, J.A.; Park, J.H.; Song, K.Y.; Kim, D.H. The neuroprotective effects of carnosine in early stage of focal ischemia rodent model. J. Korean Neurosurg. Soc. 2014, 55, 125–130. [Google Scholar] [CrossRef]
- Noguchi, K.; Ali, T.F.S.; Miyoshi, J.; Orito, K.; Negoto, T.; Biswas, T.; Taira, N.; Koga, R.; Okamoto, Y.; Fujita, M.; et al. Neuroprotective effects of a novel carnosine-hydrazide derivative on hippocampal CA1 damage after transient cerebral ischemia. Eur. J. Med. Chem. 2019, 163, 207–214. [Google Scholar] [CrossRef]
- Davis, C.K.; Laud, P.J.; Bahor, Z.; Rajanikant, G.K.; Majid, A. Systematic review and stratified meta-analysis of the efficacy of carnosine in animal models of ischemic stroke. J. Cereb. Blood Flow Metab. 2016, 36, 1686–1694. [Google Scholar] [CrossRef] [Green Version]
- Tomonaga, S.; Hayakawa, T.; Yamane, H.; Maemura, H.; Sato, M.; Takahata, Y.; Morimatsu, F.; Furuse, M. Oral administration of chicken breast extract increases brain carnosine and anserine concentrations in rats. Nutr. Neurosci. 2007, 10, 181–186. [Google Scholar] [CrossRef]
- Hoffman, J.R.; Rathmacher, J.A.; Robinson, J.; Gepner, Y.; Cohen, H. Effect of β-alanine supplementation on carnosine and histidine content in the hippocampus of 14-month-old rats. Appl. Physiol. Nutr. Metab. 2019, 44, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, H.; Heinrich, M.; Birkemeyer, C.; Meixensberger, J.; Gaunitz, F. The proton-coupled oligopeptide transporters PEPT2, PHT1 and PHT2 mediate the uptake of carnosine in glioblastoma cells. Amino Acids. 2019, 51, 999–1008. [Google Scholar] [CrossRef]
- Stuerenburg, H.J. The roles of carnosine in aging of skeletal muscle and in neuromuscular diseases. Biochemistry 2000, 65, 862–865. [Google Scholar] [PubMed]
- Hata, J.; Ohara, T.; Katakura, Y.; Shimizu, K.; Yamashita, S.; Yoshida, D.; Honda, T.; Hirakawa, Y.; Shibata, M.; Sakata, S.; et al. Association between serum β-alanine and risk of dementia. Am. J. Epidemiol. 2019, 188, 1637–1645. [Google Scholar] [PubMed]
- Hisatsune, T.; Kaneko, J.; Kurashige, H.; Cao, Y.; Satsu, H.; Totsuka, M.; Katakura, Y.; Imabayashi, E.; Matsuda, H. Effect of anserine/carnosine supplementation on verbal episodic memory in elderly people. J. Alzheimers Dis. 2016, 50, 149–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuoka, N.; Yoshimine, C.; Hori, M.; Tanaka, M.; Asada, T.; Abe, K.; Hisatsune, T. Effects of anserine/carnosine Supplementation on mild cognitive impairment with APOE4. Nutrients 2019, 11, 1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawahara, M.; Sadakane, Y.; Mizuno, K.; Kato-Negishi, M.; Tanaka, K.-i. Carnosine as a Possible Drug for Zinc-Induced Neurotoxicity and Vascular Dementia. Int. J. Mol. Sci. 2020, 21, 2570. https://doi.org/10.3390/ijms21072570
Kawahara M, Sadakane Y, Mizuno K, Kato-Negishi M, Tanaka K-i. Carnosine as a Possible Drug for Zinc-Induced Neurotoxicity and Vascular Dementia. International Journal of Molecular Sciences. 2020; 21(7):2570. https://doi.org/10.3390/ijms21072570
Chicago/Turabian StyleKawahara, Masahiro, Yutaka Sadakane, Keiko Mizuno, Midori Kato-Negishi, and Ken-ichiro Tanaka. 2020. "Carnosine as a Possible Drug for Zinc-Induced Neurotoxicity and Vascular Dementia" International Journal of Molecular Sciences 21, no. 7: 2570. https://doi.org/10.3390/ijms21072570
APA StyleKawahara, M., Sadakane, Y., Mizuno, K., Kato-Negishi, M., & Tanaka, K. -i. (2020). Carnosine as a Possible Drug for Zinc-Induced Neurotoxicity and Vascular Dementia. International Journal of Molecular Sciences, 21(7), 2570. https://doi.org/10.3390/ijms21072570