Increased Protein S-Glutathionylation in Leber’s Hereditary Optic Neuropathy (LHON)

 ,
, 
Abstract
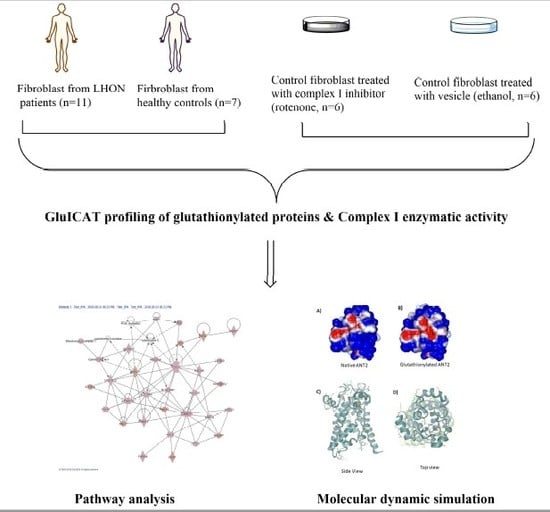
:
1. Introduction
2. Results
2.1. Population
2.2. Complex I Impairment in LHON and Treated Fibroblasts
2.3. Complex I Deficiency Induces ROS Overproduction.
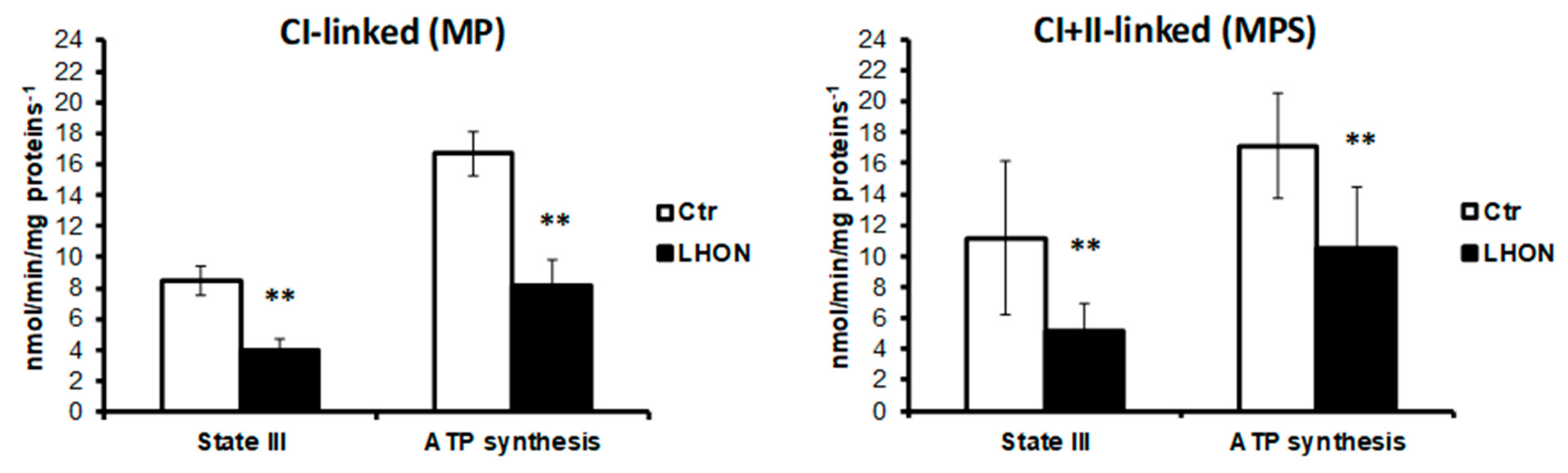
2.4. Phosphorylating Respiration and ATP Production in LHON Permeabilized Fibroblasts
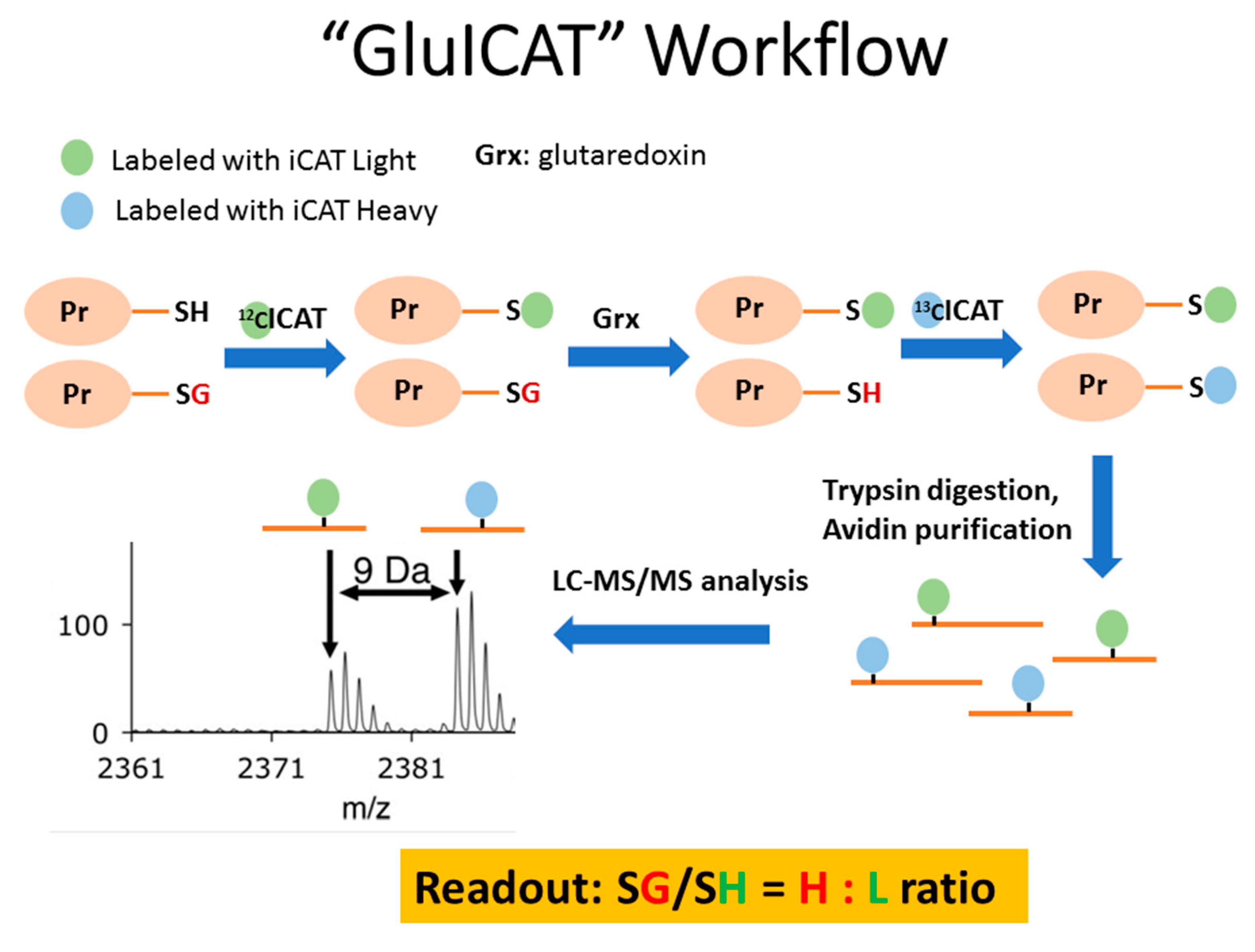
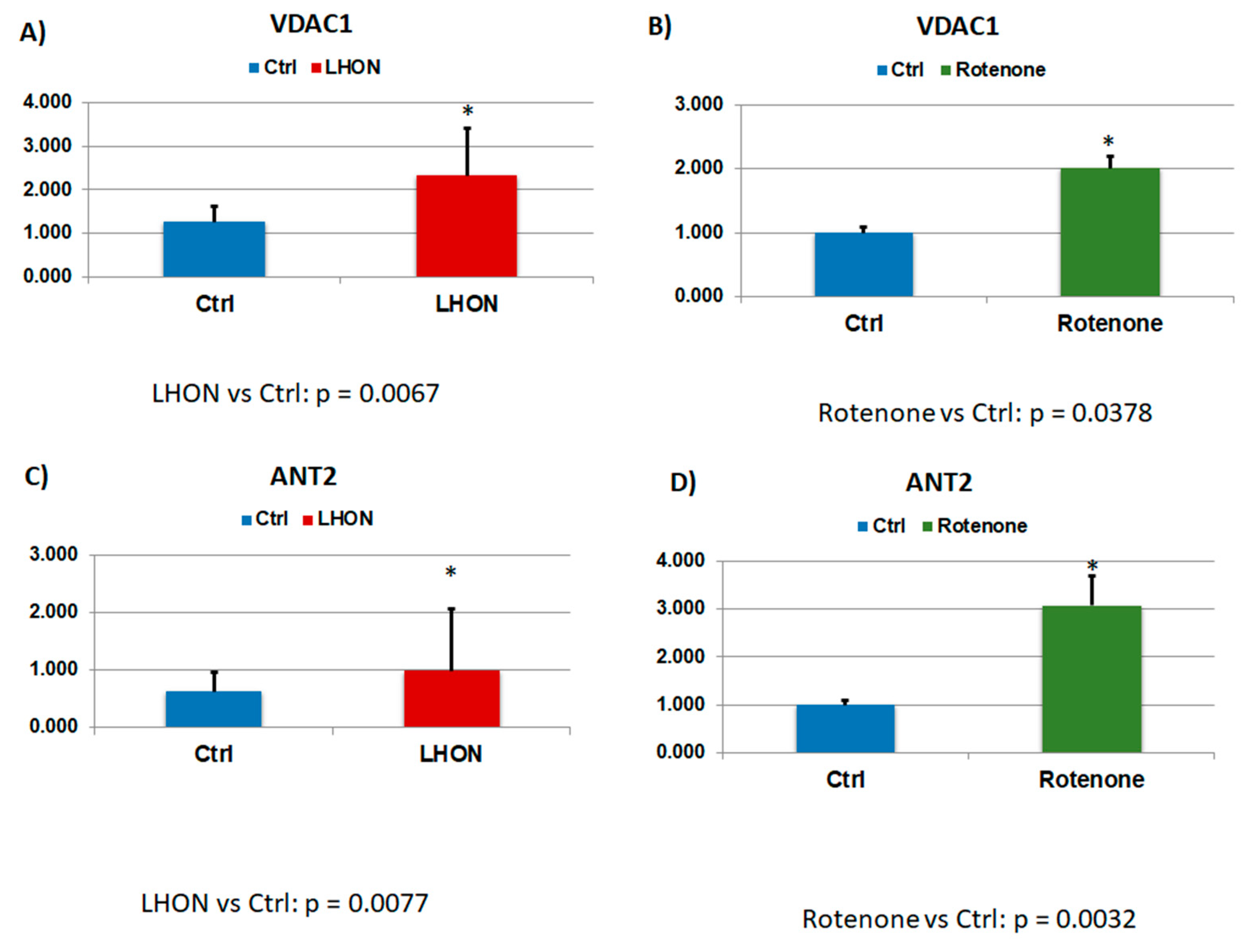
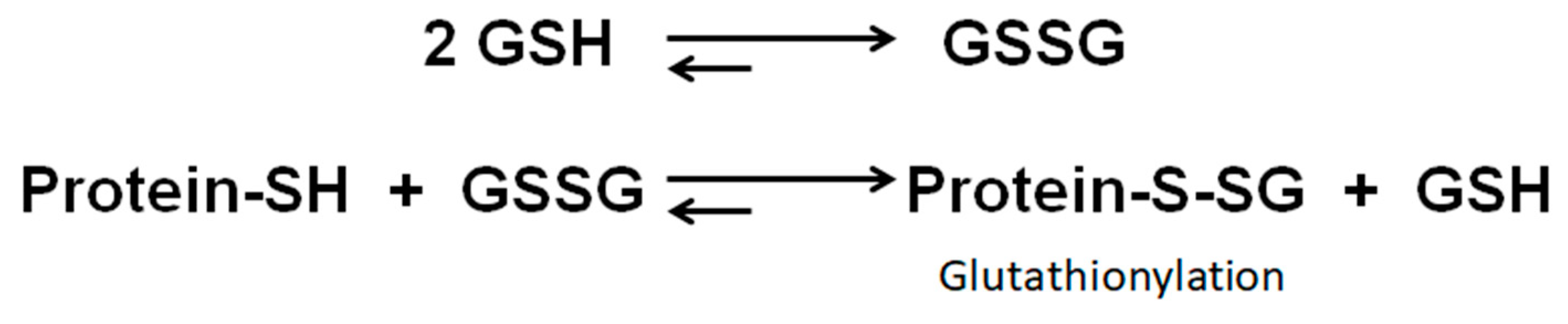
2.5. Protein S-glutathionylation profile in LHON fibroblasts
2.6. Protein S-Glutathionylation Profile in Fibroblasts with Complex I Inhibition
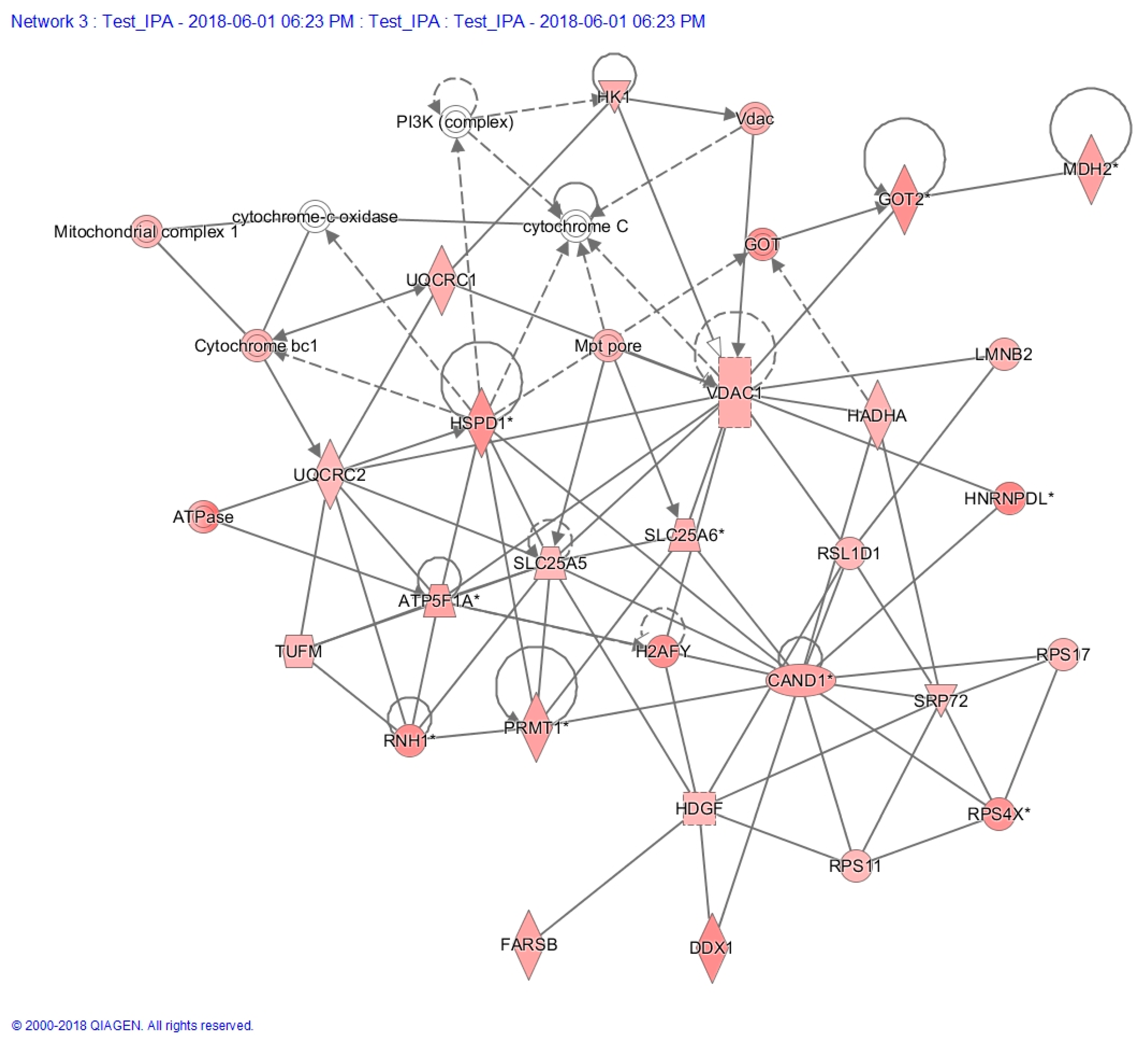
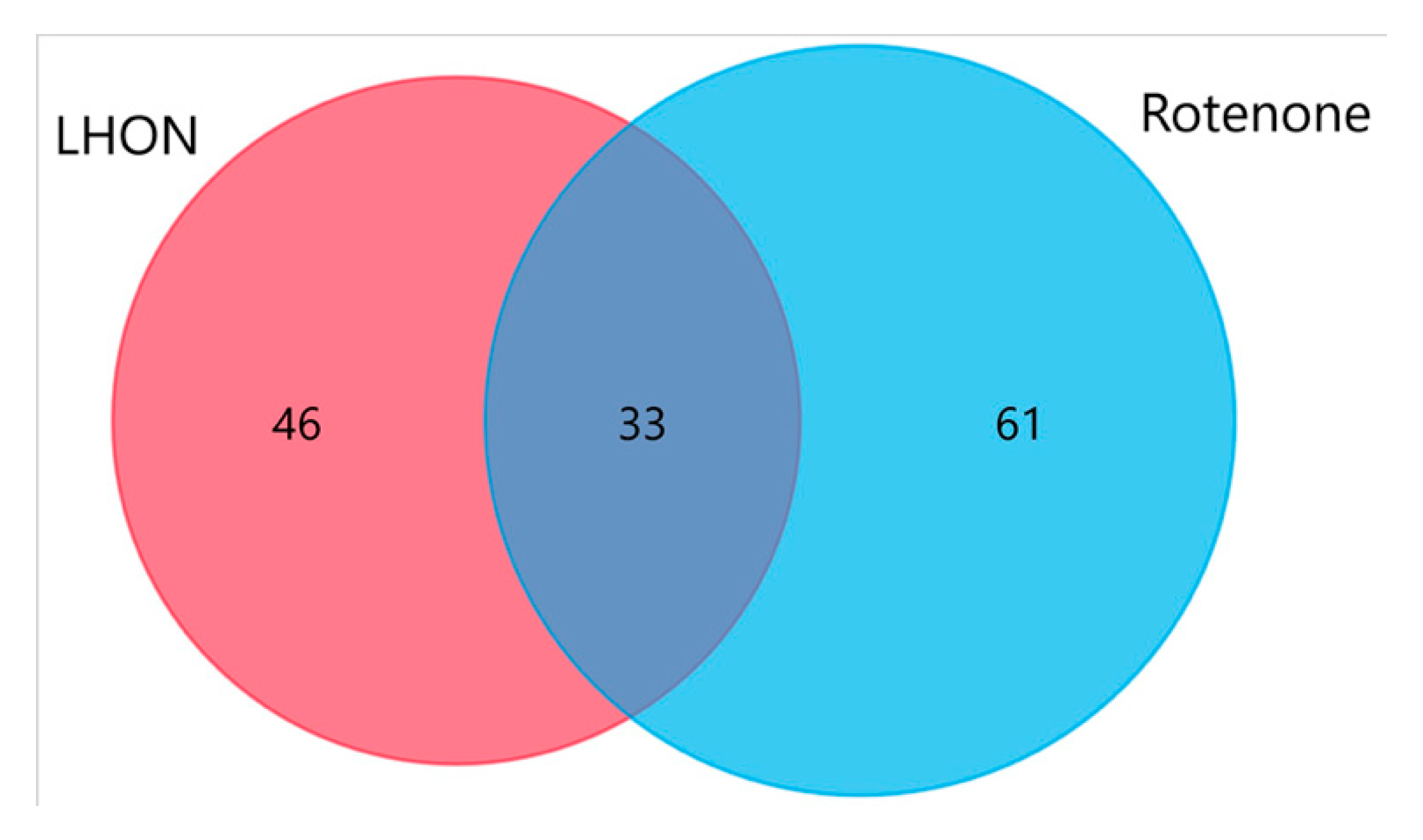
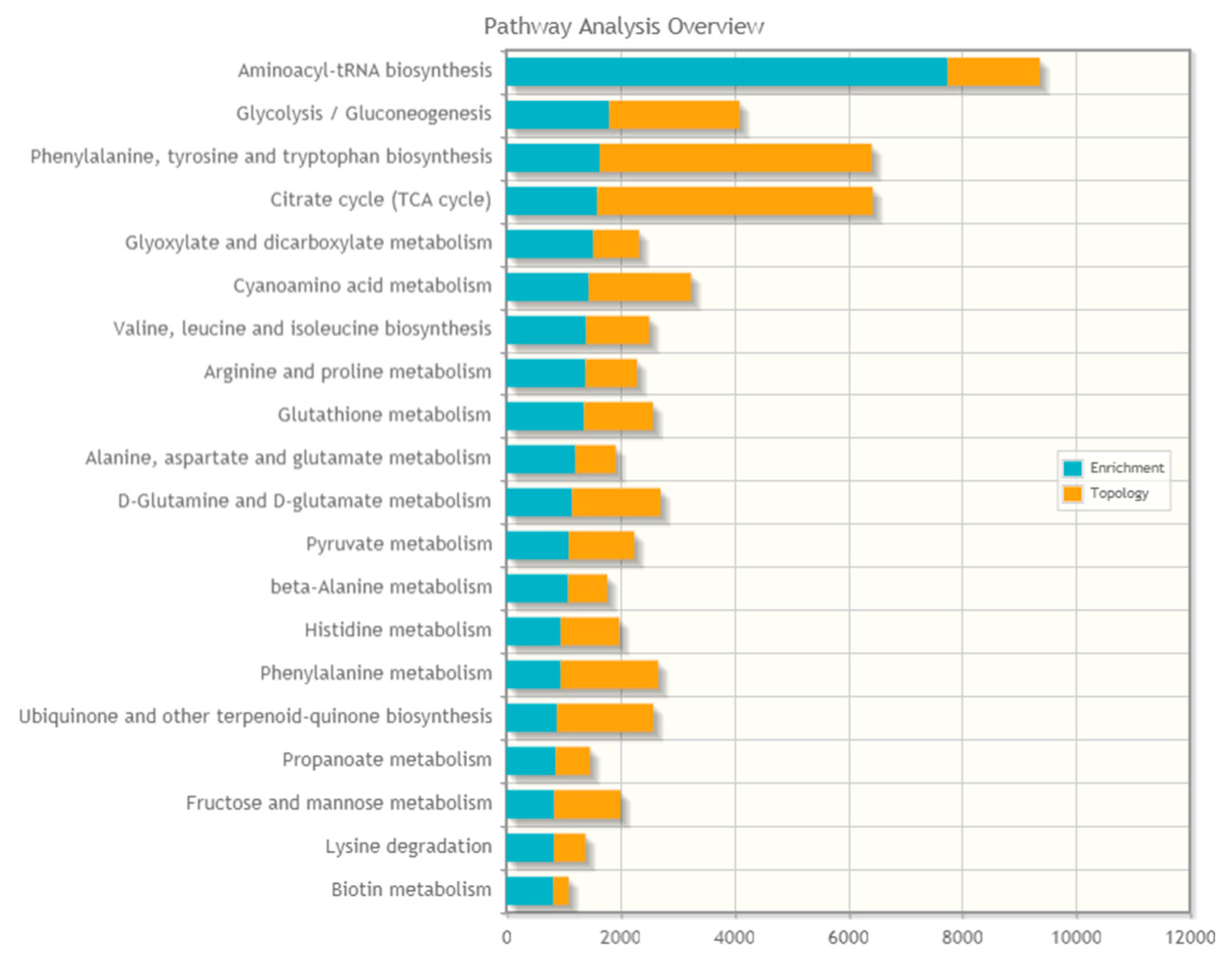
2.7. Proteo-Metabolomic Mapping of S-Glutathionylation in LHON
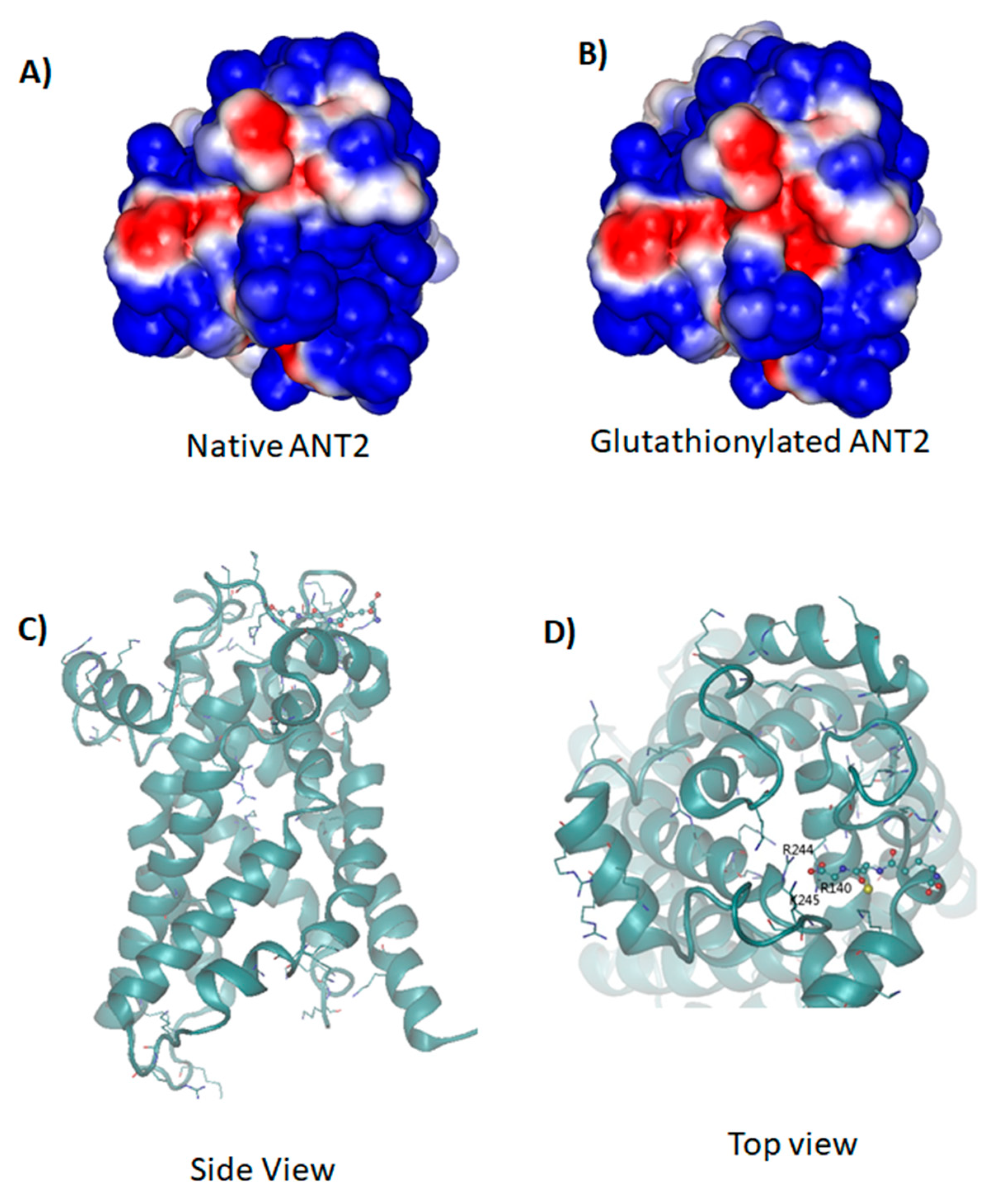
2.8. Molecular Dynamic Simulations of Native and Glutathionylated ANT2
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Cell Cultures
4.3. Complex I Enzymatic Activity and Inhibition
4.4. Mitochondrial ROS Production
4.5. Measurements of Phosphorylating Respiration and ATP Production in LHON-Permeabilized Fibroblasts
4.6. Enrichment of S-glutathionylated Proteins
4.7. LC-MS/MS Analysis
4.8. Data Analysis
4.9. Simulations of the Molecular Dynamics (MD) of Native and Glutathionylated ANT2
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIF | Apoptosis inducing factor |
| ANT2 | Adenine nucleotide translocase 2 |
| ATP | Adenosine triphosphate |
| DNA | Deoxyribonucleic acid |
