Skin Barrier Abnormalities and Immune Dysfunction in Atopic Dermatitis

 , ,
, , 
 and
and
Abstract
:1. Introduction
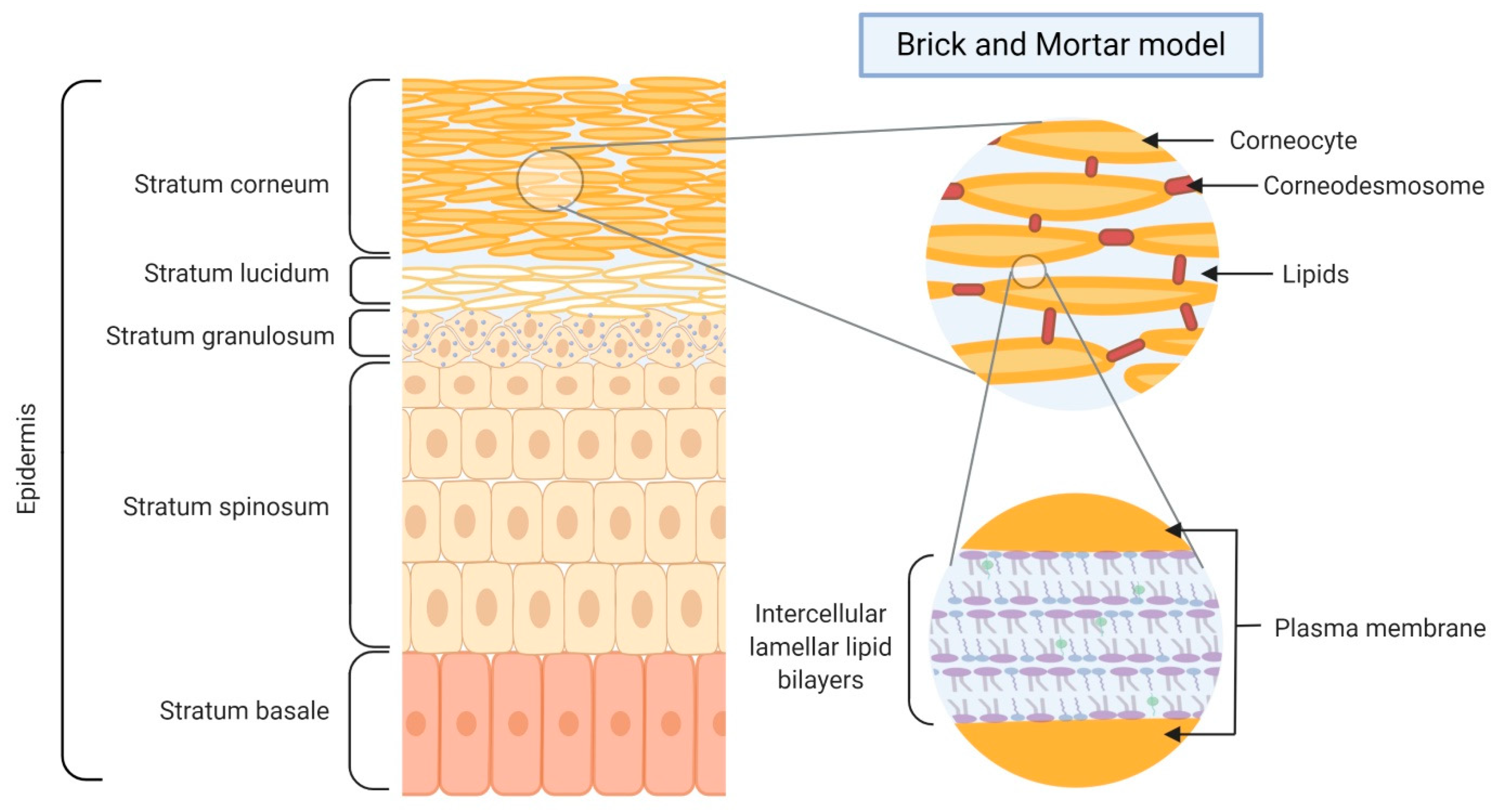
2. Skin Barrier Formation and Function
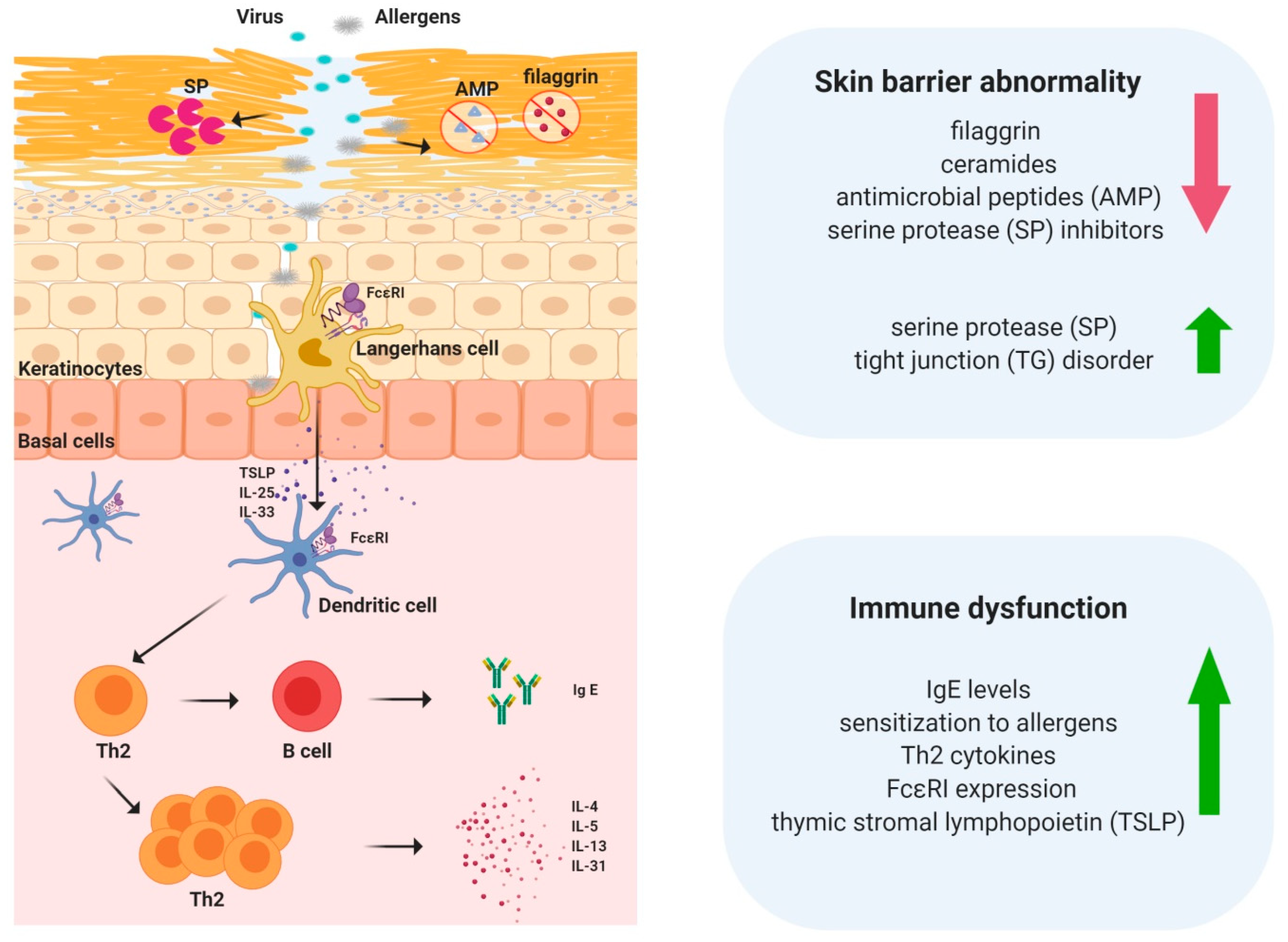
3. Skin Barrier Abnormalities in Atopic Dermatitis
3.1. Lipids
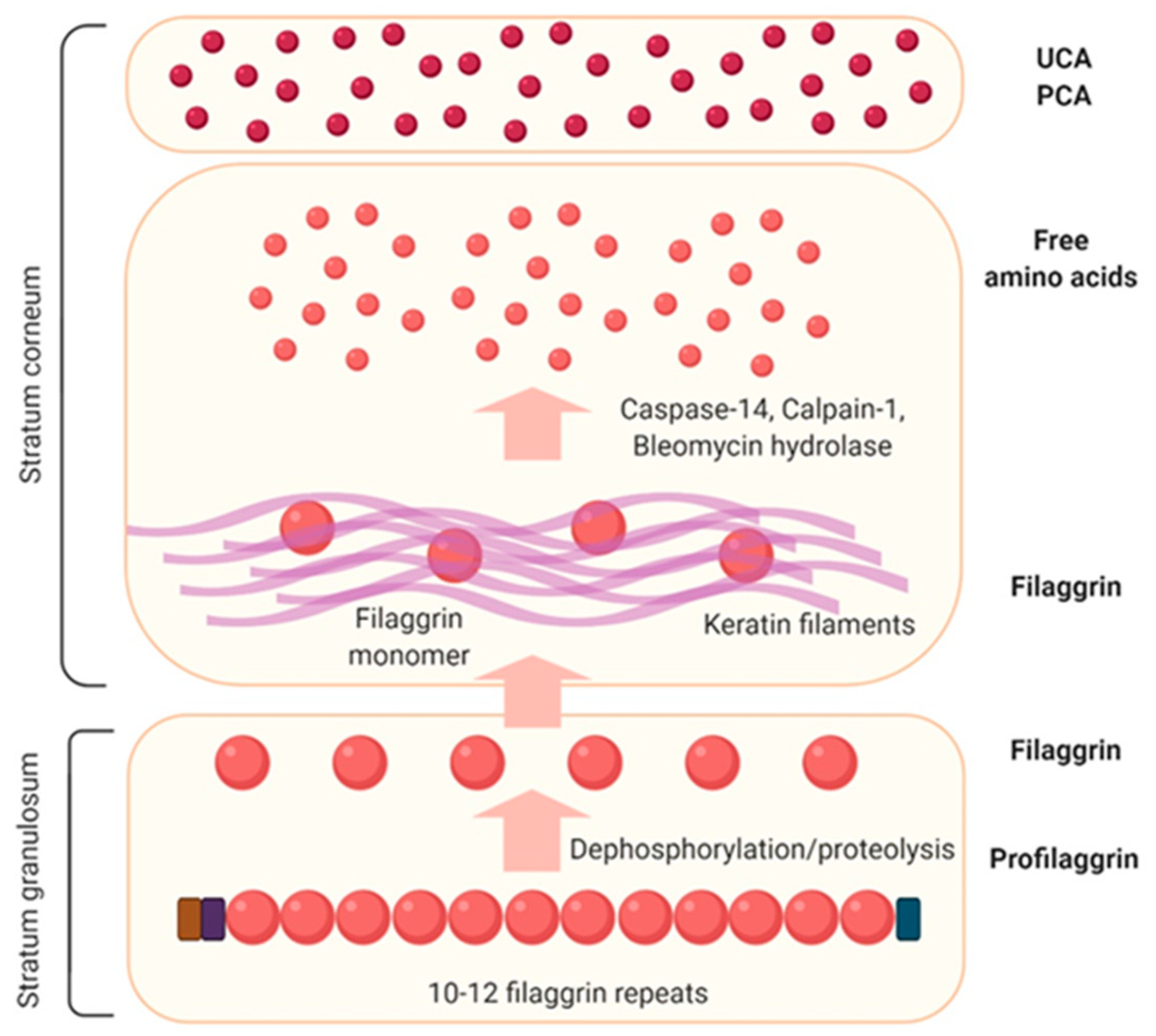
3.2. Filaggrin
3.3. Tight Junctions (TJs)
4. Immune Dysfunction in Atopic Dermatitis
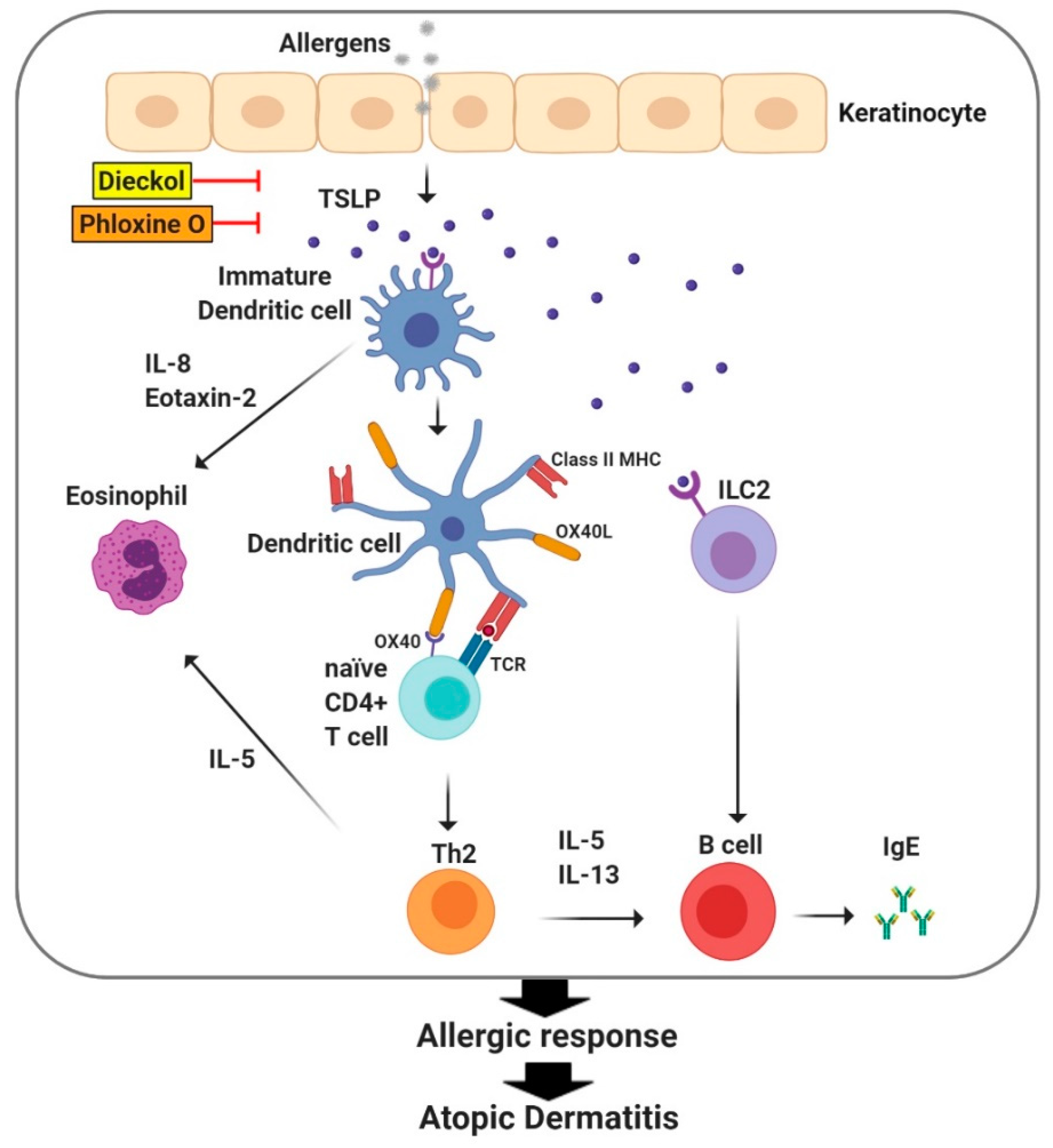
5. TSLP Regulates Immune Responses in Atopic Dermatitis
6. The Involvement of ILC2s in the Pathogenesis of Atopic Dermatitis
7. Toll-Like Receptors and Atopic Dermatitis
8. The Emerging Role of Inflammasomes in Atopic Dermatitis Symptoms
9. Crosstalk between the Skin Barrier and Immune System in Atopic Dermatitis
10. Development of Treatment Restoring Skin Barrier Abnormalities and Immune Dysfunction
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Spergel, J.M.; Paller, A.S. Atopic dermatitis and the atopic march. J. Allergy Clin. Immunol. 2003, 112, S118–S127. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.N.; Mancini, A.J.; Paller, A.S.; Simpson, E.L.; Eichenfield, L.F. Understanding and managing atopic dermatitis in adult patients. Semin. Cutan. Med. Surg. 2012, 31, S18–S22. [Google Scholar] [CrossRef] [PubMed]
- Novak, N.; Leung, D.Y. Advances in atopic dermatitis. Curr. Opin. Immunol. 2011, 23, 778–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaniboni, M.C.; Samorano, L.P.; Orfali, R.L.; Aoki, V. Skin barrier in atopic dermatitis: Beyond filaggrin. An. Bras. Dermatol. 2016, 91, 472–478. [Google Scholar] [CrossRef]
- Lee, S.H.; Jeong, S.K.; Ahn, S.K. An update of the defensive barrier function of skin. Yonsei Med. J. 2006, 47, 293–306. [Google Scholar] [CrossRef] [Green Version]
- Darlenski, R.; Kazandjieva, J.; Tsankov, N. Skin barrier function: Morphological basis and regulatory mechanisms. J. Clin. Med. 2011, 4, 36–45. [Google Scholar]
- Wickett, R.R.; Visscher, M.O. Structure and function of the epidermal barrier. Am. J. Infect. Control. 2006, 34, S98–S110. [Google Scholar] [CrossRef]
- Pouillot, A.; Dayan, N.; Polla, A.S.; Polla, L.L.; Polla, B.S. The stratum corneum: A double paradox. J. Cosmet. Dermatol. 2008, 7, 143–148. [Google Scholar] [CrossRef]
- Elsholz, F.; Harteneck, C.; Muller, W.; Friedland, K. Calcium—A central regulator of keratinocyte differentiation in health and disease. Eur. J. Dermatol. EJD 2014, 24, 650–661. [Google Scholar] [CrossRef] [Green Version]
- Menon, G.K.; Cleary, G.W.; Lane, M.E. The structure and function of the stratum corneum. Int. J. Pharm. 2012, 435, 3–9. [Google Scholar] [CrossRef]
