The p38 MAPK Signaling Activation in Colorectal Cancer upon Therapeutic Treatments


 and
and
Abstract
:1. Introduction
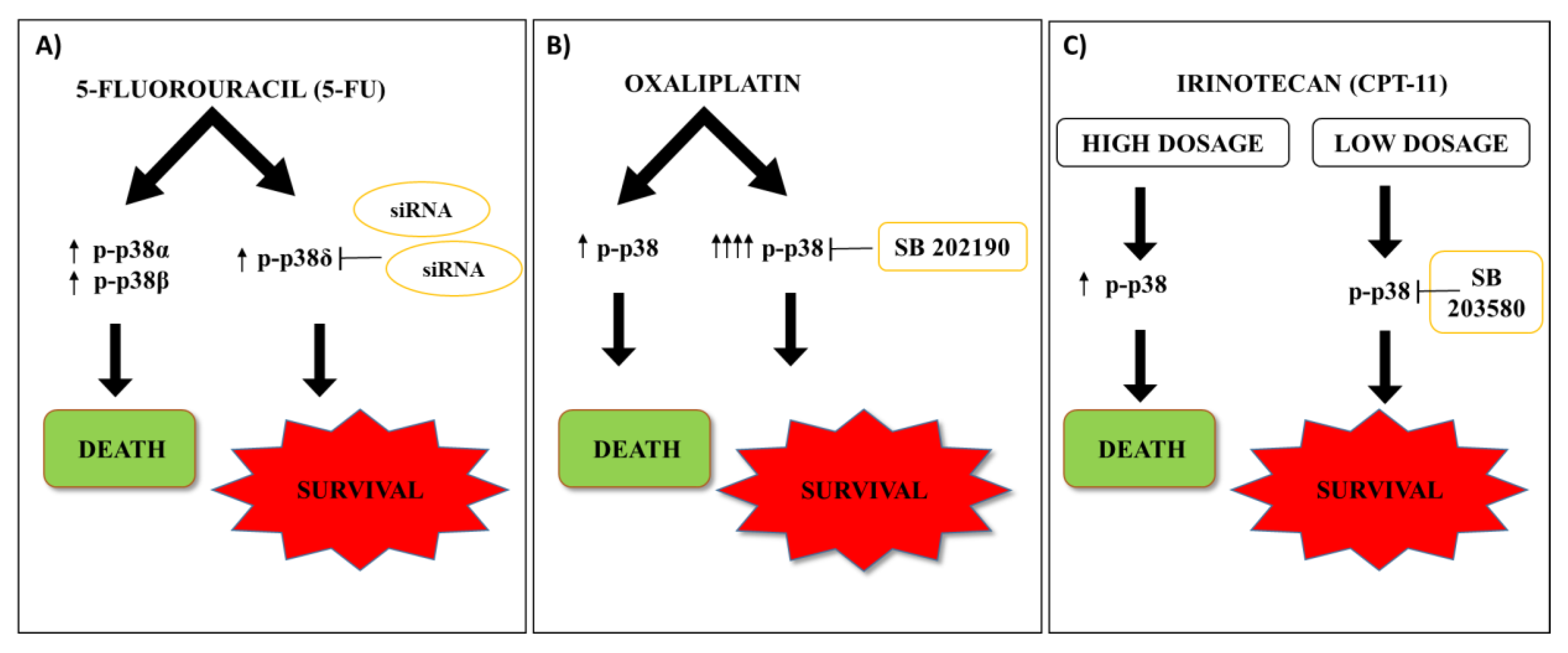
2. 5-Fluorouracil Effects on the p38 MAPK Signaling Pathway in CRC
3. Oxaliplatin Effects on the p38 MAPK Signaling Pathway in CRC
4. Irinotecan Effects on the p38 MAPK Signaling Pathway in CRC
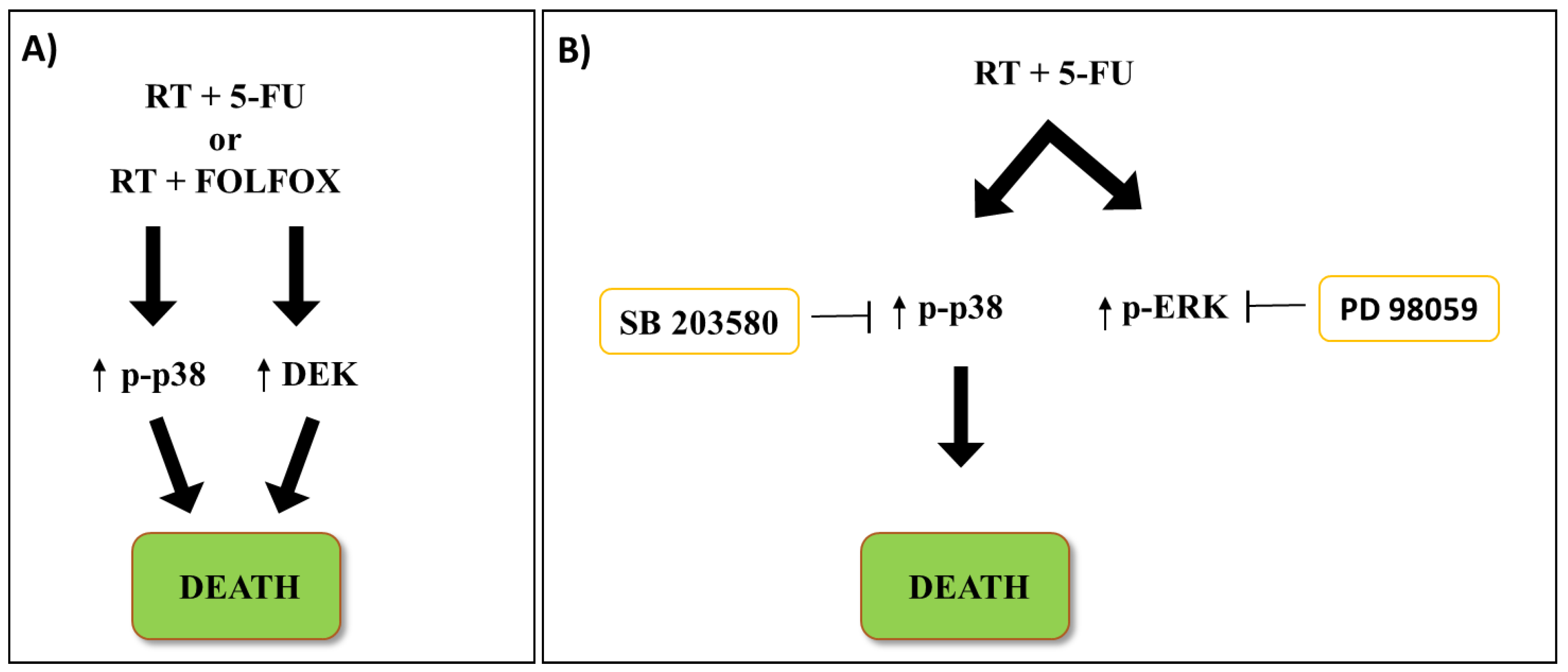
5. Radiotherapy Effects on the p38 MAPK Signaling Pathway in CRC
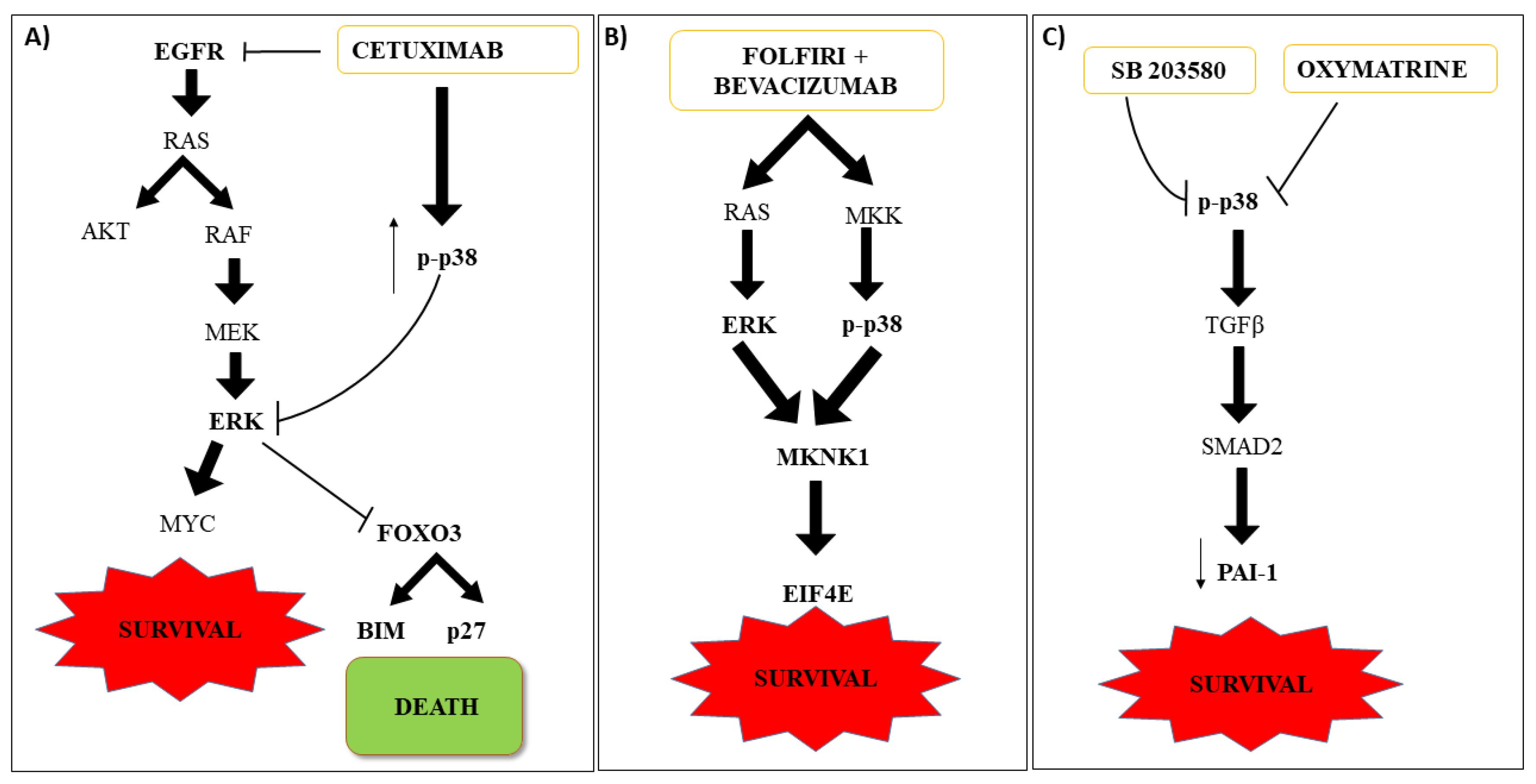
6. Role of the p38 MAPK Signaling Pathway in the Response to Other Therapeutic Strategies
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-FU | 5-fluorouracil |
| 4E-BP1 | eIF4E binding protein-1 |
| AKT ara-c ASK1 | Protein kinase B Cytosine arabinoside Apoptosis signal-regulating kinase 1 |
| ATM | Ataxia-telangiectasia mutated |
| CFZ | Carfilzomib |
| CPT-11 | Irinotecan/Camptothecin-11 |
| eIF4E | Eukaryotic translation initiation factor 4E |
| ERK1/2 | Extracellular signal-regulated-1/2-ER2 |
| FOLFOX | Folinic acid, 5-fluorouracil, Oxaliplatin |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| HuR | Hu antigen R |
| MAPKK/MKK | Mitogen-activated protein kinase kinase |
| MEKK3 | Mitogen-activated protein kinase kinase kinase 3 |
| MKP-1 | Mitogen-activated protein kinase 1 |
| MKNK1 | MAPK-interacting kinase 1 |
| OM | Oxymatrine |
| mTOR | Mammalian phosphorylation target of rapamycin |
| p70S6K | Ribosomal protein S6 kinase |
| PAI-1 | Plasminogen activator inhibitor 1 |
| PCNA | Proliferating Cell Nuclear Antigen |
| PFS | Progression-free survival |
| PP2AC | Serine/threonine-protein phosphatase 2A catalytic subunit alpha isoform |
| PXR | Pregnane X receptor |
| RT | Radiotherapy |
| SAPK/JNK | c-Jun N-terminal kinases |
| SN-38 | 7-etil-10-idrossi-camptotecina |
| SMAD | Small mother against decapentaplegic |
| TGFβ-1 | Transforming growth factor beta 1 |
| TP53 | Tumor protein p53 |
| TPL2 | Tumor progression locus 2 |
References
- Steelman, L.S.; Fitzgerald, T.; Lertpiriyapong, K.; Cocco, L.; Follo, M.Y.; Martelli, A.M.; Neri, L.M.; Marmiroli, S.; Libra, M.; Candido, S.; et al. Critical Roles of EGFR Family Members in Breast Cancer and Breast Cancer Stem Cells: Targets for Therapy. Curr. Pharm. Des. 2016, 22, 2358–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, M.J.; Morris, A.M.; Sun, W. Precision Medicine Versus Population Medicine in Colon Cancer: From Prospects of Prevention, Adjuvant Chemotherapy, and Surveillance. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Raftery, L.; Sanoff, H.K.; Goldberg, R. Colon cancer in older adults. Semin Oncol. 