| GPx | Glutathione peroxidase |
| Grx1 | Glutaredoxin 1 |
| GSH | Glutathione |
| GSSG | Glutathione disulfide |
| ICAT | Isotope-coded affinity tag |
| LHON | Leber’s Hereditary Optic Neuropathy |
| MPTP | Mitochondrial permeability transition pore |
| mtDNA | Mitochondrial DNA |
| ND | NADH dehydrogenase |
| PPARα | Peroxisome proliferator-activated receptor α |
| PTMs | Post-translational modifications |
| RMSD | Root mean square deviation |
| UCP | Uncoupling protein |
| VDAC1 | Voltage-dependent anion-selective channel protein 1 |
References
- Mascialino, B.; Leinonen, M.; Meier, T. Meta-analysis of the prevalence of Leber hereditary optic neuropathy mtDNA mutations in Europe. Eur. J. Ophthalmol. 2012, 22, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Milea, D.; Amati-Bonneau, P.; Reynier, P.; Bonneau, D. Genetically determined optic neuropathies. Curr. Opin. Neurol. 2010, 23, 24–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carelli, V.; La Morgia, C.; Ross-Cisneros, F.N.; Sadun, A.A. Optic neuropathies: The tip of the neurodegeneration iceberg. Hum. Mol. Genet. 2017, 26, R139–R150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu-Wai-Man, P.; Newman, N.J. Inherited eye-related disorders due to mitochondrial dysfunction. Hum. Mol. Genet. 2017, 26, R12–R20. [Google Scholar] [CrossRef] [Green Version]
- Chevrollier, A.; Guillet, V.; Loiseau, D.; Gueguen, N.; de Crescenzo, M.A.; Verny, C.; Ferre, M.; Dollfus, H.; Odent, S.; Milea, D.; et al. Hereditary optic neuropathies share a common mitochondrial coupling defect. Ann. Neurol. 2008, 63, 794–798. [Google Scholar] [CrossRef]
