Genetic Dissection of Hypertrophic Cardiomyopathy with Myocardial RNA-Seq


Abstract
:
1. Introduction
2. Results
2.1. Statistics of RNA-Seq Data
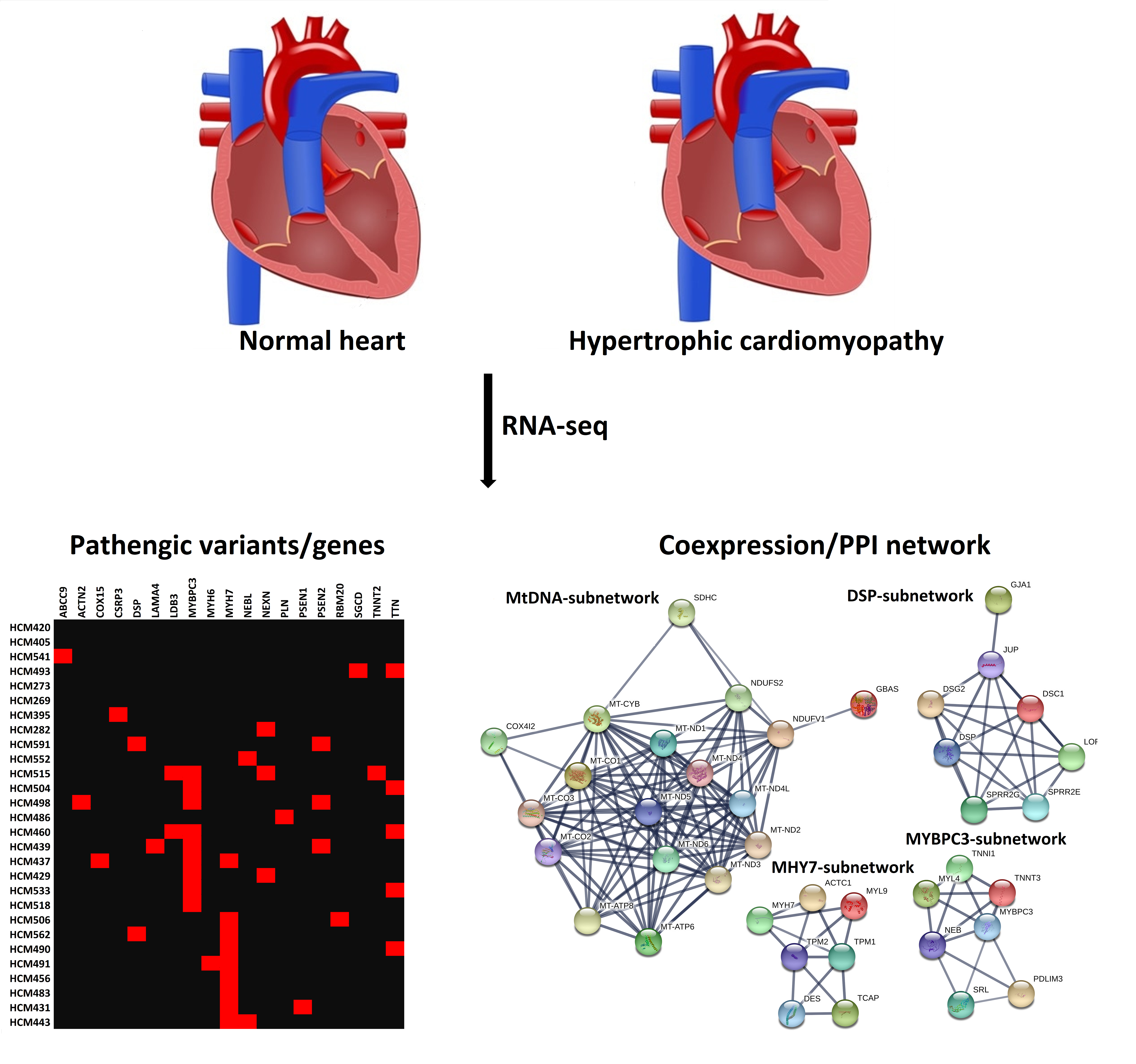
