Electrophoretic Deposition of Copper(II)–Chitosan Complexes for Antibacterial Coatings



Abstract
:
1. Introduction
2. Results and Discussion
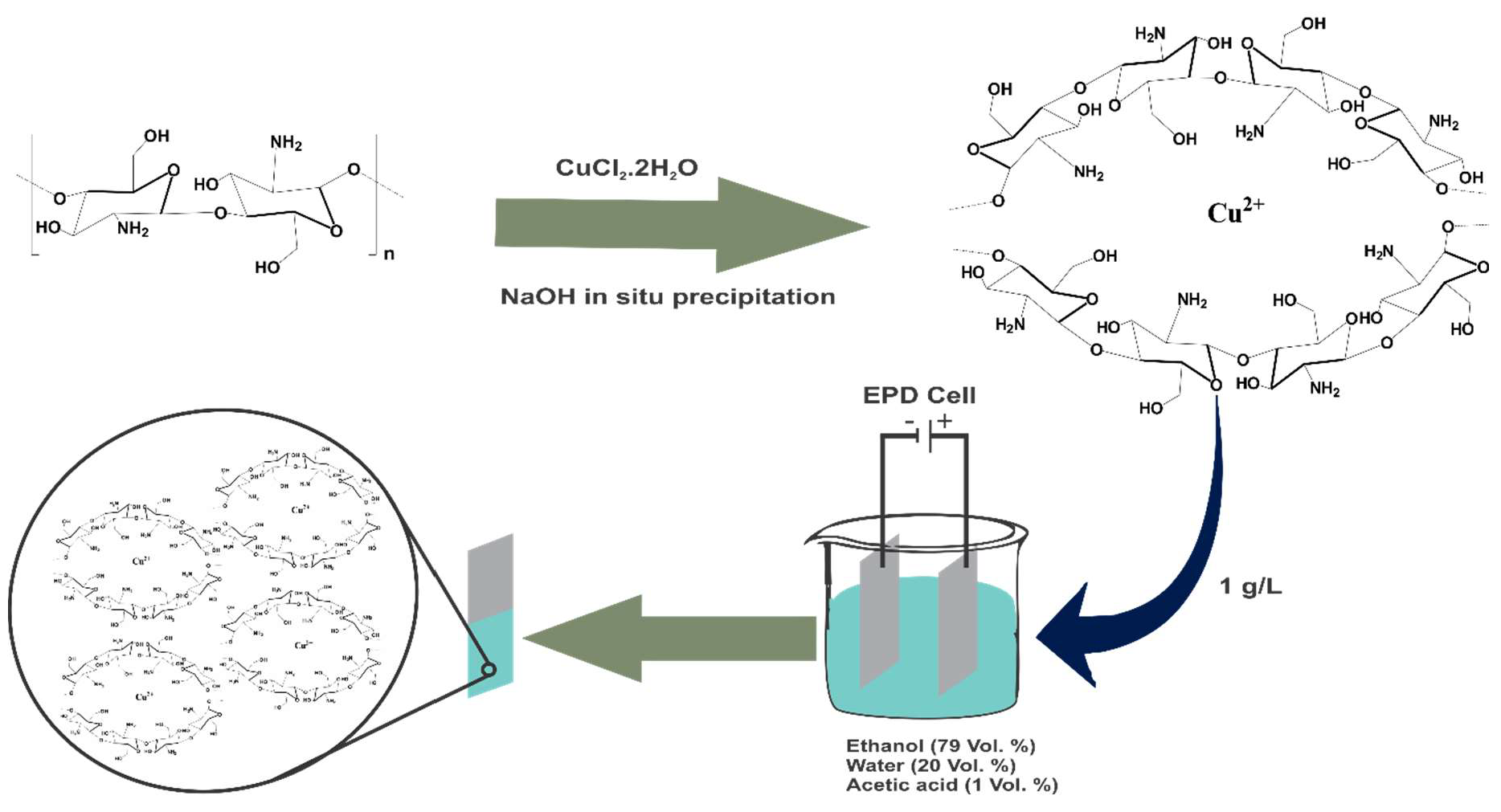
2.1. EPD Process of Cu(II)–CS
2.2. Morphological Analysis
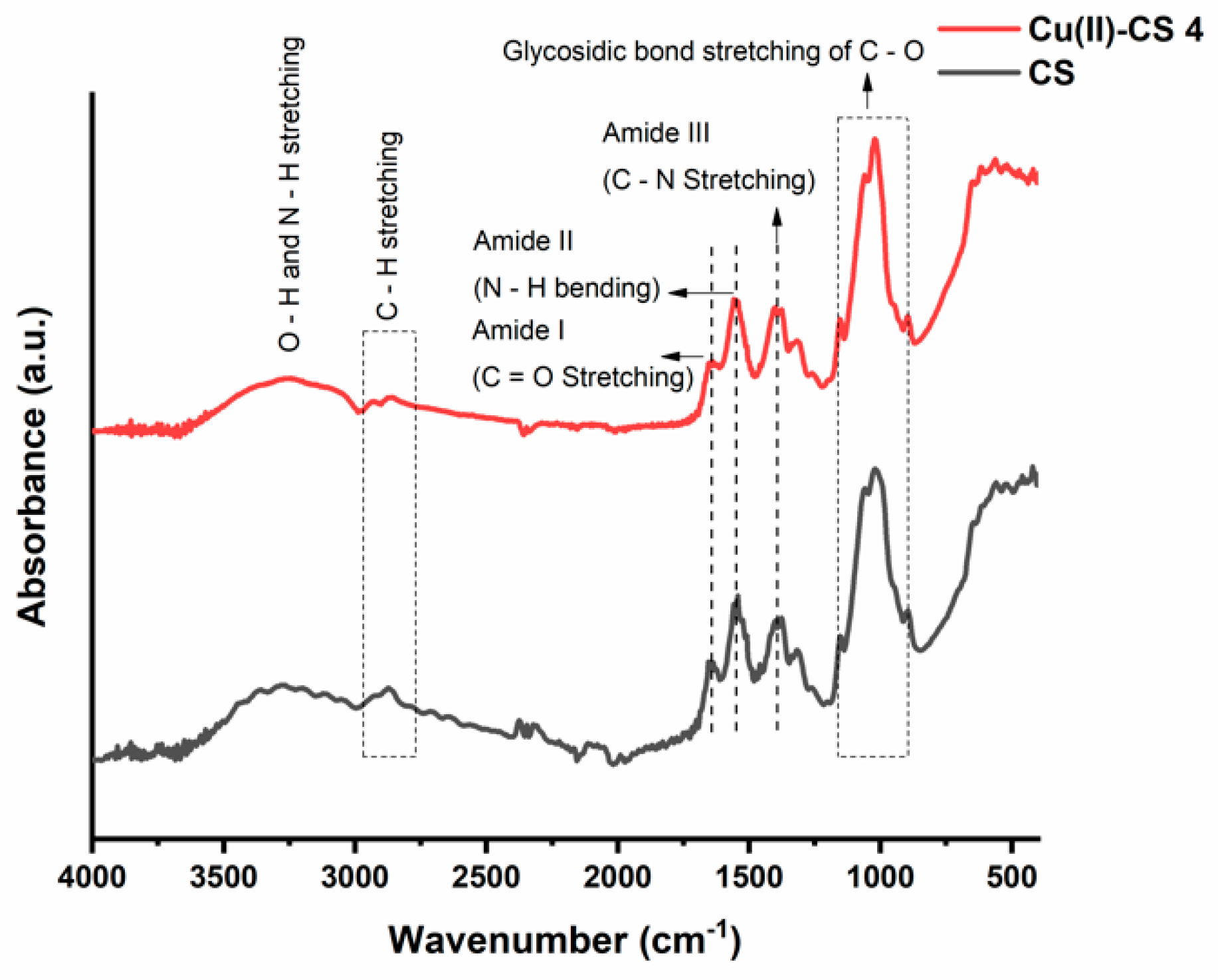
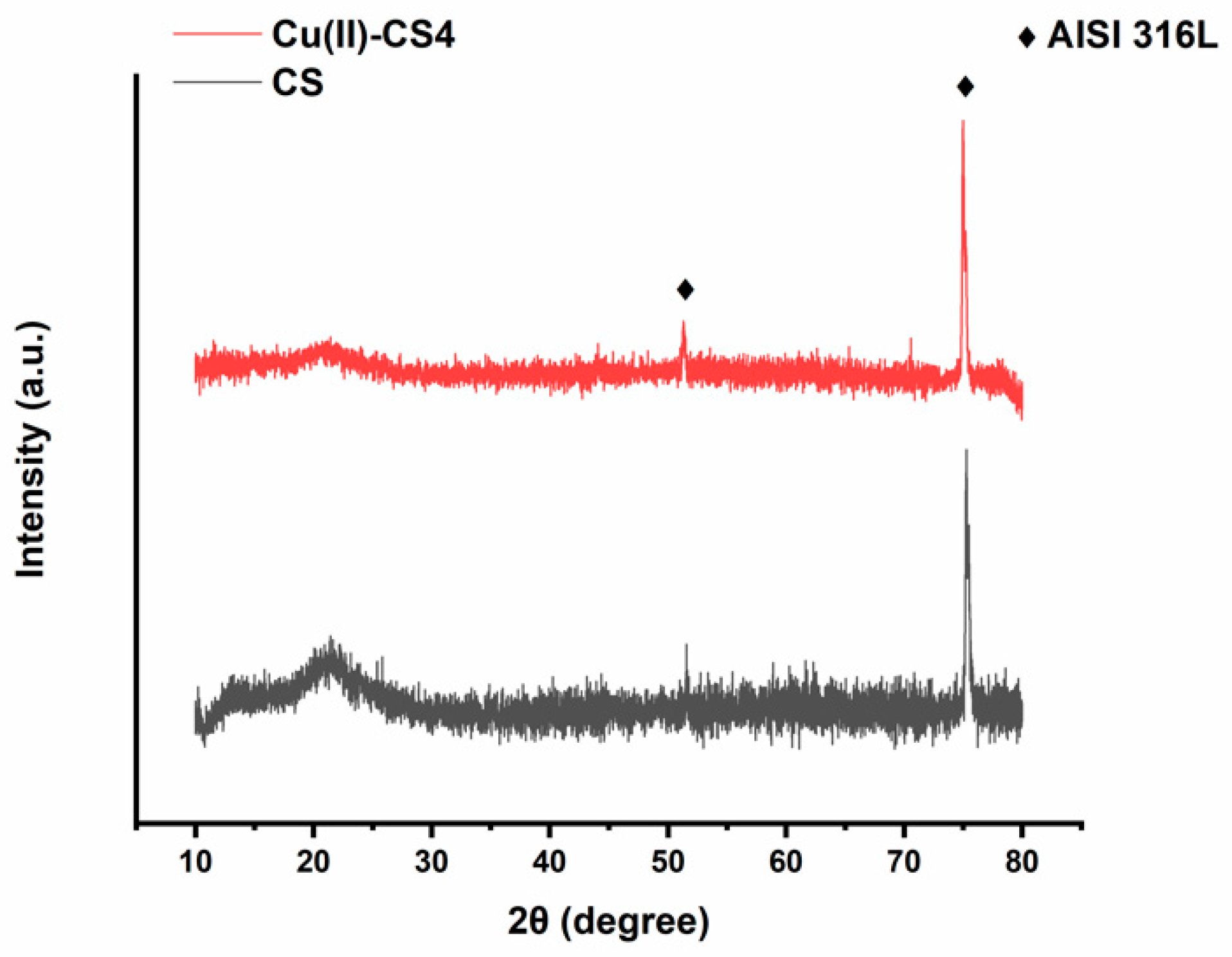
2.3. Chemical and Structural Characterization
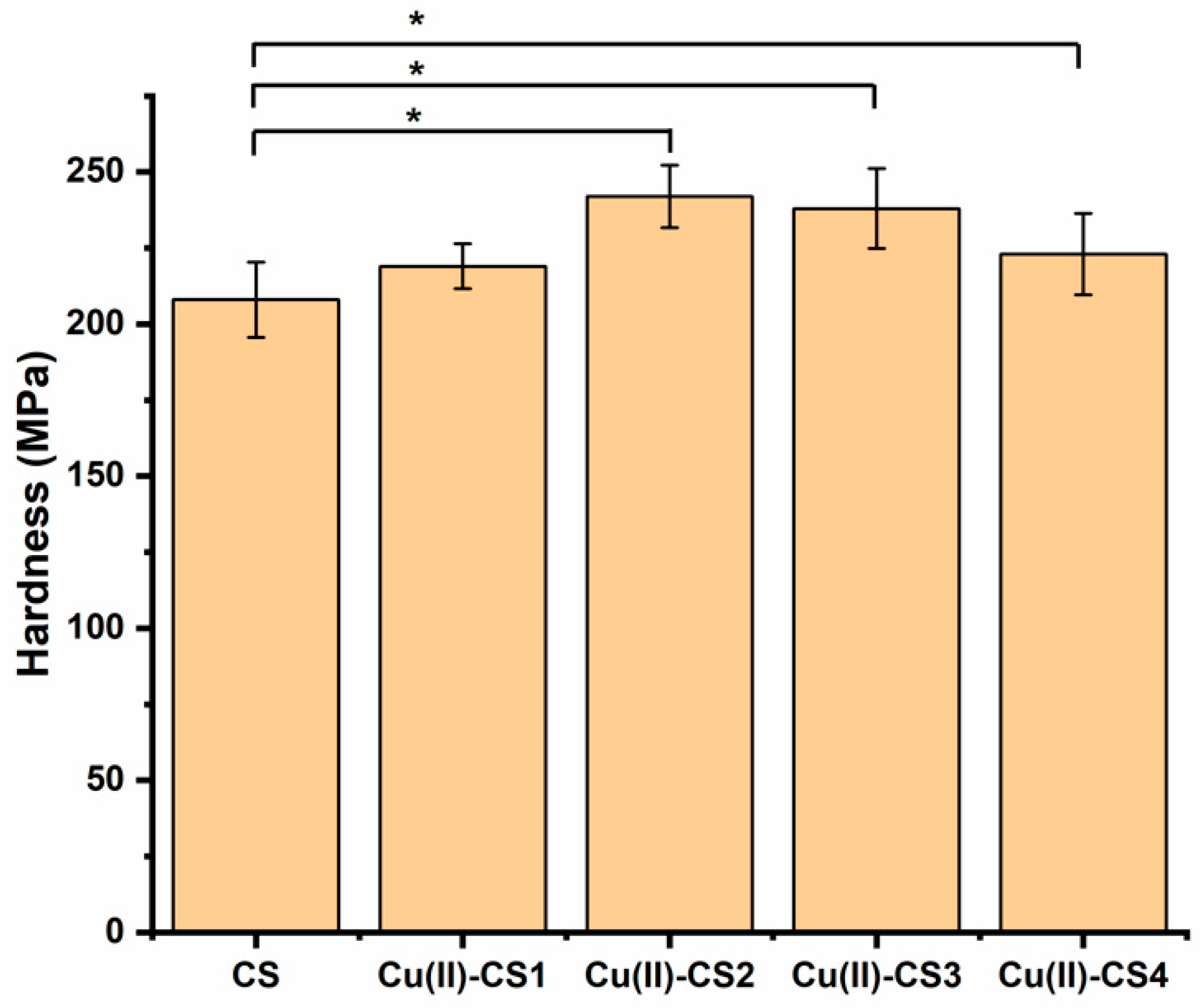
2.4. Nano Indentation
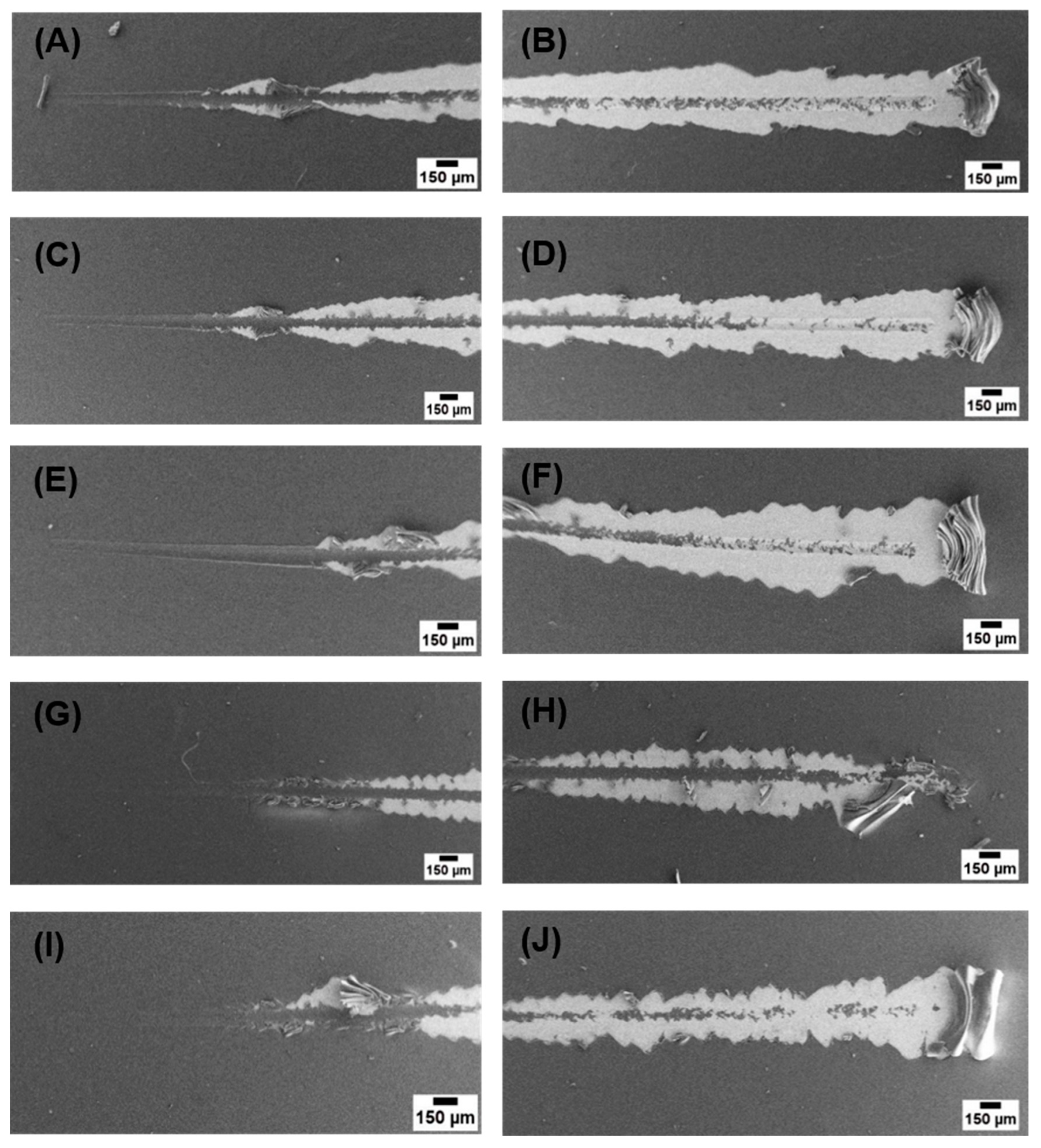
2.5. Scratch Test
2.6. Swelling Ratio
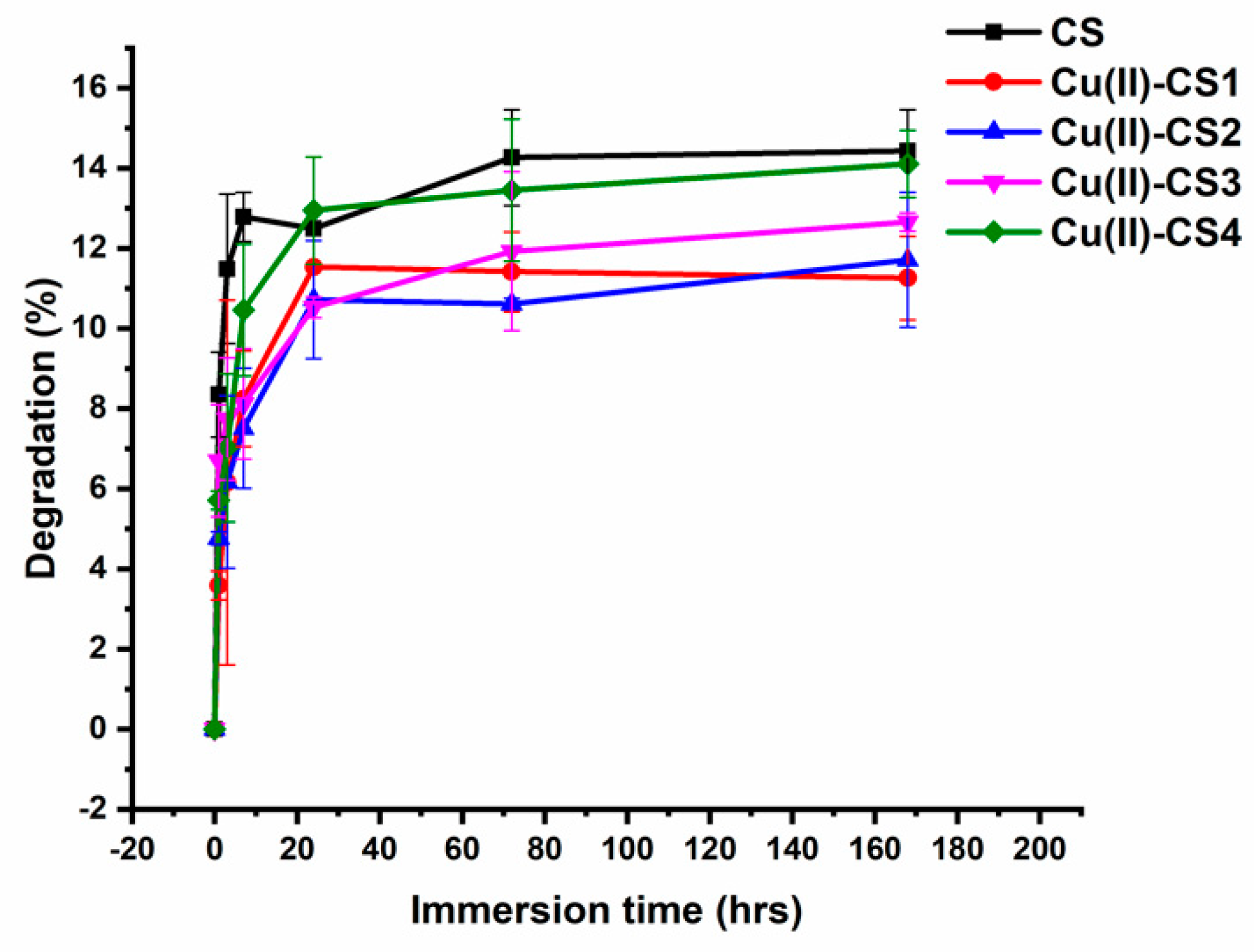
2.7. In Vitro Degradation
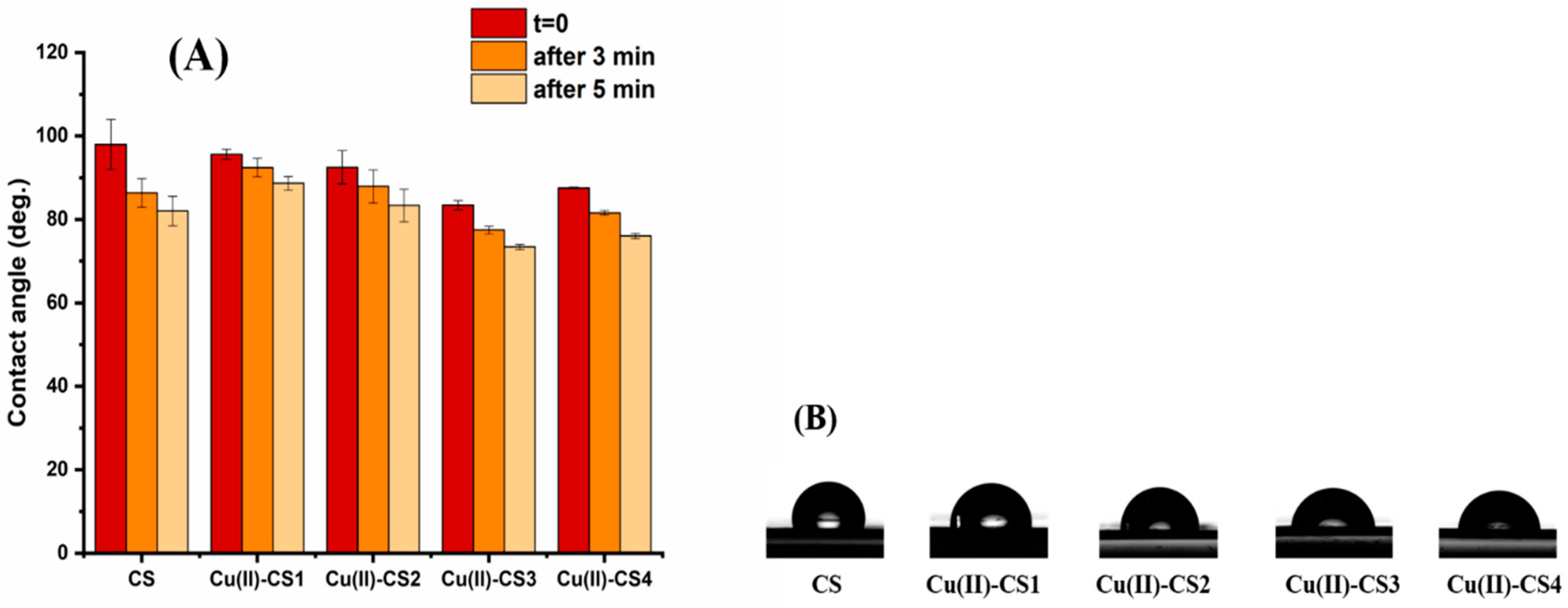
2.8. Wettability
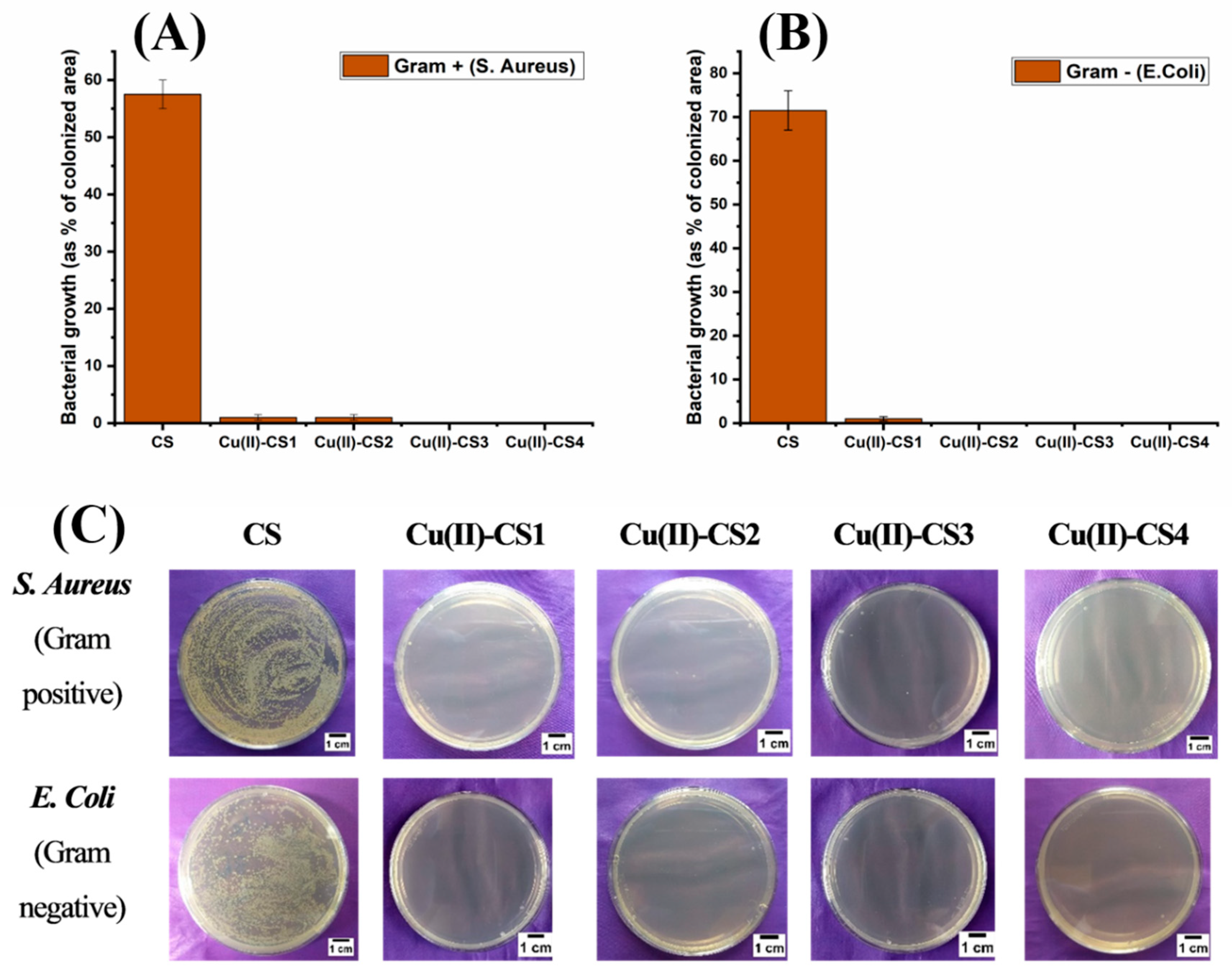
2.9. Bacterial Culture
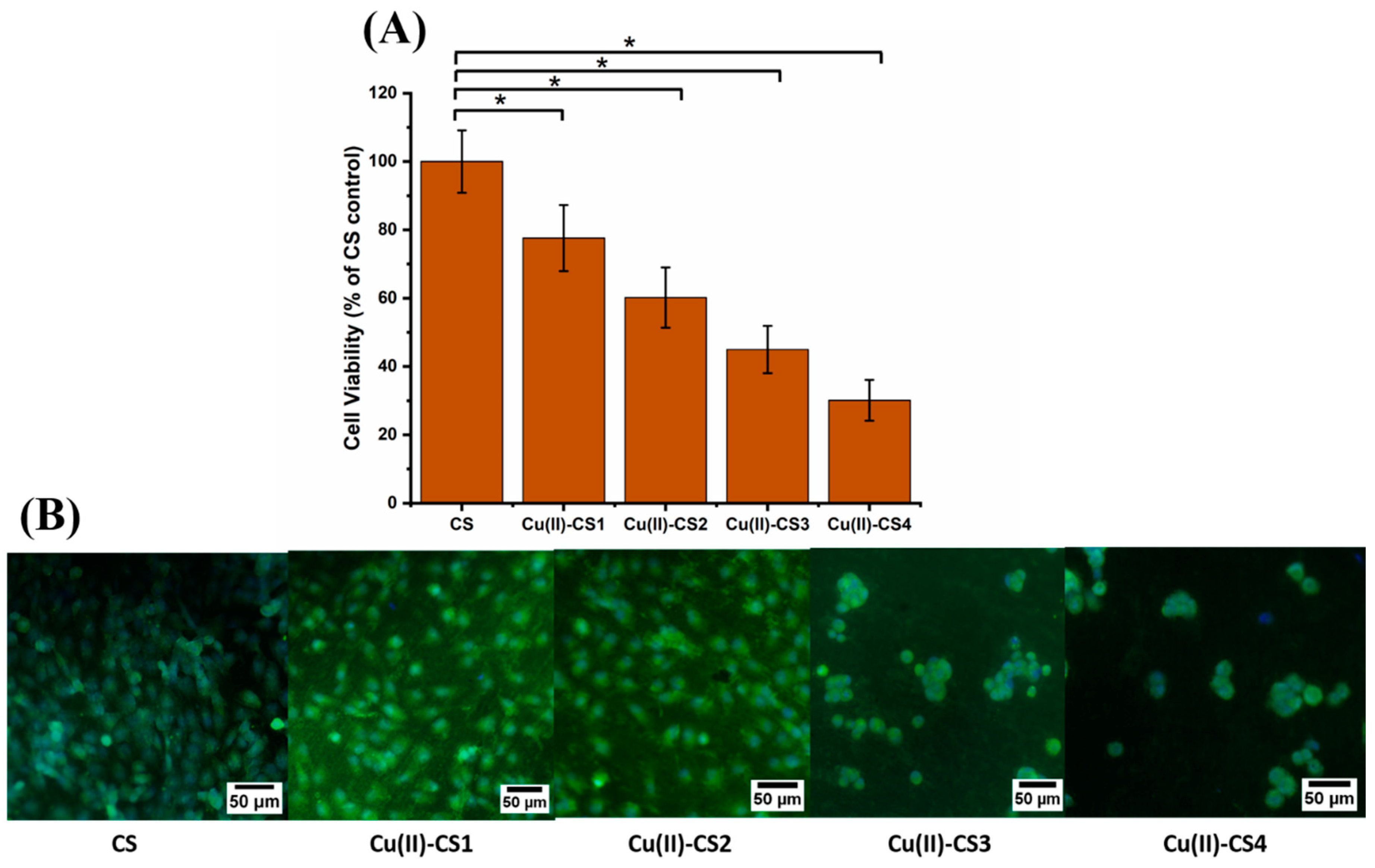
2.10. Cell Biology
3. Experimental Section
3.1. Material
3.2. Synthesis of Cu(II)–CS Complex
3.3. EPD of Cu(II)–CS
3.4. Characterization of the Coatings
3.4.1. Morphological Analysis
3.4.2. Chemical and Structural Characterization
3.4.3. Mechanical Characterizations
3.4.4. Swelling Ratio
3.4.5. In Vitro Degradation
3.4.6. Wettability
3.4.7. Bacterial Culture
3.4.8. In Vitro Cell Culture Test
3.5. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pishbin, F.; Mouriño, V.; Gilchrist, J.; McComb, D.; Kreppel, S.; Salih, V.; Ryan, M.P.; Boccaccini, A. Single-step electrochemical deposition of antimicrobial orthopaedic coatings based on a bioactive glass/chitosan/nano-silver composite system. Acta Biomater. 2013, 9, 7469–7479. [Google Scholar] [CrossRef] [PubMed]
- Pishbin, F.; Mouriño, V.; Flor, S.; Kreppel, S.; Salih, V.; Ryan, M.P.; Boccaccini, A.R.; Mourino, V.; Flor, S.; Kreppel, S.; et al. Electrophoretic Deposition of Gentamicin-Loaded Bioactive Glass/Chitosan Composite Coatings for Orthopaedic Implants. ACS Appl. Mater. Interfaces 2014, 6, 8796–8806. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Rivera, L.R.; Ellis, T.; Qi, J.; Ryan, M.P.; Boccaccini, A. Bioactive and Antibacterial Coatings Based on Zein/Bioactive Glass Composites by Electrophoretic Deposition. Coatings 2018, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Chen, Q.; Zhang, Y.; Diba, M.; Kolwijck, E.; Shao, J.; Jansen, J.A.; Yang, F.; Boccaccini, A.; Leeuwenburgh, S.C.G. Electrophoretic Deposition of Chitosan Coatings Modified with Gelatin Nanospheres To Tune the Release of Antibiotics. ACS Appl. Mater. Interfaces 2016, 8, 13785–13792. [Google Scholar] [CrossRef] [PubMed]
- Michael, C.A.; Dominey-Howes, D.; Labbate, M. The Antimicrobial Resistance Crisis: Causes, Consequences, and Management. Front. Public Health. 2014, 2, 145. [Google Scholar] [CrossRef]
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 2: Management Strategies and New Agents. P T 2015, 40, 344–352. [Google Scholar]
- Cloutier, M.; Mantovani, D.; Rosei, F. Antibacterial Coatings: Challenges, Perspectives, and Opportunities. Trends Biotechnol. 2015, 33, 637–652. [Google Scholar] [CrossRef]
- Vasilev, K.; Cook, J.; Griesser, H. Antibacterial surfaces for biomedical devices. Expert Rev. Med. Devices 2009, 6, 553–567. [Google Scholar] [CrossRef]
- Zhao, L.; Chu, P.K.; Zhang, Y.; Wu, Z. Antibacterial coatings on titanium implants. J. Biomed. Mater. Res. Part B: Appl. Biomater. 2009, 91, 470–480. [Google Scholar] [CrossRef]
- Ghalayani Esfahani, A.; Lazazzera, B.; Draghi, L.; Farè, S.; Chiesa, R.; De Nardo, L.; Billi, F.; Fare, S.; Chiesa, R.; De Nardo, L.; et al. Bactericidal activity of gallium-doped chitosan coatings against staphylococcal infection. J. Appl. Microbiol. 2019, 126, 87–101. [Google Scholar] [CrossRef] [Green Version]
- Kong, M.; Chen, X.; Xing, K.; Park, H.J. Antimicrobial properties of chitosan and mode of action: A state of the art review. Int. J. Food Microbiol. 2010, 144, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.H.M.; El-Hady, M.F.; Sayed, W.; Hefni, H. Preparation of some chitosan heavy metal complexes and study of its properties. Polym. Sci. Ser. A 2012, 54, 113–124. [Google Scholar] [CrossRef]