- Sala, G.; Trombin, F.; Beretta, S.; Tremolizzo, L.; Presutto, P.; Montopoli, M.; Fantin, M.; Martinuzzi, A.; Carelli, V.; Ferrarese, C. Antioxidants partially restore glutamate transport defect in leber hereditary optic neuropathy cybrids. J. Neurosci. Res. 2008, 86, 3331–3337. [Google Scholar] [CrossRef]
- Ghelli, A.; Porcelli, A.M.; Zanna, C.; Vidoni, S.; Mattioli, S.; Barbieri, A.; Iommarini, L.; Pala, M.; Achilli, A.; Torroni, A.; et al. The background of mitochondrial DNA haplogroup J increases the sensitivity of Leber’s hereditary optic neuropathy cells to 2,5-hexanedione toxicity. PLoS ONE 2009, 4, e7922. [Google Scholar] [CrossRef]
- Giordano, C.; Iommarini, L.; Giordano, L.; Maresca, A.; Pisano, A.; Valentino, M.L.; Caporali, L.; Liguori, R.; Deceglie, S.; Roberti, M.; et al. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain 2014, 137 (Pt 2), 335–353. [Google Scholar] [CrossRef] [Green Version]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. Mitochondrial Thiols in Antioxidant Protection and Redox Signaling: Distinct Roles for Glutathionylation and Other Thiol Modifications. Antioxid. Redox Signal. 2012, 16, 476–495. [Google Scholar] [CrossRef]
- Pimentel, D.; Haeussler, D.J.; Matsui, R.; Burgoyne, J.R.; Cohen, R.A.; Bachschmid, M.M. Regulation of Cell Physiology and Pathology by Protein S-Glutathionylation: Lessons Learned from the Cardiovascular System. Antioxid. Redox Signal. 2012, 16, 524–542. [Google Scholar] [CrossRef] [Green Version]