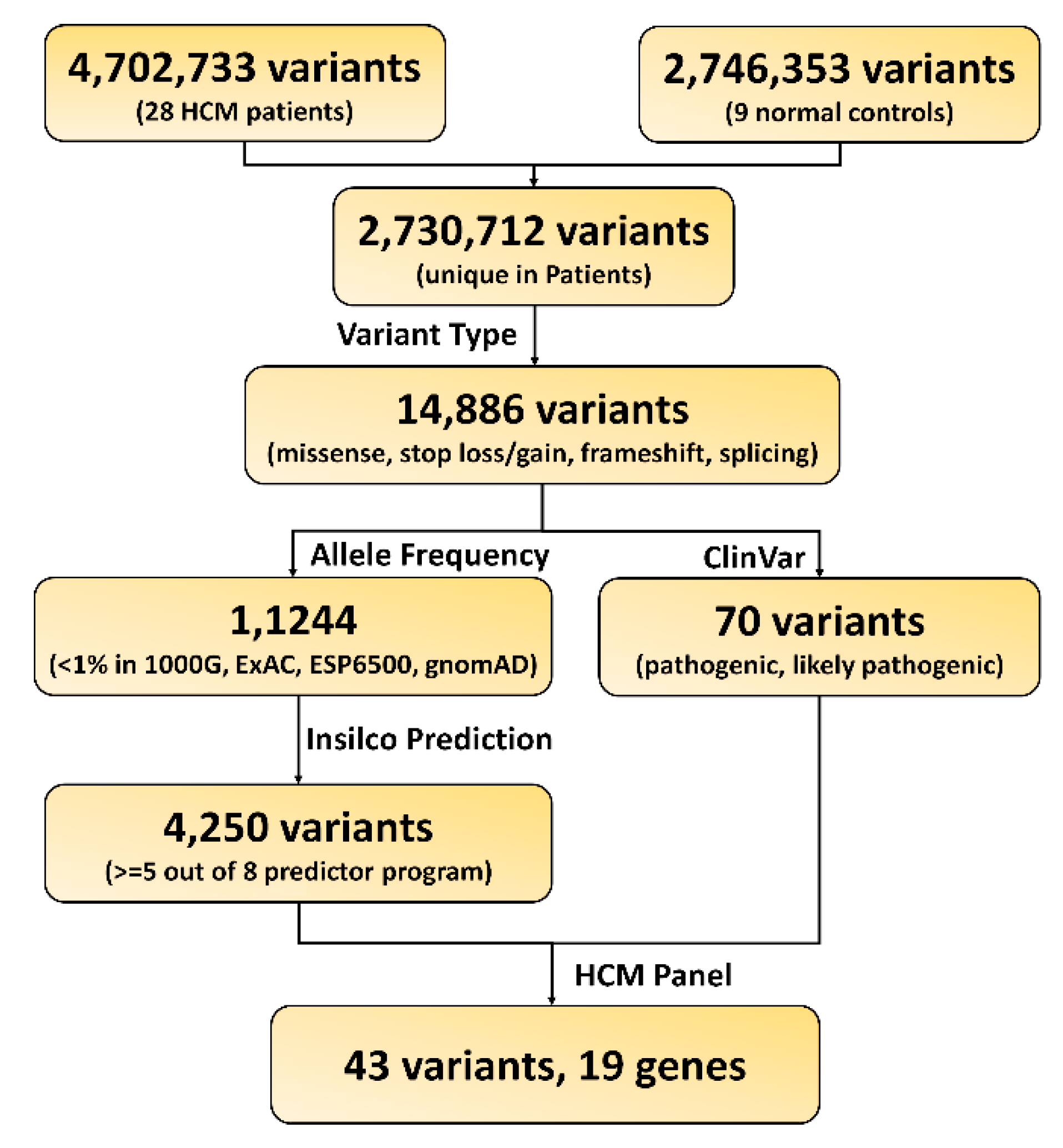
2.2. Pathogenic Variants and Genes Prioritization
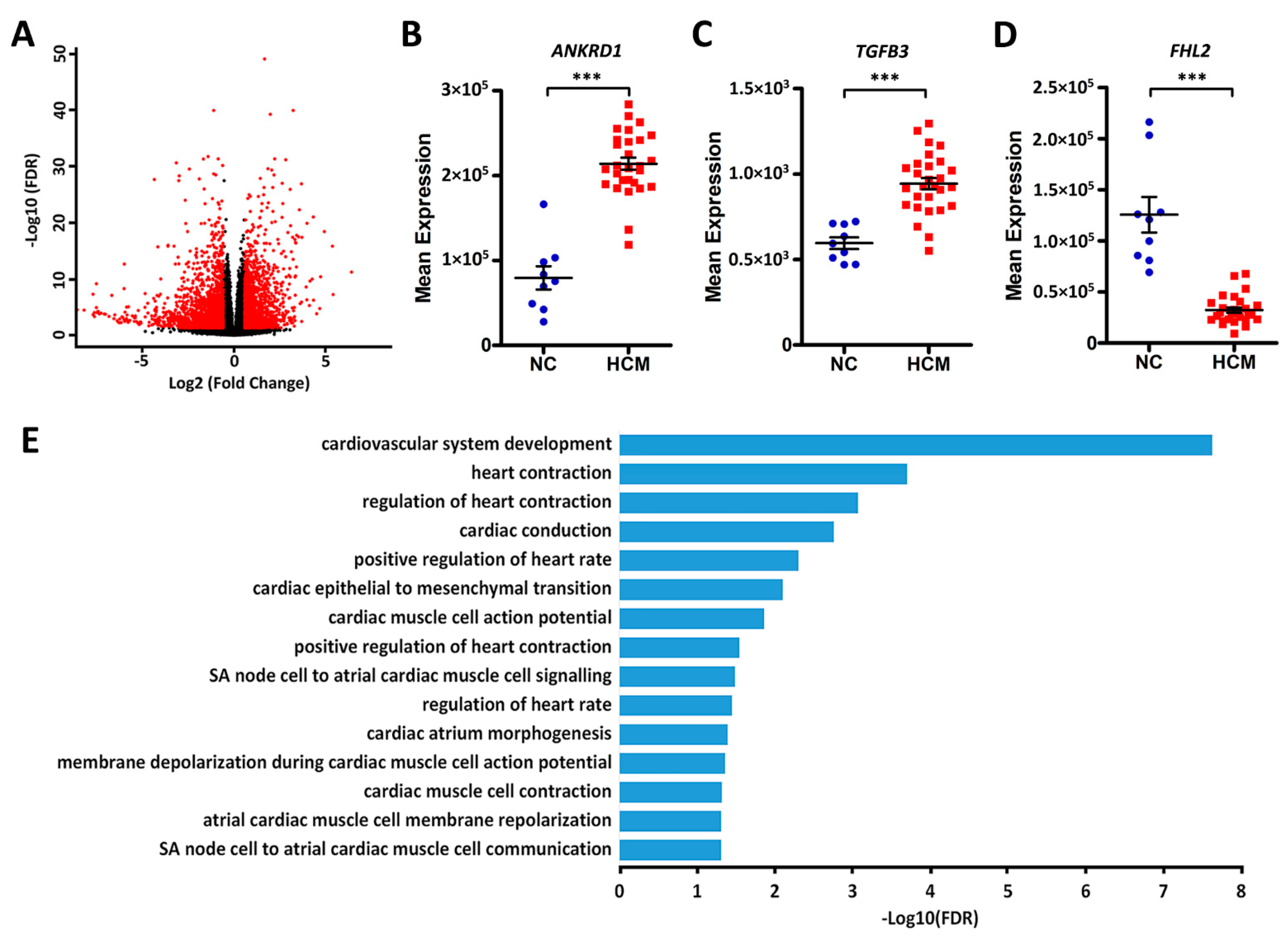
