Electrophysiological Abnormalities in VLCAD Deficient hiPSC-Cardiomyocytes Can Be Improved by Lowering Accumulation of Fatty Acid Oxidation Intermediates

 , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. VLCADD Patients
2.2. Generation of Patient-Specific VLCADD-CM
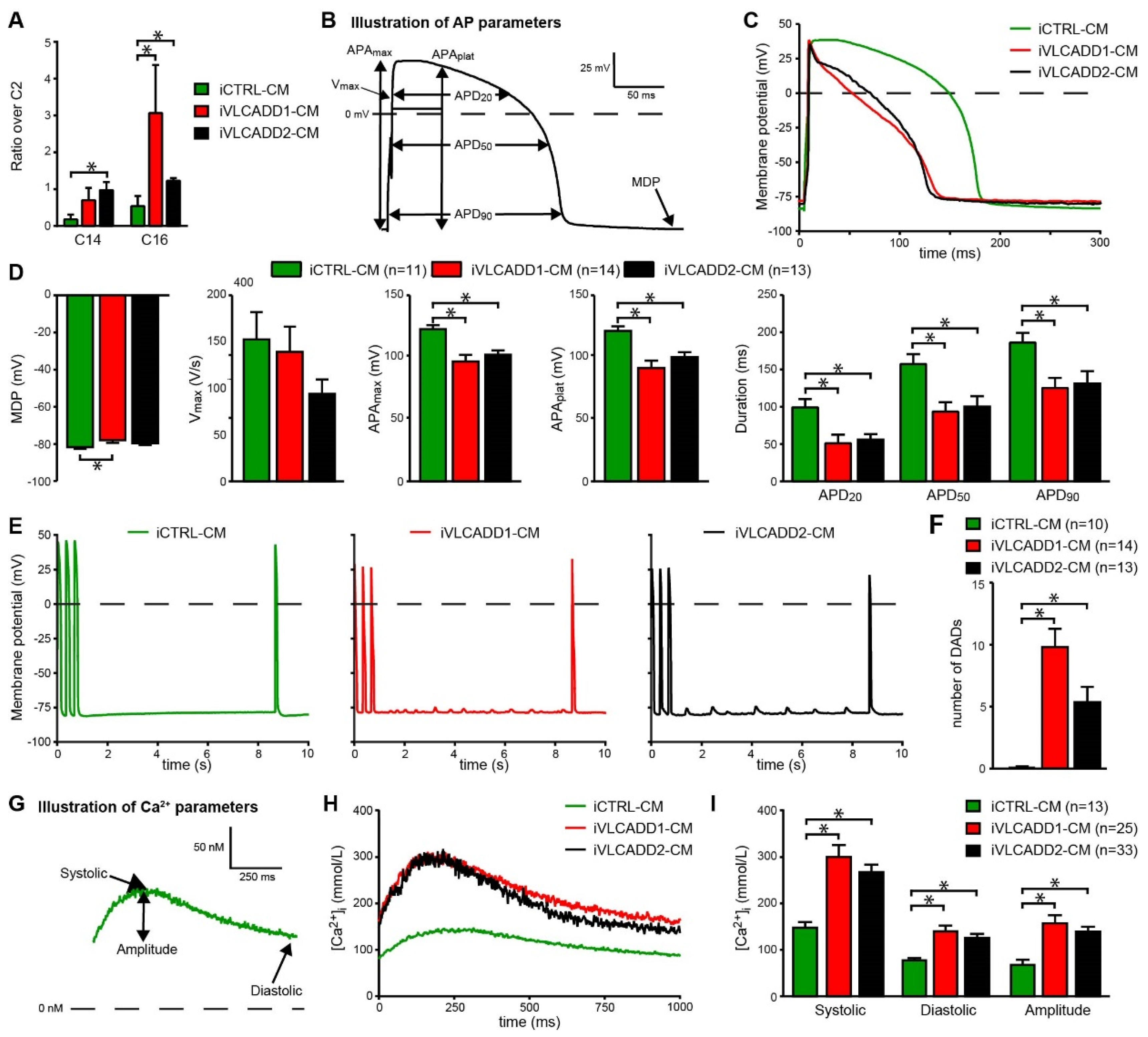
2.3. VLCADD-CMs Accumulate Long-Chain Acylcarnitine Species
2.4. VLCADD-CMs Exhibit Shortened Action Potentials and Delayed after Depolarizations
2.5. VLCADD-CMs Show Increased Ca2+ Concentration
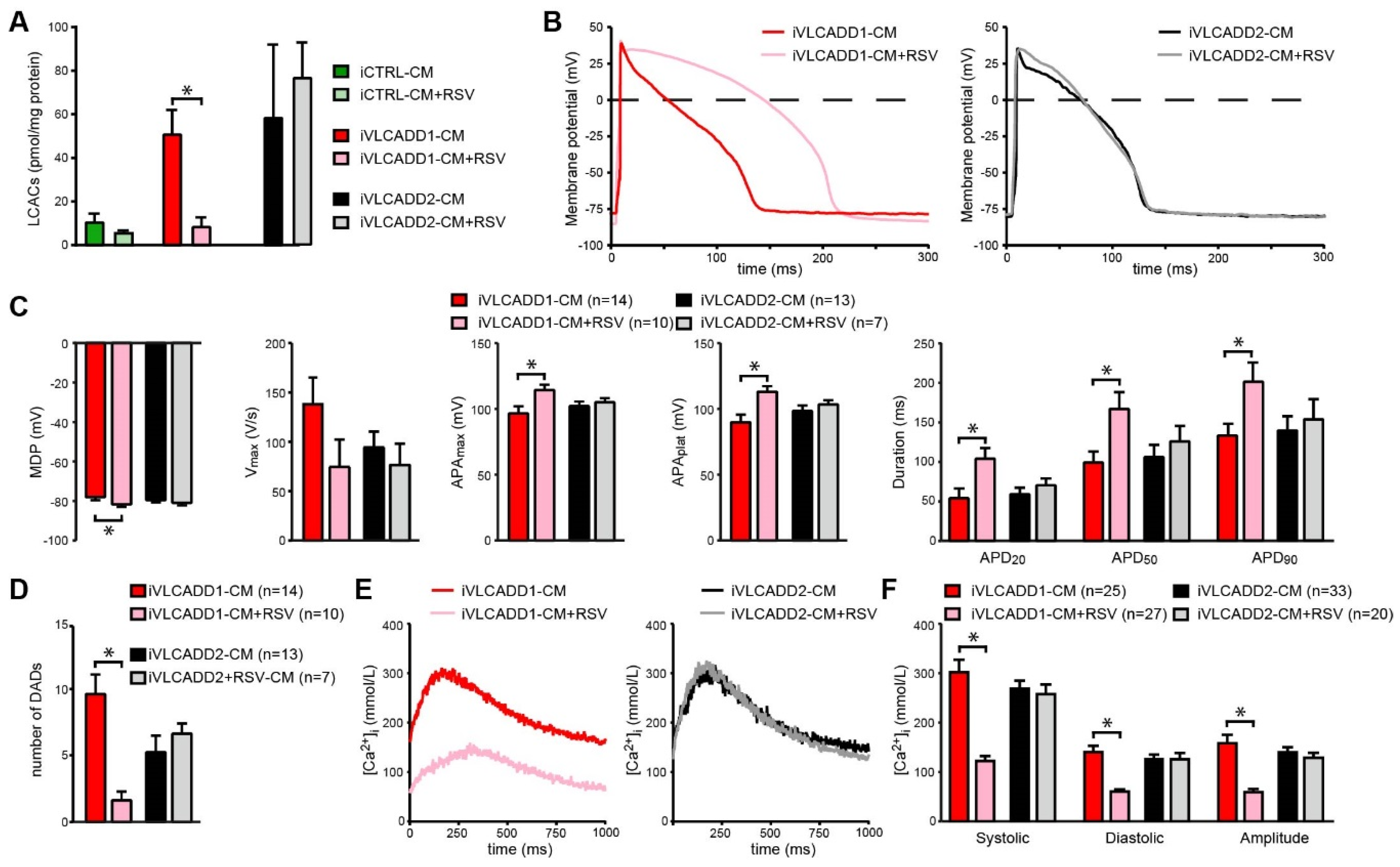
2.6. Resveratrol Improves Biochemical and Electrophysiological Derangements in VLCADD-CM of Patient 1
2.7. Blocking of lcFAO Flux and Consequently Reducing Accumulation of lcFAO Intermediates Mitigated Electrophysiological Abnormalities in VLCADD-CM
3. Discussion
4. Materials and Methods
4.1. Patient Selection and Clinical Data Analysis
4.2. Fibroblast Culture
4.3. VLCAD Activity
4.4. Measurement of Long-Chain Fatty Acid Oxidation (lcFAO) Flux
4.5. Acylcarnitine Profiling
4.6. Generation and Maintenance of hiPSC Lines
4.7. Differentiation of hiPSC Lines into Cardiomyocytes
4.8. Preparation of hiPSC-CM for Electrophysiology
4.9. Cellular Electrophysiology in hiPSC-CMs
4.10. Cytoplasmic Calcium Measurements
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mathur, A.; Sims, H.F.; Gopalakrishnan, D.; Gibson, B.; Rinaldo, P.; Vockley, J.; Hug, G.; Strauss, A.W. Molecular heterogeneity in very-long-chain acyl-CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation 1999, 99, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Knottnerus, S.J.G.; Bleeker, J.C.; Wust, R.C.I.; Ferdinandusse, S.; Ijist, L.; Wijburg, F.A.; Wanders, R.J.A.; Visser, G.; Houtkooper, R.H. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev. Endocr. Metab. Disord. 2018, 19, 93–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vianey-Saban, C.; Divry, P.; Brivet, M.; Nada, M.; Zabot, M.T.; Mathieu, M.; Roe, C. Mitochondrial very-long-chain acyl-coenzyme A dehydrogenase deficiency: Clinical characteristics and diagnostic considerations in 30 patients. Clin. Chim. Acta Int. J. Clin. Chem. 1998, 269, 43–62. [Google Scholar] [CrossRef]
- Bonnet, D.; Martin, D.; Pascale De, L.; Villain, E.; Jouvet, P.; Rabier, D.; Brivet, M.; Saudubray, J.M. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation 1999, 100, 2248–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluge, S.; Kuhnelt, P.; Block, A.; Merkel, M.; Gocht, A.; Lukacs, Z.; Kohlschutter, A.; Kreymann, G. A young woman with persistent hypoglycemia, rhabdomyolysis, and coma: Recognizing fatty acid oxidation defects in adults. Crit. Care Med. 2003, 31, 1273–1276. [Google Scholar] [CrossRef]