- Grek, C.L.; Zhang, J.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Causes and consequences of cysteine S-glutathionylation. J. Biol. Chem. 2013, 288, 26497–26504. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, P. Protein glutathionylation in health and disease. Biochim. Biophys. Acta 2013, 1830, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Piemonte, F. Protein glutathionylation in cardiovascular diseases. Int. J. Mol. Sci. 2013, 14, 20845–20876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.; Gill, R.; Mailloux, R.J. Protein S-glutathionylation: The linchpin for the transmission of regulatory information on redox buffering capacity in mitochondria. Chem. Biol. Interact. 2019, 299, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.L.; Brookes, P.S. Oxygen sensitivity of mitochondrial reactive oxygen species generation depends on metabolic conditions. J. Biol. Chem. 2009, 284, 16236–16245. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.C.Y.; Soh, A.C.K.; Kioh, D.Y.Q.; Li, J.; Verma, C.; Koh, S.K.; Beuerman, R.W.; Zhou, L.; Chan, E.C.Y. Reactive Metabolite-induced Protein Glutathionylation: A Potentially Novel Mechanism Underlying Acetaminophen Hepatotoxicity. Mol. Cell. Proteom. 2018, 17, 2034–2050. [Google Scholar] [CrossRef] [Green Version]
- Chao de la Barca, J.M.; Simard, G.; Amati-Bonneau, P.; Safiedeen, Z.; Prunier-Mirebeau, D.; Chupin, S.; Gadras, C.; Tessier, L.; Gueguen, N.; Chevrollier, A.; et al. The metabolomic signature of Leber’s hereditary optic neuropathy reveals endoplasmic reticulum stress. Brain 2016, 139, 2864–2876. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tajkhorshid, E. Electrostatic funneling of substrate in mitochondrial inner membrane carriers. Proc. Natl. Acad. Sci. USA 2008, 105, 9598–9603. [Google Scholar] [CrossRef] [Green Version]