2008, 35, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Avruch, J. MAP kinase pathways: The first twenty years. Biochim. Biophys. Acta 2007, 1773, 1150–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stramucci, L.; Pranteda, A.; Bossi, G. Insights of Crosstalk between p53 Protein and the MKK3/MKK6/p38 MAPK Signaling Pathway in Cancer. Cancers 2018, 10, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Keshet, Y.; Seger, R. The MAP kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar] [CrossRef]
- Lee, J.C.; Laydon, J.T.; McDonnell, P.C.; Gallagher, T.F.; Kumar, S.; Green, D.; McNulty, D.; Blumenthal, M.J.; Heys, J.R.; Landvatter, S.W.; et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994, 372, 739–746. [Google Scholar] [CrossRef]
- Cuenda, A.; Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef] [Green Version]
- Olson, J.M.; Hallahan, A.R. p38 MAP kinase: A convergence point in cancer therapy. Trends Mol. Med. 2004, 10, 125–129. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-Morcillo, M.A.; Valero, M.L.; Callejas-Valera, J.L.; Arias-González, L.; Melgar-Rojas, P.; Galán-Moya, E.M.; García-Gil, E.; García-Cano, J.; Sánchez-Prieto, R. P38MAPK is a major determinant of the balance between apoptosis and autophagy triggered by 5-fluorouracil: Implication in resistance. Oncogene 2012, 31, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Hernández Losa, J.; Parada Cobo, C.; Guinea Viniegra, J.; Sánchez-Arevalo Lobo, V.J.; Ramón y Cajal, S.; Sánchez-Prieto, R. Role of the p38 MAPK pathway in cisplatin-based therapy. Oncogene 2003, 22, 3998–4006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habiro, A.; Tanno, S.; Koizumi, K.; Izawa, T.; Nakano, Y.; Osanai, M.; Mizukami, Y.; Okumura, T.; Kohgo, Y. Involvement of p38 mitogen-activated protein kinase in gemcitabine-induced apoptosis in human pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2004, 316, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koizumi, K.; Tanno, S.; Nakano, Y.; Habiro, A.; Izawa, T.; Mizukami, Y.; Okumura, T.; Kohgo, Y. Activation of p38 mitogen-activated protein kinase is necessary for gemcitabine-induced cytotoxicity in human pancreatic cancer cells. Anticancer Res. 2005, 25, 3347–3353. [Google Scholar]
- Sánchez-Arévalo Lobo, V.J.; Aceves Luquero, C.I.; Alvarez-Vallina, L.; Tipping, A.J.; Viniegra, J.G.; Hernández Losa, J.; Parada Cobo, C.; Galán Moya, E.M.; Gayoso Cruz, J.; Melo, J.V.; et al. Modulation of the p38 MAPK (mitogen-activated protein kinase) pathway through Bcr/Abl: Implications in the cellular response to Ara-C. Biochem. J. 2005, 387, 231–238. [Google Scholar] [CrossRef]