- Elias, P.M. Skin barrier function. Curr. Allergy Asthma Rep. 2008, 8, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Nemes, Z.; Steinert, P.M. Bricks and mortar of the epidermal barrier. Exp. Mol. Med. 1999, 31, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Imokawa, G.; Abe, A.; Jin, K.; Higaki, Y.; Kawashima, M.; Hidano, A. Decreased level of ceramides in stratum corneum of atopic dermatitis: An etiologic factor in atopic dry skin? J. Investig. Dermatol. 1991, 96, 523–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, M.D.; Kim, B.E.; Gao, P.; Grant, A.V.; Boguniewicz, M.; Debenedetto, A.; Schneider, L.; Beck, L.A.; Barnes, K.C.; Leung, D.Y. Cytokine modulation of atopic dermatitis filaggrin skin expression. J. Allergy Clin. Immunol. 2007, 120, 150–155. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.E.; Leung, D.Y.; Boguniewicz, M.; Howell, M.D. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin. Immunol. 2008, 126, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Jungersted, J.M.; Scheer, H.; Mempel, M.; Baurecht, H.; Cifuentes, L.; Hogh, J.K.; Hellgren, L.I.; Jemec, G.B.; Agner, T.; Weidinger, S. Stratum corneum lipids, skin barrier function and filaggrin mutations in patients with atopic eczema. Allergy 2010, 65, 911–918. [Google Scholar] [CrossRef]
- Elias, P.M.; Wakefield, J.S. Mechanisms of abnormal lamellar body secretion and the dysfunctional skin barrier in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Wolf, R.; Wolf, D. Abnormal epidermal barrier in the pathogenesis of atopic dermatitis. Clin. Dermatol. 2012, 30, 329–334. [Google Scholar] [CrossRef]
- Janssens, M.; van Smeden, J.; Gooris, G.S.; Bras, W.; Portale, G.; Caspers, P.J.; Vreeken, R.J.; Hankemeier, T.; Kezic, S.; Wolterbeek, R.; et al. Increase in short-chain ceramides correlates with an altered lipid organization and decreased barrier function in atopic eczema patients. J. Lipid Res. 2012, 53, 2755–2766. [Google Scholar] [CrossRef] [Green Version]
- Malik, K.; Heitmiller, K.D.; Czarnowicki, T. An Update on the Pathophysiology of Atopic Dermatitis. Dermatol. Clin. 2017, 35, 317–326. [Google Scholar] [CrossRef]
- Quiroz, F.G.; Fiore, V.F.; Levorse, J.; Polak, L.; Wong, E.; Pasolli, H.A.; Fuchs, E. Liquid-liquid phase separation drives skin barrier formation. Science 2020, 367, 6483. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.N.; Irvine, A.D.; Terron-Kwiatkowski, A.; Zhao, Y.; Liao, H.; Lee, S.P.; Goudie, D.R.; Sandilands, A.; Campbell, L.E.; Smith, F.J.; et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 2006, 38, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Szegedi, A. Filaggrin mutations in early- and late-onset atopic dermatitis. Br. J. Dermatol. 2015, 172, 320–321. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Henry, J.; Hsu, C.Y.; Balica, S.; Jean-Decoster, C.; Mechin, M.C.; Hansmann, B.; Rodriguez, E.; Weindinger, S.; Schmitt, A.M.; et al. Defects of filaggrin-like proteins in both lesional and nonlesional atopic skin. J. Allergy Clin. Immunol. 2013, 131, 1094–1102. [Google Scholar] [CrossRef]