- Yamamoto, A.; Nakamura, K.; Matsumoto, S.; Iwai, M.; Shigematsu, Y.; Tajima, G.; Tsumura, M.; Okada, S.; Mitsubuchi, H.; Endo, F. VLCAD deficiency in a patient who recovered from ventricular fibrillation, but died suddenly of a respiratory syncytial virus infection. Pediatrics Int. Official J. Jpn. Pediatric Soc. 2013, 55, 775–778. [Google Scholar] [CrossRef]
- Bleeker, J.C.; Kok, I.L.; Ferdinandusse, S.; van der Pol, W.L.; Cuppen, I.; Bosch, A.M.; Langeveld, M.; Derks, T.G.J.; Williams, M.; de Vries, M.; et al. Impact of NBS for VLCAD deficiency on genetic, enzymatic and clinical outcomes. J. Inherit. Metab. Dis. 2019, 42, 414–423. [Google Scholar] [CrossRef]
- Pena, L.D.; van Calcar, S.C.; Hansen, J.; Edick, M.J.; Walsh Vockley, C.; Leslie, N.; Cameron, C.; Mohsen, A.W.; Berry, S.A.; Arnold, G.L.; et al. Outcomes and genotype-phenotype correlations in 52 individuals with VLCAD deficiency diagnosed by NBS and enrolled in the IBEM-IS database. Mol. Genet. Metab. 2016. [Google Scholar] [CrossRef] [Green Version]
- Xiong, D.; He, H.; James, J.; Tokunaga, C.; Powers, C.; Huang, Y.; Osinska, H.; Towbin, J.A.; Purevjav, E.; Balschi, J.A.; et al. Cardiac-specific VLCAD deficiency induces dilated cardiomyopathy and cold intolerance. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, 326–338. [Google Scholar] [CrossRef] [Green Version]
- Diekman, E.; de Sain-van der Velden, M.; Waterham, H.; Kluijtmans, L.; Schielen, P.; van Veen, E.B.; Ferdinandusse, S.; Wijburg, F.; Visser, G. The Newborn Screening Paradox: Sensitivity vs. Overdiagnosis in VLCAD Deficiency. JIMD Rep. 2016, 27, 101–106. [Google Scholar] [CrossRef]
- Knabb, M.T.; Saffitz, J.E.; Corr, P.B.; Sobel, B.E. The dependence of electrophysiological derangements on accumulation of endogenous long-chain acyl carnitine in hypoxic neonatal rat myocytes. Circ. Res. 1986, 58, 230–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corr, P.B.; Creer, M.H.; Yamada, K.A.; Saffitz, J.E.; Sobel, B.E. Prophylaxis of early ventricular fibrillation by inhibition of acylcarnitine accumulation. J. Clin. Investig. 1989, 83, 927–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meszaros, J.; Pappano, A.J. Electrophysiological effects of L-palmitoylcarnitine in single ventricular myocytes. Am. J. Physiol. 1990, 258, H931–H938. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Corr, P.B. Palmitoyl carnitine modifies sodium currents and induces transient inward current in ventricular myocytes. Am. J. Physiol. 1994, 266, H1034–H1046. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Corr, P.B. Palmitoylcarnitine increases [Na+]i and initiates transient inward current in adult ventricular myocytes. Am. J. Physiol. 1995, 268, H2405–H2417. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.A.; Kanter, E.M.; Newatia, A. Long-Chain Acylcarnitine Induces Ca2+ Efflux from the Sarcoplasmic Reticulum. J. Cardiovasc. Pharmacol. 2000, 36, 14–21. [Google Scholar] [CrossRef]
- Pei, Z.; Xiao, Y.; Meng, J.; Hudmon, A.; Cummins, T.R. Cardiac sodium channel palmitoylation regulates channel availability and myocyte excitability with implications for arrhythmia generation. Nat. Commun. 2016, 7, 12035. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.A.; McHowat, J.; Yan, G.X.; Donahue, K.; Peirick, J.; Kleber, A.G.; Corr, P.B. Cellular uncoupling induced by accumulation of long-chain acylcarnitine during ischemia. Circ. Res. 1994, 74, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, B.F.; Rosen, M.R. Cellular mechanisms for cardiac arrhythmias. Circ. Res. 1981, 49, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Devalla, H.D.; Gelinas, R.; Aburawi, E.H.; Beqqali, A.; Goyette, P.; Freund, C.; Chaix, M.A.; Tadros, R.; Jiang, H.; Le Bechec, A.; et al. TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol. Med. 2016, 8, 1390–1408. [Google Scholar] [CrossRef]
- Bastin, J.; Lopes-Costa, A.; Djouadi, F. Exposure to resveratrol triggers pharmacological correction of fatty acid utilization in human fatty acid oxidation-deficient fibroblasts. Hum. Mol. Genet. 2011, 20, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aires, V.; Delmas, D.; Le Bachelier, C.; Latruffe, N.; Schlemmer, D.; Benoist, J.F.; Djouadi, F.; Bastin, J. Stilbenes and resveratrol metabolites improve mitochondrial fatty acid oxidation defects in human fibroblasts. Orphanet J. Rare Dis. 2014, 9, 79. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Wall, S.R.; Olley, P.M.; Davies, N.J. Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ. Res. 1988, 63, 1036–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanders, R.J.; Ruiter, J.P.; Ijist, L.; Waterham, H.R.; Houten, S.M. The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J. Inherit. Metab. Dis. 2010, 33, 479–494. [Google Scholar] [CrossRef] [Green Version]