- Galan-Moya, E.M.; Hernandez-Losa, J.; Aceves Luquero, C.I.; de la Cruz-Morcillo, M.A.; Ramírez-Castillejo, C.; Callejas-Valera, J.L.; Arriaga, A.; Aranburo, A.F.; Ramón y Cajal, S.; Silvio Gutkind, J.; et al. c-Abl activates p38 MAPK independently of its tyrosine kinase activity: Implications in cisplatin-based therapy. Int. J. Cancer 2008, 122, 289–297. [Google Scholar] [CrossRef]
- Stramucci, L.; Pranteda, A.; Stravato, A.; Amoreo, C.A.; Pennetti, A.; Diodoro, M.G.; Bartolazzi, A.; Milella, M.; Bossi, G. MKK3 sustains cell proliferation and survival through p38DELTA MAPK activation in colorectal cancer. Cell Death Dis. 2019, 10, 842. [Google Scholar] [CrossRef]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Primers. 2015, 1, 15065. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- García-Cano, J.; Roche, O.; Cimas, F.J.; Pascual-Serra, R.; Ortega-Muelas, M.; Fernández-Aroca, D.M.; Sánchez-Prieto, R. p38MAPK and Chemotherapy: We Always Need to Hear Both Sides of the Story. Front. Cell Dev. Biol. 2016, 4, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossi, V.; Peserico, A.; Tezil, T.; Simone, C. p38α MAPK pathway: A key factor in colorectal cancer therapy and chemoresistance. World J. Gastroenterol. 2014, 20, 9744–9758. [Google Scholar] [CrossRef] [PubMed]
- Can, G.; Akpinar, B.; Baran, Y.; Zhivotovsky, B.; Olsson, M. 5-Fluorouracil signaling through a calcium-calmodulin-dependent pathway is required for p53 activation and apoptosis in colon carcinoma cells. Oncogene 2013, 32, 4529–4538. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.Y.; Miah, A.; Sales, K.M.; Fuller, B.; Seifalian, A.M.; Winslet, M. Inhibition of the p38 MAPK pathway sensitises human colon cancer cells to 5-fluorouracil treatment. Int. J. Oncol. 2011, 38, 1695–1702. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.L.; Huang, W.S.; Lee, K.C.; Tung, S.Y.; Chen, C.N.; Chang, S.F. Effect of 5-fluorouracil on excision repair cross-complementing 1 expression and consequent cytotoxicity regulation in human gastric cancer cells. J. Cell Biochem. 2018, 119, 8472–8480. [Google Scholar] [CrossRef]
- Bracht, K.; Nicholls, A.M.; Liu, Y.; Bodmer, W.F. 5-Fluorouracil response in a large panel of colorectal cancer cell lines is associated with mismatch repair deficiency. Br. J. Cancer 2010, 103, 340–346. [Google Scholar] [CrossRef] [Green Version]
- Stramucci, L.; Bossi, G. Approaching the challenges of MKK3/p38delta MAPK targeting for therapeutic purpose in colorectal cancer. J. Exp. Clin. Cancer Res. 2019, 38, 504. [Google Scholar] [CrossRef]
- Wils, J. Adjuvant treatment of colon cancer: Past, present and future. J. Chemother. 2007, 19, 115–122. [Google Scholar] [CrossRef]