- Thyssen, J.P.; Kezic, S. Causes of epidermal filaggrin reduction and their role in the pathogenesis of atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 792–799. [Google Scholar] [CrossRef]
- De Benedetto, A.; Rafaels, N.M.; McGirt, L.Y.; Ivanov, A.I.; Georas, S.N.; Cheadle, C.; Berger, A.E.; Zhang, K.; Vidyasagar, S.; Yoshida, T. Tight junction defects in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2011, 127, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Furuse, M.; Hata, M.; Furuse, K.; Yoshida, Y.; Haratake, A.; Sugitani, Y.; Noda, T.; Kubo, A.; Tsukita, S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: A lesson from claudin-1-deficient mice. J. Cell Biol. 2002, 156, 1099–1111. [Google Scholar] [CrossRef]
- Bergmann, S.; von Buenau, B.; Vidal, Y.S.S.; Haftek, M.; Wladykowski, E.; Houdek, P.; Lezius, S.; Duplan, H.; Basler, K.; Dahnhardt-Pfeiffer, S.; et al. Claudin-1 decrease impacts epidermal barrier function in atopic dermatitis lesions dose-dependently. Sci. Rep. 2020, 10, 2024. [Google Scholar] [CrossRef]
- Tokumasu, R.; Yamaga, K.; Yamazaki, Y.; Murota, H.; Suzuki, K.; Tamura, A.; Bando, K.; Furuta, Y.; Katayama, I.; Tsukita, S. Dose-dependent role of claudin-1 in vivo in orchestrating features of atopic dermatitis. Proc. Natl. Acad. Sci. USA 2016, 113, E4061–E4068. [Google Scholar] [CrossRef] [Green Version]
- Yuki, T.; Komiya, A.; Kusaka, A.; Kuze, T.; Sugiyama, Y.; Inoue, S. Impaired tight junctions obstruct stratum corneum formation by altering polar lipid and profilaggrin processing. J. Dermatol. Sci. 2013, 69, 148–158. [Google Scholar] [CrossRef]
- Sheikhi, A.; Giti, H.; Heibor, M.R.; Jafarzadeh, A.; Shakerian, M.; Baharifar, N.; Niruzad, F.; Moghaddam, A.S.; Kokhaei, P.; Baghaeifar, M. Lactobacilus delbrueckii subsp. bulgaricus modulates the secretion of Th1/Th2 and Treg cell-related cytokines by PBMCs from patients with atopic dermatitis. Drug Res. 2017, 67, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Roesner, L.M.; Werfel, T.; Heratizadeh, A. The adaptive immune system in atopic dermatitis and implications on therapy. Expert Rev. Clin. Immunol. 2016, 12, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Moosbrugger-Martinz, V.; Tripp, C.H.; Clausen, B.E.; Schmuth, M.; Dubrac, S. Atopic dermatitis induces the expansion of thymus-derived regulatory T cells exhibiting a Th2-like phenotype in mice. J. Cell. Mol. Med. 2016, 20, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Brunner, P.M.; Guttman-Yassky, E.; Leung, D.Y. The immunology of atopic dermatitis and its reversibility with broad-spectrum and targeted therapies. J. Allergy Clin. Immunol. 2017, 139, S65–S76. [Google Scholar] [CrossRef] [Green Version]