- Rhazi, M.; Desbrières, J.; Tolaimate, A.; Rinaudo, M.; Vottero, P.; Alagui, A.; Rhazi, M.; Desbriéres, J.; Tolaimate, A.; Rinaudo, M.; et al. Contribution to the study of the complexation of copper by chitosan and oligomers. Polymer (Guildf) 2002, 43, 1267–1276. [Google Scholar] [CrossRef]
- Higazy, A.; Hashem, M.; El Shafei, A.; Shaker, N.; Hady, M.A. Development of antimicrobial jute packaging using chitosan and chitosan–metal complex. Carbohydr. Polym. 2010, 79, 867–874. [Google Scholar] [CrossRef]
- Wang, X.; Du, Y.; Fan, L.; Liu, H.; Hu, Y. Chitosan- metal complexes as antimicrobial agent: Synthesis, characterization and Structure-activity study. Polym. Bull. 2005, 55, 105–113. [Google Scholar] [CrossRef]
- Gritsch, L.; Lovell, C.; Goldmann, W.H.; Boccaccini, A. Fabrication and characterization of copper(II)-chitosan complexes as antibiotic-free antibacterial biomaterial. Carbohydr. Polym. 2018, 179, 370–378. [Google Scholar] [CrossRef]
- Li, P.; Zhanga, X.; Xu, R.; Wang, W.; Liu, X.; Yeung, K.; Chu, P.K. Electrochemically deposited chitosan/Ag complex coatings on biomedical NiTi alloy for antibacterial application. Surf. Coat. Technol. 2013, 232, 370–375. [Google Scholar] [CrossRef]
- Gritsch, L.; Maqbool, M.; Mouriño, V.; Ciraldo, F.E.; Cresswell, M.; Jackson, P.R.; Lovell, C.; Boccaccini, A. Chitosan/hydroxyapatite composite bone tissue engineering scaffolds with dual and decoupled therapeutic ion delivery: Copper and strontium. J. Mater. Chem. B 2019, 7, 6109–6124. [Google Scholar] [CrossRef] [Green Version]
- Mouriño, V.; Cattalini, J.P.; Boccaccini, A. Metallic ions as therapeutic agents in tissue engineering scaffolds: An overview of their biological applications and strategies for new developments. J. R. Soc. Interface 2012, 9, 401–419. [Google Scholar] [CrossRef] [Green Version]