- Casini, S.; Verkerk, A.O.; Remme, C.A. Human iPSC-Derived Cardiomyocytes for Investigation of Disease Mechanisms and Therapeutic Strategies in Inherited Arrhythmia Syndromes: Strengths and Limitations. Cardiovasc. Drugs Ther. 2017, 31, 325–344. [Google Scholar] [CrossRef] [Green Version]
- Verkerk, A.O.; Veldkamp, M.W.; Bouman, L.N.; van Ginneken, A.C. Calcium-activated Cl- current contributes to delayed afterdepolarizations in single Purkinje and ventricular myocytes. Circulation 2000, 101, 2639–2644. [Google Scholar] [CrossRef] [Green Version]
- Ferro, F.; Ouille, A.; Tran, T.A.; Fontanaud, P.; Bois, P.; Babuty, D.; Labarthe, F.; Le Guennec, J.Y. Long-chain acylcarnitines regulate the hERG channel. PLoS ONE 2012, 7, e41686. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.P.; Yin, J.X.; Liu, Z.; Zhang, Y.; Wang, Q.S.; Zhao, J. Effect of resveratrol on L-type calcium current in rat ventricular myocytes. Acta Pharmacol. Sin. 2006, 27, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Corr, P.B. Influence of long-chain acylcarnitines on voltage-dependent calcium current in adult ventricular myocytes. Am. J. Physiol. 1992, 263, H410–H417. [Google Scholar] [CrossRef]
- Janse, M.J.; D’Alnoncourt, C.N. Reflections on reentry and focal activity. Am. J. Cardiol. 1987, 60, 21f–26f. [Google Scholar] [CrossRef]
- Keating, M.T.; Sanguinetti, M.C. Molecular and Cellular Mechanisms of Cardiac Arrhythmias. Cell 2001, 104, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Wit, A.L. Afterdepolarizations and triggered activity as a mechanism for clinical arrhythmias. Pacing Clin. Electrophysiol. 2018, 41, 883–896. [Google Scholar] [CrossRef]
- Diekman, E.F.; Ferdinandusse, S.; van der Pol, L.; Waterham, H.R.; Ruiter, J.P.; Ijlst, L.; Wanders, R.J.; Houten, S.M.; Wijburg, F.A.; Blank, A.C.; et al. Fatty acid oxidation flux predicts the clinical severity of VLCAD deficiency. Genet. Med. Off. J. Am. Coll. Med Genet. 2015, 17, 989–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, I.Y.; Matsa, E.; Wu, J.C. Induced pluripotent stem cells: At the heart of cardiovascular precision medicine. Nat. Rev. Cardiol. 2016, 13, 333–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molkentin, J.D.; Lu, J.-R.; Antos, C.L.; Markham, B.; Richardson, J.; Robbins, J.; Grant, S.R.; Olson, E.N. A Calcineurin-Dependent Transcriptional Pathway for Cardiac Hypertrophy. Cell 1998, 93, 215–228. [Google Scholar] [CrossRef] [Green Version]