- Sadun, A.A.; Morgia, C.L.; Carelli, V. Leber’s Hereditray Optic Neuropathy. Curr. Treat. Opt. Neurol. 2011, 13, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Manevich, Y.; He, L.; Hutchens, S.; Pazoles, C.J.; Tew, K.D. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J. Biol. Chem. 2009, 284, 436–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonellis, A.; Green, E.D. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genomics Hum. Genet. 2008, 9, 87–107. [Google Scholar] [CrossRef] [PubMed]
- Sissler, M.; González-Serrano, L.E.; Westhof, E. Recent Advances in Mitochondrial Aminoacyl-tRNA Synthetases and Disease. Trends. Mol. Med. 2017, 23, 693–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, N.; Martin, W. The energetics of genome complexity. Nature 2010, 467, 929–934. [Google Scholar] [CrossRef]
- Cortopassi, G.; Danielson, S.; Alemi, M.; Zhan, S.S.; Tong, T.; Carelli, V.; Martinuzzi, A.; Marzuki, S.; Majamaa, K.; Wong, A. Mitochondrial disease activates transcripts of the unfolded protein response and cell cycle and inhibits vesicular secretion and oligodendrocyte-specific transcripts. Mitochondrion 2006, 6, 161–175. [Google Scholar] [CrossRef]
- Tun, A.W.; Chaiyarit, S.; Kaewsutthi, S.; Katanyoo, W.; Chuenkongkaew, W.; Kuwano, M.; Tomonaga, T.; Peerapittayamongkol, C.; Thongboonkerd, V.; Lertrit, P. Profiling the mitochondrial proteome of Leber’s Hereditary Optic Neuropathy (LHON) in Thailand: Downregulation of bioenergetics and mitochondrial protein quality control pathways in fibroblasts with the 11778G4A mutation. PLoS ONE 2014, 9, e106779. [Google Scholar] [CrossRef] [Green Version]