- Cianferoni, A.; Spergel, J. The importance of TSLP in allergic disease and its role as a potential therapeutic target. Expert Rev. Clin. Immunol. 2014, 10, 1463–1474. [Google Scholar] [CrossRef] [Green Version]
- Quentmeier, H.; Drexler, H.; Fleckenstein, D.; Zaborski, M.; Armstrong, A.; Sims, J.; Lyman, S. Cloning of human thymic stromal lymphopoietin (TSLP) and signaling mechanisms leading to proliferation. Leukemia 2001, 15, 1286–1292. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Wang, Y.-H.; Duramad, O.; Hori, T.; Delespesse, G.J.; Watanabe, N.; Qin, F.X.-F.; Yao, Z.; Cao, W.; Liu, Y.-J. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J. Exp. Med. 2005, 202, 1213–1223. [Google Scholar] [CrossRef] [Green Version]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A. Human epithelial cells trigger dendritic cell–mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef]
- Liu, Y.-J. Thymic stromal lymphopoietin and OX40 ligand pathway in the initiation of dendritic cell–mediated allergic inflammation. J. Allergy Clin. Immunol. 2007, 120, 238–244. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, X.; Zhou, B. DC-derived TSLP promotes T h2 polarization in LPS-primed allergic airway inflammation. Eur. J. Immunol. 2012, 42, 1735–1743. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Oh, J.W.; Lee, H.E.; Lee, B.H.; Lim, K.M.; Lee, J.Y. Topical Application of Dieckol Ameliorates Atopic Dermatitis in NC/Nga Mice by Suppressing Thymic Stromal Lymphopoietin Production. J. Investig. Dermatol. 2016, 136, 1062–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.E.; Yang, G.; Kim, K.B.; Lee, B.M.; Lee, J.Y. Phloxine O, a Cosmetic Colorant, Suppresses the Expression of Thymic Stromal Lymphopoietin and Acute Dermatitis Symptoms in Mice. Biomol. Ther. 2018, 26, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.-I.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate production of TH 2 cytokines by adipose tissue-associated c-Kit+ Sca-1+ lymphoid cells. Nature 2010, 463, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef] [Green Version]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.; et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef]
- Imai, Y.; Yasuda, K.; Sakaguchi, Y.; Haneda, T.; Mizutani, H.; Yoshimoto, T.; Nakanishi, K.; Yamanishi, K. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 13921–13926. [Google Scholar] [CrossRef] [Green Version]