- Mehmood, R.K.; Parker, J.; Ahmed, S.; Qasem, E.; Mohammed, A.A.; Zeeshan, M.; Jehangir, E. Review of Cisplatin and Oxaliplatin in Current Immunogenic and Monoclonal Antibodies Perspective. World J. Oncol. 2014, 5, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Alcindor, T.; Beauger, N. Oxaliplatin: A review in the era of molecularly targeted therapy. Curr. Oncol. 2011, 18, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Dey, H.; Liu, Z.R. Phosphorylation of p68 RNA helicase by p38 MAP kinase contributes to colon cancer cells apoptosis induced by oxaliplatin. BMC Cell Biol. 2012, 13, 27. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.F.; Hu, H.C.; Chao, J.I. Oxaliplatin down-regulates survivin by p38 MAP kinase and proteasome in human colon cancer cells. Chem. Biol. Interact. 2010, 188, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Perfettini, J.L.; Castedo, M.; Nardacci, R.; Ciccosanti, F.; Boya, P.; Roumier, T.; Larochette, N.; Piacentini, M.; Kroemer, G. Essential role of p53 phosphorylation by p38 MAPK in apoptosis induction by the HIV-1 envelope. J. Exp. Med. 2005, 201, 279–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakitina, T.V.; Vasilevskaya, I.A.; O’Dwyer, P.J. Additive interaction of oxaliplatin and 17-allylamino-17-demethoxygeldanamycin in colon cancer cell lines results from inhibition of nuclear factor kappaB signaling. Cancer Res. 2003, 63, 8600–8605. [Google Scholar] [PubMed]
- Chocry, M.; Leloup, L.; Kovacic, H. Reversion of resistance to oxaliplatin by inhibition of p38 MAPK in colorectal cancer cell lines: Involvement of the calpain / Nox1 pathway. Oncotarget 2017, 8, 103710–103730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saltz, L.B. Irinotecan in the first-line treatment of colorectal cancer. Oncology (Williston Park) 1998, 12, 54–58. [Google Scholar]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Rudolf, E.; Kralova, V.; Rudolf, K.; John, S. The role of p38 in irinotecan-induced DNA damage and apoptosis of colon cancer cells. Mutat. Res. 2013, 741, 27–34. [Google Scholar] [CrossRef]
- Tang, W.; Su, G.; Li, J.; Liao, J.; Chen, S.; Huang, C.; Liu, F.; Chen, Q.; Ye, Y. Enhanced anti-colorectal cancer effects of carfilzomib combined with CPT-11 via downregulation of nuclear factor-κB in vitro and in vivo. Int. J. Oncol 2014, 45, 995–1010. [Google Scholar] [CrossRef] [Green Version]