- Du, W.-L.; Niu, S.-S.; Xu, Y.-L.; Xu, Z.-R.; Fan, C.-L. Antibacterial activity of chitosan tripolyphosphate nanoparticles loaded with various metal ions. Carbohydr. Polym. 2009, 75, 385–389. [Google Scholar] [CrossRef]
- Deng, C.; Zhang, P.; Vulesevic, B.; Kuraitis, D.; Li, F.; Yang, A.F.; Griffith, M.; Ruel, M.; Suuronen, E.J. A Collagen–Chitosan Hydrogel for Endothelial Differentiation and Angiogenesis. Tissue Eng. Part A 2010, 16, 3099–3109. [Google Scholar] [CrossRef] [PubMed]
- Avcu, E.; Baştan, F.E.; Abdullah, H.Z.; Rehman, M.A.U.; Avcu, Y.Y.; Boccaccini, A. Electrophoretic deposition of chitosan-based composite coatings for biomedical applications: A review. Prog. Mater. Sci. 2019, 103, 69–108. [Google Scholar] [CrossRef]
- Simchi, A.; Pishbin, F.; Boccaccini, A.R. Electrophoretic deposition of chitosan. Mater. Lett. 2009, 63, 2253–2256. [Google Scholar] [CrossRef]
- Ma, K.; Gong, L.; Cai, X.; Huang, P.; Cai, J.; Huang, D.; Jiang, T. A green single-step procedure to synthesize Ag-containing nanocomposite coatings with low cytotoxicity and efficient antibacterial properties. Int. J. Nanomed. 2017, 12, 3665. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Ma, K.; Cai, X.; Yang, X.; Hu, Y.; Huang, P.; Wang, F.; Jiang, T.; Wang, Y. Evaluation of antibacterial, angiogenic, and osteogenic activities of green synthesized gap-bridging copper-doped nanocomposite coatings. Int. J. Nanomed. 2017, 12, 7483–7500. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Cai, J.; Ma, K.; Huang, P.; Gong, L.; Huang, D.; Jiang, T.; Wang, Y. Fabrication and characterization of Mg-doped chitosan–gelatin nanocompound coatings for titanium surface functionalization. J. Biomater. Sci. Polym. Ed. 2016, 27, 954–971. [Google Scholar] [CrossRef]