- Sussman, M.A.; Lim, H.W.; Gude, N.; Taigen, T.; Olson, E.N.; Robbins, J.; Colbert, M.C.; Gualberto, A.; Wieczorek, D.F.; Molkentin, J.D. Prevention of Cardiac Hypertrophy in Mice by Calcineurin Inhibition. Science 1998, 281, 1690. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.H.; Baczko, I.; Jones, L.; Fercho, M.; Light, P.E. Inhibition of cardiac voltage-gated sodium channels by grape polyphenols. Br. J. Pharmacol. 2006, 149, 657–665. [Google Scholar] [CrossRef] [Green Version]
- Gillingham, M.B.; Scott, B.; Elliott, D.; Harding, C.O. Metabolic control during exercise with and without medium-chain triglycerides (MCT) in children with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol. Genet. Metab. 2006, 89, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Tucci, S.; Behringer, S.; Spiekerkoetter, U. De novo fatty acid biosynthesis and elongation in very long-chain acyl-CoA dehydrogenase-deficient mice supplemented with odd or even medium-chain fatty acids. FEBS J. 2015, 282, 4242–4253. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.M.; Butt, Y.; Messmer, B.; Boriak, R.; Bennett, M.J. Medium-chain fatty acids undergo elongation before beta-oxidation in fibroblasts. Biochem. Biophys. Res. Commun. 2006, 346, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Carnielli, V.P.; Sulkers, E.J.; Moretti, C.; Wattimena, J.L.; van Goudoever, J.B.; Degenhart, H.J.; Zacchello, F.; Sauer, P.J. Conversion of octanoic acid into long-chain saturated fatty acids in premature infants fed a formula containing medium-chain triglycerides. Metabolism 1994, 43, 1287–1292. [Google Scholar] [CrossRef]
- Primassin, S.; Tucci, S.; Herebian, D.; Seibt, A.; Hoffmann, L.; ter Veld, F.; Spiekerkoetter, U. Pre-exercise medium-chain triglyceride application prevents acylcarnitine accumulation in skeletal muscle from very-long-chain acyl-CoA-dehydrogenase-deficient mice. J. Inherit. Metab. Dis. 2010, 33, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Streckfuss-Bomeke, K.; Wolf, F.; Azizian, A.; Stauske, M.; Tiburcy, M.; Wagner, S.; Hubscher, D.; Dressel, R.; Chen, S.; Jende, J.; et al. Comparative study of human-induced pluripotent stem cells derived from bone marrow cells, hair keratinocytes, and skin fibroblasts. Eur. Heart J. 2013, 34, 2618–2629. [Google Scholar] [CrossRef] [Green Version]
- Olpin, S.E.; Manning, N.J.; Pollitt, R.J.; Clarke, S. Improved detection of long-chain fatty acid oxidation defects in intact cells using [9,10-3H]oleic acid. J. Inherit. Metab. Dis. 1997, 20, 415–419. [Google Scholar] [CrossRef]