- Paillas, S.; Boissière, F.; Bibeau, F.; Denouel, A.; Mollevi, C.; Causse, A.; Denis, V.; Vezzio-Vié, N.; Marzi, L.; Cortijo, C.; et al. Targeting the p38 MAPK pathway inhibits irinotecan resistance in colon adenocarcinoma. Cancer Res. 2011, 71, 1041–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paillas, S.; Causse, A.; Marzi, L.; de Medina, P.; Poirot, M.; Denis, V.; Vezzio-Vie, N.; Espert, L.; Arzouk, H.; Coquelle, A.; et al. MAPK14/p38α confers irinotecan resistance to TP53-defective cells by inducing survival autophagy. Autophagy 2012, 8, 1098–1112. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.C.; Lee, N.H.; Hsu, H.H.; Ho, T.J.; Tu, C.C.; Hsieh, D.J.; Lin, Y.M.; Chen, L.M.; Kuo, W.W.; Huang, C.Y. Thymoquinone induces caspase-independent, autophagic cell death in CPT-11-resistant lovo colon cancer via mitochondrial dysfunction and activation of JNK and p38. J. Agric. Food Chem. 2015, 63, 1540–1546. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.Y.; Wu, V.W.C. A Review on the Special Radiotherapy Techniques of Colorectal Cancer. Front. Oncol. 2019, 9, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Stat Facts: Colorectal Cancer. Available online: https://seer.cancer.gov/statfacts/html/colorect.html (accessed on 14 January 2020).
- Fazeli, M.S.; Keramati, M.R. Rectal cancer: A review. Med. J. Islam Repub. Iran 2015, 29, 171. [Google Scholar] [PubMed]
- Dahlberg, M.; Glimelius, B.; Påhlman, L. Improved survival and reduction in local failure rates after preoperative radiotherapy: Evidence for the generalizability of the results of Swedish Rectal Cancer Trial. Ann. Surg. 1999, 229, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Useros, J.; Moreno, I.; Fernandez-Aceñero, M.J.; Rodriguez-Remirez, M.; Borrero-Palacios, A.; Cebrian, A.; Gomez Del Pulgar, T.; Del Puerto-Nevado, L.; Li, W.; Puime-Otin, A.; et al. The potential predictive value of DEK expression for neoadjuvant chemoradiotherapy response in locally advanced rectal cancer. BMC Cancer 2018, 18, 144. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, Y.; Yang, L.; Yin, D. Intermittent low dose irradiation enhances the effectiveness of radio- and chemo-therapy for human colorectal adenocarcinoma cell line HT-29. Oncol. Rep. 2017, 38, 591–597. [Google Scholar] [CrossRef] [Green Version]
- De la Cruz-Morcillo, M.A.; García-Cano, J.; Arias-González, L.; García-Gil, E.; Artacho-Cordón, F.; Ríos-Arrabal, S.; Valero, M.L.; Cimas, F.J.; Serrano-Oviedo, L.; Villas, M.V.; et al. Abrogation of the p38 MAPK α signaling pathway does not promote radioresistance but its activity is required for 5-Fluorouracil-associated radiosensitivity. Cancer Lett. 2013, 335, 66–74. [Google Scholar] [CrossRef]
- Lafarga, V.; Cuadrado, A.; Lopez de Silanes, I.; Bengoechea, R.; Fernandez-Capetillo, O.; Nebreda, A.R. p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol. Cell Biol. 2009, 29, 4341–4351. [Google Scholar] [CrossRef] [Green Version]