- Ma, K.; Huang, D.; Cai, J.; Cai, X.; Gong, L.; Huang, P.; Wang, Y.; Jiang, T. Surface functionalization with strontium-containing nanocomposite coatings via EPD. Colloids Surf. B: Biointerfaces 2016, 146, 97–106. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, B.; Gray, K.M.; Cheng, Y.; Kim, E.; Rubloff, G.; Bentley, W.E.; Wang, Q.; Payne, G.F. Electrodeposition of a weak polyelectrolyte hydrogel: Remarkable effects of salt on kinetics, structure and properties. Soft Matter 2013, 9, 2703–2710. [Google Scholar] [CrossRef]
- Lei, M.; Qu, X.; Liu, H.; Liu, Y.; Wang, S.; Wu, S.; Bentley, W.E.; Payne, G.F.; Liu, C. Programmable Electrofabrication of Porous Janus Films with Tunable Janus Balance for Anisotropic Cell Guidance and Tissue Regeneration. Adv. Funct. Mater. 2019, 29, 1900065. [Google Scholar] [CrossRef]
- Brunel, F.; El Gueddari, N.E.; Moerschbacher, B.M. Complexation of copper(II) with chitosan nanogels: Toward control of microbial growth. Carbohydr. Polym. 2013, 92, 1348–1356. [Google Scholar] [CrossRef]
- Wu, F.-C.; Tseng, R.-L.; Juang, R.-S. Role of pH in Metal Adsorption from Aqueous Solutions Containing Chelating Agents on Chitosan. Ind. Eng. Chem. Res. 1999, 38, 270–275. [Google Scholar] [CrossRef]
- Schmuhl, R.; Krieg, H.; Keizer, K. Adsorption of Cu(II) and Cr(VI) ions by chitosan: Kinetics and equilibrium studies. Water SA 2001, 27, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Juang, R.-S.; Shao, H.-J. Effect of pH on Competitive Adsorption of Cu(II), Ni(II), and Zn(II) from Water onto Chitosan Beads. Adsorption 2002, 8, 71–78. [Google Scholar] [CrossRef]
- Unagolla, J.; Adikary, S. Adsorption characteristics of cadmium and lead heavy metals into locally synthesized Chitosan Biopolymer. Trop. Agric. Res. 2015, 26, 395. [Google Scholar] [CrossRef]
- Cárdenas, G.; Miranda, S.P. FTIR and TGA studies of chitosan composite fims. J. Chil. Chem. Soc. 2004, 49, 291–295. [Google Scholar] [CrossRef]
- Qu, J.; Hu, Q.; Shen, K.; Zhang, K.; Li, Y.; Li, H.; Zhang, Q.; Wang, J.; Quan, W. The preparation and characterization of chitosan rods modified with Fe3+ by a chelation mechanism. Carbohydr. Res. 2011, 346, 822–827. [Google Scholar] [CrossRef]