- Ben Jehuda, R.; Eisen, B.; Shemer, Y.; Mekies, L.N.; Szantai, A.; Reiter, I.; Cui, H.; Guan, K.; Haron-Khun, S.; Freimark, D.; et al. CRISPR correction of the PRKAG2 gene mutation in the patient’s induced pluripotent stem cell-derived cardiomyocytes eliminates electrophysiological and structural abnormalities. Heart Rhythm. 2018, 15, 267–276. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Dudek, J.; Cheng, I.F.; Balleininger, M.; Vaz, F.M.; Streckfuss-Bomeke, K.; Hubscher, D.; Vukotic, M.; Wanders, R.J.; Rehling, P.; Guan, K. Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome. Stem Cell Res. 2013, 11, 806–819. [Google Scholar] [CrossRef] [Green Version]
- Dambrot, C.; Braam, S.R.; Tertoolen, L.G.; Birket, M.; Atsma, D.E.; Mummery, C.L. Serum supplemented culture medium masks hypertrophic phenotypes in human pluripotent stem cell derived cardiomyocytes. J. Cell. Mol. Med. 2014, 18, 1509–1518. [Google Scholar] [CrossRef]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 2013, 12, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Meijer van Putten, R.M.; Mengarelli, I.; Guan, K.; Zegers, J.G.; van Ginneken, A.C.; Verkerk, A.O.; Wilders, R. Ion channelopathies in human induced pluripotent stem cell derived cardiomyocytes: A dynamic clamp study with virtual IK1. Front. Physiol. 2015, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, P.H.; Lynch, J.W. Liquid junction potentials and small cell effects in patch-clamp analysis. J. Membr. Biol. 1991, 121, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, M.; Mummery, C.L.; Wilde, A.A.; Bezzina, C.R.; Verkerk, A.O. Induced pluripotent stem cell derived cardiomyocytes as models for cardiac arrhythmias. Front. Physiol. 2012, 3, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baartscheer, A.; Schumacher, C.A.; Fiolet, J.W. Cytoplasmic sodium, calcium and free energy change of the Na+/Ca2+-exchanger in rat ventricular myocytes. J. Mol. Cell. Cardiol. 1998, 30, 2437–2447. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| PID (Name of hiPSC Line) | Patient 1 (iVLCADD1) | Patient 2 (iVLCADD2) |
|---|---|---|
| Age of presentation (current age) | 1.25 y (23 y) | 0.1 y (22 y) |
| Sex | female | female |
| Mutations in ACADVL gene | c. 848T >C (p.Val283Ala) c.1141_1143delGAG (p.Glu381del) | c.104delC (p.Pro35Leufs*26) c.104delC (p.Pro35Leufs*26) |
| VLCAD activity (in nmol/min/mg and % of controls) | Fibroblasts: 0.10 (3%) Lymphocytes: 0.18 (5%) | Fibroblasts: <0.06 (0%) Lymphocytes <0.15 (0%) |
| lcFAO flux (% of control) | 32 | 7 |