- Zlobec, I. Novel biomarkers for the prediction of metastasis in colorectal cancer. Expert Opin. Med. Diagn. 2013, 7, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Shin-ya, T.; Nishio, K.; Ito, F. Mitogen-activated protein kinase phosphatase-1 modulated JNK activation is critical for apoptosis induced by inhibitor of epidermal growth factor receptor-tyrosine kinase. FEBS J. 2009, 276, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Montagut, C.; Iglesias, M.; Arumi, M.; Bellosillo, B.; Gallen, M.; Martinez-Fernandez, A.; Martinez-Aviles, L.; Cañadas, I.; Dalmases, A.; Moragon, E.; et al. Mitogen-activated protein kinase phosphatase-1 (MKP-1) impairs the response to anti-epidermal growth factor receptor (EGFR) antibody cetuximab in metastatic colorectal cancer patients. Br. J. Cancer 2010, 102, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Lam, F.; Proud, C.; Wang, S. Targeting Mnks for cancer therapy. Oncotarget 2012, 3, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Wheater, M.J.; Johnson, P.W.; Blaydes, J.P. The role of MNK proteins and eIF4E phosphorylation in breast cancer cell proliferation and survival. Cancer Biol. Ther. 2010, 10, 728–735. [Google Scholar] [CrossRef] [Green Version]
- Furic, L.; Rong, L.; Larsson, O.; Koumakpayi, I.H.; Yoshida, K.; Brueschke, A.; Petroulakis, E.; Robichaud, N.; Pollak, M.; Gaboury, L.A.; et al. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc. Natl. Acad. Sci. USA 2010, 107, 14134–14139. [Google Scholar] [CrossRef] [Green Version]
- Berger, M.D.; Stintzing, S.; Heinemann, V.; Yang, D.; Cao, S.; Sunakawa, Y.; Ning, Y.; Matsusaka, S.; Okazaki, S.; Miyamoto, Y.; et al. Impact of genetic variations in the MAPK signaling pathway on outcome in metastatic colorectal cancer patients treated with first-line FOLFIRI and bevacizumab: Data from FIRE-3 and TRIBE trials. Ann. Oncol. 2017, 28, 2780–2785. [Google Scholar] [CrossRef]
- Han, Z.; Zhu, S.; Han, X.; Wang, Z.; Wu, S.; Zheng, R. Baicalein inhibits hepatocellular carcinoma cells through suppressing the expression of CD24. Int. Immunopharmacol. 2015, 29, 416–422. [Google Scholar] [CrossRef]
- Chung, H.; Choi, H.S.; Seo, E.K.; Kang, D.H.; Oh, E.S. Baicalin and baicalein inhibit transforming growth factor-β1-mediated epithelial-mesenchymal transition in human breast epithelial cells. Biochem. Biophys. Res. Commun. 2015, 458, 707–713. [Google Scholar] [CrossRef]
- Choi, E.O.; Park, C.; Hwang, H.J.; Hong, S.H.; Kim, G.Y.; Cho, E.J.; Kim, W.J.; Choi, Y.H. Baicalein induces apoptosis via ROS-dependent activation of caspases in human bladder cancer 5637 cells. Int. J. Oncol. 2016, 49, 1009–1018. [Google Scholar] [CrossRef]