- Niebuhr, M.; Langnickel, J.; Draing, C.; Renz, H.; Kapp, A.; Werfel, T. Dysregulation of toll-like receptor-2 (TLR-2)-induced effects in monocytes from patients with atopic dermatitis: Impact of the TLR-2 R753Q polymorphism. Allergy 2008, 63, 728–734. [Google Scholar] [CrossRef]
- Salpietro, C.; Rigoli, L.; Miraglia Del Giudice, M.; Cuppari, C.; Di Bella, C.; Salpietro, A.; Maiello, N.; La Rosa, M.; Marseglia, G.L.; Leonardi, S.; et al. TLR2 and TLR4 gene polymorphisms and atopic dermatitis in Italian children: A multicenter study. Int. J. Immunopathol. Pharm. 2011, 24, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Can, C.; Yazicioglu, M.; Gurkan, H.; Tozkir, H.; Gorgulu, A.; Sut, N.H. Lack of Association between Toll-like Receptor 2 Polymorphisms (R753Q and A-16934T) and Atopic Dermatitis in Children from Thrace Region of Turkey. Balkan Med. J. 2017, 34, 232–238. [Google Scholar] [CrossRef]
- Novak, N.; Yu, C.F.; Bussmann, C.; Maintz, L.; Peng, W.M.; Hart, J.; Hagemann, T.; Diaz-Lacava, A.; Baurecht, H.J.; Klopp, N.; et al. Putative association of a TLR9 promoter polymorphism with atopic eczema. Allergy 2007, 62, 766–772. [Google Scholar] [CrossRef]
- Dai, X.; Sayama, K.; Tohyama, M.; Shirakata, Y.; Hanakawa, Y.; Tokumaru, S.; Yang, L.; Hirakawa, S.; Hashimoto, K. Mite allergen is a danger signal for the skin via activation of inflammasome in keratinocytes. J. Allergy Clin. Immunol. 2011, 127, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Niebuhr, M.; Baumert, K.; Heratizadeh, A.; Satzger, I.; Werfel, T. Impaired NLRP3 inflammasome expression and function in atopic dermatitis due to Th2 milieu. Allergy 2014, 69, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Tanaka, M.; Tsutsui, H.; Kupper, T.S.; Asahi, K.; Okamura, H.; Nakanishi, K.; Suzuki, M.; Kayagaki, N.; Black, R.A.; et al. Skin-specific caspase-1-transgenic mice show cutaneous apoptosis and pre-endotoxin shock condition with a high serum level of IL-18. J. Immunol. 2000, 165, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuepbach-Mallepell, S.; Philippe, V.; Bruggen, M.C.; Watanabe, H.; Roques, S.; Baldeschi, C.; Gaide, O. Antagonistic effect of the inflammasome on thymic stromal lymphopoietin expression in the skin. J. Allergy Clin. Immunol. 2013, 132, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J.P. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc. Natl. Acad. Sci. USA 2009, 106, 9021–9026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Koning, H.D.; Bergboer, J.G.; van den Bogaard, E.H.; van Vlijmen-Willems, I.M.; Rodijk-Olthuis, D.; Simon, A.; Zeeuwen, P.L.; Schalkwijk, J. Strong induction of AIM2 expression in human epidermis in acute and chronic inflammatory skin conditions. Exp. Dermatol. 2012, 21, 961–964. [Google Scholar] [CrossRef]