- Leceta, I.; Guerrero, P.; De La Caba, K. Functional properties of chitosan-based films. Carbohydr. Polym. 2013, 93, 339–346. [Google Scholar] [CrossRef]
- Jayaramudu, T.; Varaprasad, K.; Pyarasani, R.D.; Reddy, K.K.; Kumar, K.D.; Akbari-Fakhrabadi, A.; Mangalaraja, R.; Amalraj, J. Chitosan capped copper oxide/copper nanoparticles encapsulated microbial resistant nanocomposite films. Int. J. Biol. Macromol. 2019, 128, 499–508. [Google Scholar] [CrossRef]
- Broitman, E. Indentation Hardness Measurements at Macro-, Micro-, and Nanoscale: A Critical Overview. Tribol. Lett. 2017, 65, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Aryaei, A.; Jayatissa, A.H.; Jayasuriya, A.C. Nano and micro mechanical properties of uncross-linked and cross-linked chitosan films. J. Mech. Behav. Biomed. Mater. 2012, 5, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Fahim, I.S.; Aboulkhair, N.; Everitt, N.M. Nanoindentation Investigation on Chitosan Thin Films with Different Types of Nano Fillers. J. Mater. Sci. Res. 2018, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Bull, S.J. Failure Mode Maps in the Thin Film Scratch Adhesion Test. Tribol. Int. 1997, 30, 491–498. [Google Scholar] [CrossRef]
- Siegel, R.A.; Rathbone, M.J. Overview of Controlled Release Mechanisms. In Fundamentals and Applications of Controlled Release Drug Delivery; Siepmann, J., Siegel, R.A., Rathbone, M.J., Eds.; Springer US: Boston, MA, USA, 2012; pp. 19–43. [Google Scholar]
- Timur, M.; Paşa, A. Synthesis, Characterization, Swelling, and Metal Uptake Studies of Aryl Cross-Linked Chitosan Hydrogels. ACS Omega 2018, 3, 17416–17424. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, E.P.; Da Silva, H.N.; Barbosa, F.C.; Da Silva, H.N.; Andrade, A.L.S.; Fook, M.V.L.; Silva, S.M.D.L.; Leite, I.F. Chitosan/Essential Oils Formulations for Potential Use as Wound Dressing: Physical and Antimicrobial Properties. Materials (Basel) 2019, 12, 2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baran, E.T.; Tuzlakoglu, K.; Mano, J.F.; Reis, R.L. Enzymatic degradation behavior and cytocompatibility of silk fibroin–starch–chitosan conjugate membranes. Mater. Sci. Eng. C Mater Biol Appl. 2012, 32, 1314–1322. [Google Scholar] [CrossRef] [Green Version]
- Freier, T.; Koh, H.S.; Kazazian, K.; Shoichet, M.S. Controlling cell adhesion and degradation of chitosan films by N-acetylation. Biomaterials 2005, 26, 5872–5878. [Google Scholar] [CrossRef]
- Banerjee, A.; Chatterjee, K.; Madras, G. Enzymatic degradation of polymers: A brief review. Mater. Sci. Technol. 2014, 30, 567–573. [Google Scholar] [CrossRef]
- Azevedo, H.; Reis, R.L. Understanding the Enzymatic Degradation of Biodegradable Polymers and Strategies to Control Their Degradation Rate. In Biodegradable Systems in Tissue Engineering and Regenerative Medicine; CRC Press: Boca Raton, FL, USA, 2004; pp. 177–201. [Google Scholar]
- Pawlik, A.; Rehman, M.A.U.; Nawaz, Q.; Bastan, F.E.; Sulka, G.D.; Boccaccini, A. Fabrication and characterization of electrophoretically deposited chitosan-hydroxyapatite composite coatings on anodic titanium dioxide layers. Electrochimica Acta 2019, 307, 465–473. [Google Scholar] [CrossRef]
- Rehman, M.A.U.; Baştan, F.E.; Haider, B.; Boccaccini, A. Electrophoretic deposition of PEEK/bioactive glass composite coatings for orthopedic implants: A design of experiments (DoE) study. Mater. Des. 2017, 130, 223–230. [Google Scholar] [CrossRef]
- Brouwer, J.; Van Leeuwen-Herberts, T.; De Ruit, M.O.-V. Determination of lysozyme in serum, urine, cerebrospinal fluid and feces by enzyme immunoassay. Clin. Chim. Acta 1984, 142, 21–30. [Google Scholar] [CrossRef]