- Su, M.Q.; Zhou, Y.R.; Rao, X.; Yang, H.; Zhuang, X.H.; Ke, X.J.; Peng, G.Y.; Zhou, C.L.; Shen, B.Y.; Dou, J. Baicalein induces the apoptosis of HCT116 human colon cancer cells via the upregulation of DEPP/Gadd45a and activation of MAPKs. Int. J. Oncol. 2018, 53, 750–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langenskiöld, M.; Holmdahl, L.; Angenete, E.; Falk, P.; Nordgren, S.; Ivarsson, M.L. Differential prognostic impact of uPA and PAI-1 in colon and rectal cancer. Tumour. Biol. 2009, 30, 210–220. [Google Scholar] [CrossRef]
- Wind, T.; Jensen, J.K.; Dupont, D.M.; Kulig, P.; Andreasen, P.A. Mutational analysis of plasminogen activator inhibitor-1. Eur. J. Biochem. 2003, 270, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wu, C.; Boye, A.; Wu, J.; Wang, J.; Yang, X.; Yang, Y. MAPK inhibitors modulate Smad2/3/4 complex cyto-nuclear translocation in myofibroblasts via Imp7/8 mediation. Mol. Cell Biochem. 2015, 406, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, C.; Wang, J.; Fan, Y.; Wang, Z.; Wang, Y. Oxymatrine inhibits the migration of human colorectal carcinoma RKO cells via inhibition of PAI-1 and the TGF-β1/Smad signaling pathway. Oncol. Rep. 2017, 37, 747–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Shah, Y.M.; Ma, X.; Pang, X.; Tanaka, T.; Kodama, T.; Krausz, K.W.; Gonzalez, F.J. Therapeutic role of rifaximin in inflammatory bowel disease: Clinical implication of human pregnane X receptor activation. J. Pharmacol. Exp. Ther. 2010, 335, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Mencarelli, A.; Renga, B.; Palladino, G.; Claudio, D.; Ricci, P.; Distrutti, E.; Barbanti, M.; Baldelli, F.; Fiorucci, S. Inhibition of NF-κB by a PXR-dependent pathway mediates counter-regulatory activities of rifaximin on innate immunity in intestinal epithelial cells. Eur. J. Pharmacol. 2011, 668, 317–324. [Google Scholar] [CrossRef]
- Guzińska-Ustymowicz, K.; Pryczynicz, A.; Kemona, A.; Czyzewska, J. Correlation between proliferation markers: PCNA, Ki-67, MCM-2 and antiapoptotic protein Bcl-2 in colorectal cancer. Anticancer Res. 2009, 29, 3049–3052. [Google Scholar]
- Esposito, G.; Gigli, S.; Seguella, L.; Nobile, N.; D’Alessandro, A.; Pesce, M.; Capoccia, E.; Steardo, L.; Cirillo, C.; Cuomo, R.; et al. Rifaximin, a non-absorbable antibiotic, inhibits the release of pro-angiogenic mediators in colon cancer cells through a pregnane X receptor-dependent pathway. Int. J. Oncol. 2016, 49, 639–645. [Google Scholar] [CrossRef] [Green Version]