- Bivik, C.; Verma, D.; Winge, M.C.; Lieden, A.; Bradley, M.; Rosdahl, I.; Soderkvist, P. Genetic variation in the inflammasome and atopic dermatitis susceptibility. J. Investig. Dermatol. 2013, 133, 2486–2489. [Google Scholar] [CrossRef] [Green Version]
- Kuo, I.H.; Carpenter-Mendini, A.; Yoshida, T.; McGirt, L.Y.; Ivanov, A.I.; Barnes, K.C.; Gallo, R.L.; Borkowski, A.W.; Yamasaki, K.; Leung, D.Y.; et al. Activation of epidermal toll-like receptor 2 enhances tight junction function: Implications for atopic dermatitis and skin barrier repair. J. Investig. Dermatol. 2013, 133, 988–998. [Google Scholar] [CrossRef] [Green Version]
- Leung, D.Y. New insights into atopic dermatitis: Role of skin barrier and immune dysregulation. Allergol. Int. Off. J. Jpn. Soc. Allergol. 2013, 62, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Dainichi, T.; Kitoh, A.; Otsuka, A.; Nakajima, S.; Nomura, T.; Kaplan, D.H.; Kabashima, K. The epithelial immune microenvironment (EIME) in atopic dermatitis and psoriasis. Nat. Immunol. 2018, 19, 1286–1298. [Google Scholar] [CrossRef]
- Halim, T.Y.; Steer, C.A.; Mathä, L.; Gold, M.J.; Martinez-Gonzalez, I.; McNagny, K.M.; McKenzie, A.N.; Takei, F. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 2014, 40, 425–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, Z.; Zhao, Y.; Liu, X.; Chang, C.; Zhang, J. Molecular biology of atopic dermatitis. Clin. Rev. Allergy Immunol. 2014, 47, 193–218. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.; Moran, T.; Saunders, S.P.; Kaszlikowska, A.; Floudas, A.; Bom, J.; Nunez, G.; Iwakura, Y.; O’Neill, L.; Irvine, A.D.; et al. Spontaneous atopic dermatitis in mice with a defective skin barrier is independent of ILC2 and mediated by IL-1beta. Allergy 2019, 74, 1920–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kezic, S.; O’Regan, G.M.; Lutter, R.; Jakasa, I.; Koster, E.S.; Saunders, S.; Caspers, P.; Kemperman, P.M.; Puppels, G.J.; Sandilands, A.; et al. Filaggrin loss-of-function mutations are associated with enhanced expression of IL-1 cytokines in the stratum corneum of patients with atopic dermatitis and in a murine model of filaggrin deficiency. J. Allergy Clin. Immunol. 2012, 129, 1031–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henehan, M.; De Benedetto, A. Update on protease-activated receptor 2 in cutaneous barrier, differentiation, tumorigenesis and pigmentation, and its role in related dermatologic diseases. Exp. Dermatol. 2019, 28, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadeau, P.; Henehan, M.; De Benedetto, A. Activation of protease-activated receptor 2 leads to impairment of keratinocyte tight junction integrity. J. Allergy Clin. Immunol. 2018, 142, 281–284. [Google Scholar] [CrossRef] [Green Version]