- Zamzami, N.; Marchetti, P.; Castedo, M.; Hirsch, T.; Susin, S.A.; Masse, B.; Kroemer, G. Inhibitors of permeability transition interfere with the disruption of the mitochondrial transmembrane potential during apoptosis. FEBS Lett. 1996, 384, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Chevrollier, A.; Loiseau, D.; Reynier, P.; Stepien, G. Adenine nucleotide translocase 2 is a key mitochondrial protein in cancer metabolism. Biochim. Biophys. Acta 2011, 1807, 562–567. [Google Scholar] [CrossRef] [Green Version]
- Bano, D.; Prehn, J.H.M. Apoptosis-Inducing Factor (AIF) in Physiology and Disease: The Tale of a Repented Natural Born Killer. EBioMedicine 2018, 30, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Duan, J.; Gaffrey, M.J.; Shukla, A.K.; Thrall, B.; Qian, W. Stoichiometric Quantification of S-Glutathionylation and Total Thiol Oxidation in Mouse Macrophages. Free Radic. Biol. Med. 2016, 100, S44–S45. [Google Scholar] [CrossRef]
- Jurkute, N.; Harvey, J.; Yu-Wai-Man, P. Treatment strategies for Leber hereditary optic neuropathy. Curr. Opin. Neurol. 2019, 32, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Hutter, E.; Renner, K.; Pfister, G.; Stockl, P.; Jansen-Durr, P.; Gnaiger, E. Senescence-associated changes in respiration and oxidative phosphorylation in primary human fibroblasts. Biochem. J. 2004, 380, 919–928. [Google Scholar] [CrossRef] [Green Version]