| Maximal Creatine Kinase | 400 U/L | 99889 U/L |
| Signs at presentation | Hypoglycemia 0.2 mmol/L Hypothermia (35.9 °C) | Hypoglycemia 1.7 mmol/L, vomiting, convulsions, cardiomyopathy (reversible) |
| Cardiac history | Age 15y: Holter ECG: Normal conduction. | Age 14y: ECG: aspecific repolarization abnormalities. (flat ST-T segments in the inferior and left lateral leads). Some early repolarizations in the inferior leads. Age 15y: dilated cardiomyopathy (reversible) Age 18y: Holter ECG: single ectopy from the atrial side as well as from the ventricular side. Mobitz II AV-block during sleep. |
| Other signs or symptoms | None | Rhabdomyolysis Exercise intolerance |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knottnerus, S.J.G.; Mengarelli, I.; Wüst, R.C.I.; Baartscheer, A.; Bleeker, J.C.; Coronel, R.; Ferdinandusse, S.; Guan, K.; IJlst, L.; Li, W.; et al. Electrophysiological Abnormalities in VLCAD Deficient hiPSC-Cardiomyocytes Can Be Improved by Lowering Accumulation of Fatty Acid Oxidation Intermediates. Int. J. Mol. Sci. 2020, 21, 2589. https://doi.org/10.3390/ijms21072589
Knottnerus SJG, Mengarelli I, Wüst RCI, Baartscheer A, Bleeker JC, Coronel R, Ferdinandusse S, Guan K, IJlst L, Li W, et al. Electrophysiological Abnormalities in VLCAD Deficient hiPSC-Cardiomyocytes Can Be Improved by Lowering Accumulation of Fatty Acid Oxidation Intermediates. International Journal of Molecular Sciences. 2020; 21(7):2589. https://doi.org/10.3390/ijms21072589
Chicago/Turabian StyleKnottnerus, Suzan J. G., Isabella Mengarelli, Rob C. I. Wüst, Antonius Baartscheer, Jeannette C. Bleeker, Ruben Coronel, Sacha Ferdinandusse, Kaomei Guan, Lodewijk IJlst, Wener Li, and et al. 2020. "Electrophysiological Abnormalities in VLCAD Deficient hiPSC-Cardiomyocytes Can Be Improved by Lowering Accumulation of Fatty Acid Oxidation Intermediates" International Journal of Molecular Sciences 21, no. 7: 2589. https://doi.org/10.3390/ijms21072589
APA StyleKnottnerus, S. J. G., Mengarelli, I., Wüst, R. C. I., Baartscheer, A., Bleeker, J. C., Coronel, R., Ferdinandusse, S., Guan, K., IJlst, L., Li, W., Luo, X., Portero, V. M., Ulbricht, Y., Visser, G., Wanders, R. J. A., Wijburg, F. A., Verkerk, A. O., Houtkooper, R. H., & Bezzina, C. R. (2020). Electrophysiological Abnormalities in VLCAD Deficient hiPSC-Cardiomyocytes Can Be Improved by Lowering Accumulation of Fatty Acid Oxidation Intermediates. International Journal of Molecular Sciences, 21(7), 2589. https://doi.org/10.3390/ijms21072589