- Blauvelt, A.; de Bruin-Weller, M.; Gooderham, M.; Cather, J.C.; Weisman, J.; Pariser, D.; Simpson, E.L.; Papp, K.A.; Hong, H.C.-H.; Rubel, D. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): A 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet 2017, 389, 2287–2303. [Google Scholar] [CrossRef]
- Dina Coronado, B.; Zane, L.T. Crisaborole topical ointment, 2%: A nonsteroidal, topical, anti-inflammatory phosphodiesterase 4 inhibitor in clinical development for the treatment of atopic dermatitis. J. Drugs Dermatol. 2016, 15, 390–396. [Google Scholar]
- Bissonnette, R.; Pavel, A.B.; Diaz, A.; Werth, J.L.; Zang, C.; Vranic, I.; Purohit, V.S.; Zielinski, M.A.; Vlahos, B.; Estrada, Y.D.; et al. Crisaborole and atopic dermatitis skin biomarkers: An intrapatient randomized trial. J. Allergy Clin. Immunol. 2019, 144, 1274–1289. [Google Scholar] [CrossRef] [Green Version]
- Guttman-Yassky, E.; Bissonnette, R.; Ungar, B.; Suarez-Farinas, M.; Ardeleanu, M.; Esaki, H.; Suprun, M.; Estrada, Y.; Xu, H.; Peng, X.; et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 155–172. [Google Scholar] [CrossRef] [Green Version]
- Amano, W.; Nakajima, S.; Kunugi, H.; Numata, Y.; Kitoh, A.; Egawa, G.; Dainichi, T.; Honda, T.; Otsuka, A.; Kimoto, Y.; et al. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J. Allergy Clin. Immunol. 2015, 136, 667–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.H.; Jayawickreme, C.; Rickard, D.J.; Nicodeme, E.; Bui, T.; Simmons, C.; Coquery, C.M.; Neil, J.; Pryor, W.M.; Mayhew, D.; et al. Tapinarof Is a Natural AhR Agonist that Resolves Skin Inflammation in Mice and Humans. J. Investig. Dermatol. 2017, 137, 2110–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peppers, J.; Paller, A.S.; Maeda-Chubachi, T.; Wu, S.; Robbins, K.; Gallagher, K.; Kraus, J.E. A phase 2, randomized dose-finding study of tapinarof (GSK2894512 cream) for the treatment of atopic dermatitis. J. Am. Acad. Dermatol. 2019, 80, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Blauvelt, A.; Eichenfield, L.F.; Paller, A.S.; Armstrong, A.W.; Drew, J.; Gopalan, R.; Simpson, E.L. Efficacy and Safety of Lebrikizumab, a High-Affinity Interleukin 13 Inhibitor, in Adults With Moderate to Severe Atopic Dermatitis: A Phase 2b Randomized Clinical Trial. JAMA Dermatol. 2020, 156, 411–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollenberg, A.; Howell, M.D.; Guttman-Yassky, E.; Silverberg, J.I.; Kell, C.; Ranade, K.; Moate, R.; van der Merwe, R. Treatment of atopic dermatitis with tralokinumab, an anti–IL-13 mAb. J. Allergy Clin. Immunol. 2019, 143, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamblin, M.R.; Avci, P.; Prow, T. Nanoscience in Dermatology; Academic Press: London, UK, 2016. [Google Scholar]