- Angebault, C.; Gueguen, N.; Desquiret-Dumas, V.; Chevrollier, A.; Guillet, V.; Verny, C.; Cassereau, J.; Ferre, M.; Milea, D.; Amati-Bonneau, P.; et al. Idebenone increases mitochondrial complex I activity in fibroblasts from LHON patients while producing contradictory effects on respiration. BMC Res. Notes 2011, 4, 557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, C.; Augustin, S.; Ellouze, S.; Bénit, P.; Bouaita, A.; Rustin, P.; Sahel, J.A.; Corral-Debrinski, M. The optimized allotopic expression of ND1 or ND4 genes restores respiratory chain complex I activity in fibroblasts harboring mutations in these genes. Biochim. Biophys. Acta 2008, 1783, 1707–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, M.; Villani, G.; Mazzucchelli, F.; Bresolin, N.; Papa, S.; Attardi, G. Marked aging-related decline in efficiency of oxidative phosphorylation in human skin fibroblasts. FASEB J. 2003, 17, 1706–1708. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Jämbeck, J.P.; Lyubartsev, A.P. Derivation and systematic validation of a refined all-atom force field for phosphatidylcholine lipids. J. Phys. Chem, B. 2012, 116, 3164–3179. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subjects | Gender | Age (Years) | Passages | LHON Mutations (Rate) |
|---|---|---|---|---|
| Patient 1 | M | 20 | 15 | m.11778G>A 97% |
| Patient 2 | F | 44 | 15 | m.11778G>A 100% |
| Patient 3 | M | 26 | 13 | m.11778G>A 100% |
| Patient 4 | M | 39 | 17 | m.11778G>A 100% |
| Patient 5 | F | 58 | 8 | m.11778G>A 97% |
| Patient 6 | M | 23 | 10 | m.11778G>A 97% |
| Patient 7 | M | 22 | 17 | m.14484T>C 100% |
| Patient 8 | M | 19 | 10 | m.14484T>C 100% |
| Patient 9 | M | 38 | 12 | m.3460G>A 100% |
| Patient 10 | F | 42 | 11 | m.11778G>A 100% |