- Porstmann, B.; Jung, K.; Schmechta, H.; Evers, U.; Pergande, M.; Porstmann, T.; Kramm, H.-J.; Krause, H. Measurement of lysozyme in human body fluids: Comparison of various enzyme immunoassay techniques and their diagnostic application. Clin. Biochem. 1989, 22, 349–355. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Zeta Potential (mV) at pH 4.7 | Zeta Potential Dev. (mV) |
|---|---|---|
| CS | +29 | 6 |
| Cu(II)–CS1 | +29 | 6 |
| Cu(II)–CS2 | +21 | 6 |
| Cu(II)–CS3 | +22 | 6 |
| Cu(II)–CS4 | +22 | 7 |
| Samples | Cu(II)–CS1 | Cu(II)–CS2 | Cu(II)–CS3 | Cu(II)–CS4 |
|---|---|---|---|---|
| Theoretical X % | 3 | 6 | 12 | 18 |
| CukL/ Ckα % | 2.93 ± 0.23 | 5.82 ± 0.28 | 11.45 ± 0.22 | 16.50 ± 0.27 |
| Samples ID | CS | Cu(II)–CS1 | Cu(II)–CS2 | Cu(II)–CS3 | Cu(II)–CS4 |
|---|---|---|---|---|---|
| Critical load (N) | 2.2 | 2.8 | 3.7 | 3.7 | 3.4 |
| SD | 0.1 | 0.1 | 0.2 | 0.3 | 0.2 |
| Sample Labels | X (%) | Cu2+: NH2 |
|---|---|---|
| CS | 0 | - |
| CuCS1 | 3 | 1:33 |
| CuCS2 | 6 | 1:17 |
| CuCS3 | 12 | 1:8 |
| CuCS4 | 18 | 1:6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhtar, M.A.; Ilyas, K.; Dlouhý, I.; Siska, F.; Boccaccini, A.R. Electrophoretic Deposition of Copper(II)–Chitosan Complexes for Antibacterial Coatings. Int. J. Mol. Sci. 2020, 21, 2637. https://doi.org/10.3390/ijms21072637
Akhtar MA, Ilyas K, Dlouhý I, Siska F, Boccaccini AR. Electrophoretic Deposition of Copper(II)–Chitosan Complexes for Antibacterial Coatings. International Journal of Molecular Sciences. 2020; 21(7):2637. https://doi.org/10.3390/ijms21072637
Chicago/Turabian StyleAkhtar, Muhammad Asim, Kanwal Ilyas, Ivo Dlouhý, Filip Siska, and Aldo R. Boccaccini. 2020. "Electrophoretic Deposition of Copper(II)–Chitosan Complexes for Antibacterial Coatings" International Journal of Molecular Sciences 21, no. 7: 2637. https://doi.org/10.3390/ijms21072637
APA StyleAkhtar, M. A., Ilyas, K., Dlouhý, I., Siska, F., & Boccaccini, A. R. (2020). Electrophoretic Deposition of Copper(II)–Chitosan Complexes for Antibacterial Coatings. International Journal of Molecular Sciences, 21(7), 2637. https://doi.org/10.3390/ijms21072637