- Soriano-Ruiz, J.L.; Calpena-Capmany, A.C.; Cañadas-Enrich, C.; Bozal-de Febrer, N.; Suñer-Carbó, J.; Souto, E.B.; Clares-Naveros, B. Biopharmaceutical profile of a clotrimazole nanoemulsion: Evaluation on skin and mucosae as anticandidal agent. Int. J. Pharm. 2019, 554, 105–115. [Google Scholar] [CrossRef]
- Igawa, K. Future trends in the treatment of atopic dermatitis. Immunol. Med. 2019, 42, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Lee, H.E.; Shin, S.W.; Um, S.H.; Lee, J.D.; Kim, K.B.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Efficient Transdermal Delivery of DNA Nanostructures Alleviates Atopic Dermatitis Symptoms in NC/Nga Mice. Adv. Funct. Mater. 2018, 28, 1801918. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Cytokines Required for Development | Transcription Factors | Stimulating Cytokines | Cytokine Production | Biological Function | |
|---|---|---|---|---|---|---|
| Group 1 ILCs | NK cell | IL-15 | T-bet EOMES | IL-12 IL-18 | IFN-γ TNF | Immunity to virus and cancer Chronic inflammation |
| ILC1 | IL-7 IL-15 | T-bet | IL-12 IL-18 | IFN-γ TNF | Immunity to intracellular bacteria and protozoa Chronic inflammation | |
| Group 2 ILCs | ILC2 | IL-7 | BCL11B GFI1 EST1 GATA3 | IL-25 IL-33 TSLP | IL-4 (in humans) IL-5 IL-13 AREG | Immunity to helminths Asthma and allergic disease Metabolic homeostasis |
| Group 3 ILCs | LTi cell | IL-7 | RORγt | IL-23 IL-1β | IL-17A/IL-17F IL-22 | Lymphoid tissue developments Intestinal homeostasis Immunity to extracellular bacteria Chronic inflammation |
| NCR- ILC3 | IL-7 | AHR RORγt | IL-23 IL-1β | IL-17A/IL-17F IL-22 | ||
| NCR+ ILC3 | IL-7 | AHR RORγt T-bet | IL-23 IL-1β | IL-22 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.; Seok, J.K.; Kang, H.C.; Cho, Y.-Y.; Lee, H.S.; Lee, J.Y. Skin Barrier Abnormalities and Immune Dysfunction in Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 2867. https://doi.org/10.3390/ijms21082867
Yang G, Seok JK, Kang HC, Cho Y-Y, Lee HS, Lee JY. Skin Barrier Abnormalities and Immune Dysfunction in Atopic Dermatitis. International Journal of Molecular Sciences. 2020; 21(8):2867. https://doi.org/10.3390/ijms21082867
Chicago/Turabian StyleYang, Gabsik, Jin Kyung Seok, Han Chang Kang, Yong-Yeon Cho, Hye Suk Lee, and Joo Young Lee. 2020. "Skin Barrier Abnormalities and Immune Dysfunction in Atopic Dermatitis" International Journal of Molecular Sciences 21, no. 8: 2867. https://doi.org/10.3390/ijms21082867
APA StyleYang, G., Seok, J. K., Kang, H. C., Cho, Y. -Y., Lee, H. S., & Lee, J. Y. (2020). Skin Barrier Abnormalities and Immune Dysfunction in Atopic Dermatitis. International Journal of Molecular Sciences, 21(8), 2867. https://doi.org/10.3390/ijms21082867