| Patient 11 | M | 22 | 12 | m.11778G>A 96% |
| Control 1 | M | 8 | 21 | |
| Control 2 | F | 24 | 12 | |
| Control 3 | F | 37 | 9 | |
| Control 4 | M | 30 | 11 | |
| Control 5 | F | 28 | 9 | |
| Control 6 | F | 56 | 20 | |
| Control 7 | M | 30 | 19 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Chan, J.C.Y.; Chupin, S.; Gueguen, N.; Desquiret-Dumas, V.; Koh, S.K.; Li, J.; Gao, Y.; Deng, L.; Verma, C.; et al. Increased Protein S-Glutathionylation in Leber’s Hereditary Optic Neuropathy (LHON). Int. J. Mol. Sci. 2020, 21, 3027. https://doi.org/10.3390/ijms21083027
Zhou L, Chan JCY, Chupin S, Gueguen N, Desquiret-Dumas V, Koh SK, Li J, Gao Y, Deng L, Verma C, et al. Increased Protein S-Glutathionylation in Leber’s Hereditary Optic Neuropathy (LHON). International Journal of Molecular Sciences. 2020; 21(8):3027. https://doi.org/10.3390/ijms21083027
Chicago/Turabian StyleZhou, Lei, James Chun Yip Chan, Stephanie Chupin, Naïg Gueguen, Valérie Desquiret-Dumas, Siew Kwan Koh, Jianguo Li, Yan Gao, Lu Deng, Chandra Verma, and et al. 2020. "Increased Protein S-Glutathionylation in Leber’s Hereditary Optic Neuropathy (LHON)" International Journal of Molecular Sciences 21, no. 8: 3027. https://doi.org/10.3390/ijms21083027
APA StyleZhou, L., Chan, J. C. Y., Chupin, S., Gueguen, N., Desquiret-Dumas, V., Koh, S. K., Li, J., Gao, Y., Deng, L., Verma, C., Beuerman, R. W., Chan, E. C. Y., Milea, D., & Reynier, P. (2020). Increased Protein S-Glutathionylation in Leber’s Hereditary Optic Neuropathy (LHON). International Journal of Molecular Sciences, 21(8), 3027. https://doi.org/10.3390/ijms21083027