- Mi, C.; Ma, J.; Wang, K.S.; Zuo, H.X.; Wang, Z.; Li, M.Y.; Piao, L.X.; Xu, G.H.; Li, X.; Quan, Z.S.; et al. Imperatorin suppresses proliferation and angiogenesis of human colon cancer cell by targeting HIF-1α via the mTOR/p70S6K/4E-BP1 and MAPK pathways. J. Ethnopharmacol. 2017, 203, 27–38. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Qin, X.; Liu, F.; White, E.; Zheng, X.F. PP2AC Level Determines Differential Programming of p38-TSC-mTOR Signaling and Therapeutic Response to p38-Targeted Therapy in Colorectal Cancer. EBioMedicine 2015, 2, 1944–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Compound | Action | References |
|---|---|---|
| Cetuximab | Chimeric monoclonal antibody against epidermal growth factor receptor (EGFR). Its interaction prevents the binding to EGF inhibiting cell growth and survival. | [53,54] |
| Bevacizumab | Anti-VEGF recombinant monoclonal antibody. It inhibits VEGF receptors (VEGFR) preventing blood vessels proliferation. | [55,56,57,58] |
| Baicalein (5,6,7-trihydroxyflavone) | Flavone, type of flavonoid, originally isolated from the roots of Scutellaria baicalensis. Compound with anti-tumor activity, in several cancers, mainly due to its capacities to inhibit cyclins complexes and thus to regulate the cell cycle. | [59,60,61,62] |
| Oxymatrine | Potent monosomic alkaloid derived from the root of Sophora flavescens Ait. Compound with anti-inflammatory, anti-oxidative and hepatoprotective activities. | [63,66] |
| Rifaximin | Synthetic rifamycin derivative and anti-bacterial agent, used for the treatment of gastroenteritis by Escherichia coli infections. It may also be used in the treatment of hepatic encephalopathy. | [67,68,69,70] |
| Imperatorin | Tumor necrosis factor antagonist; furanocoumarin from West African medicinal plant Clausena anisata. | [71] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pranteda, A.; Piastra, V.; Stramucci, L.; Fratantonio, D.; Bossi, G. The p38 MAPK Signaling Activation in Colorectal Cancer upon Therapeutic Treatments. Int. J. Mol. Sci. 2020, 21, 2773. https://doi.org/10.3390/ijms21082773
Pranteda A, Piastra V, Stramucci L, Fratantonio D, Bossi G. The p38 MAPK Signaling Activation in Colorectal Cancer upon Therapeutic Treatments. International Journal of Molecular Sciences. 2020; 21(8):2773. https://doi.org/10.3390/ijms21082773
Chicago/Turabian StylePranteda, Angelina, Valentina Piastra, Lorenzo Stramucci, Deborah Fratantonio, and Gianluca Bossi. 2020. "The p38 MAPK Signaling Activation in Colorectal Cancer upon Therapeutic Treatments" International Journal of Molecular Sciences 21, no. 8: 2773. https://doi.org/10.3390/ijms21082773
APA StylePranteda, A., Piastra, V., Stramucci, L., Fratantonio, D., & Bossi, G. (2020). The p38 MAPK Signaling Activation in Colorectal Cancer upon Therapeutic Treatments. International Journal of Molecular Sciences, 21(8), 2773. https://doi.org/10.3390/ijms21082773