2.3. Identification of DEGs between HCM Patients and Healthy Controls
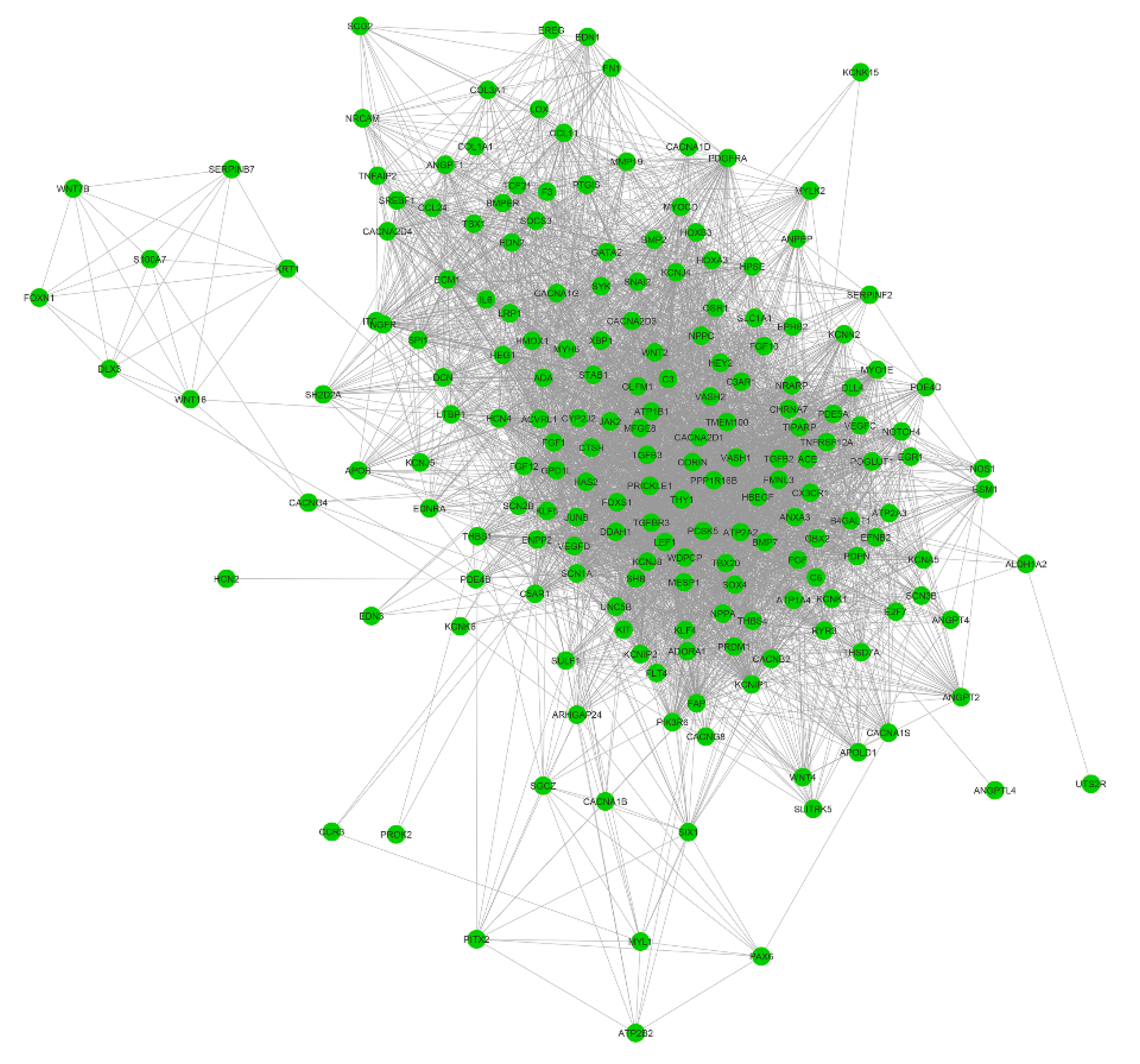
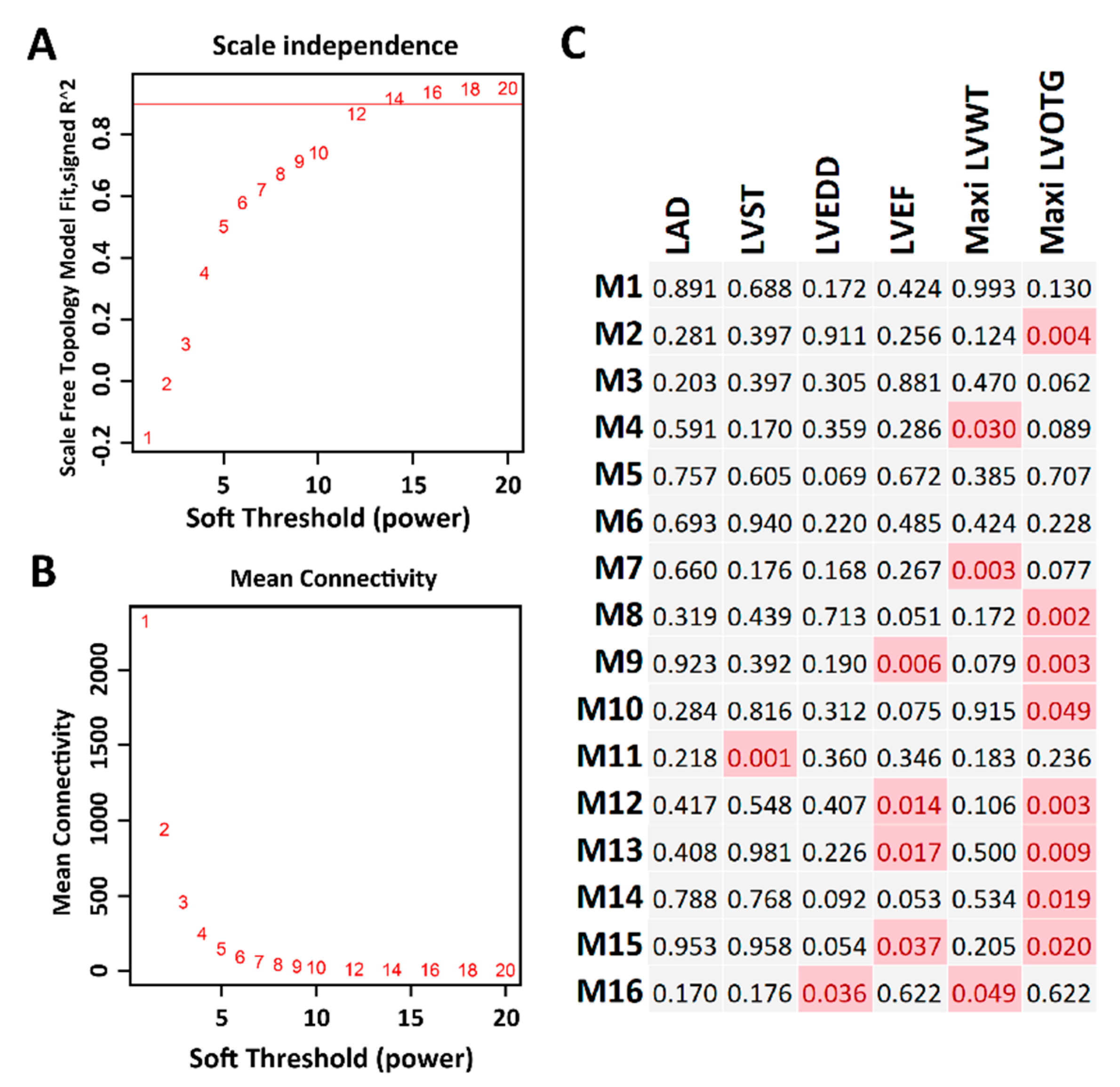
2.4. Co-Expression Network Analysis
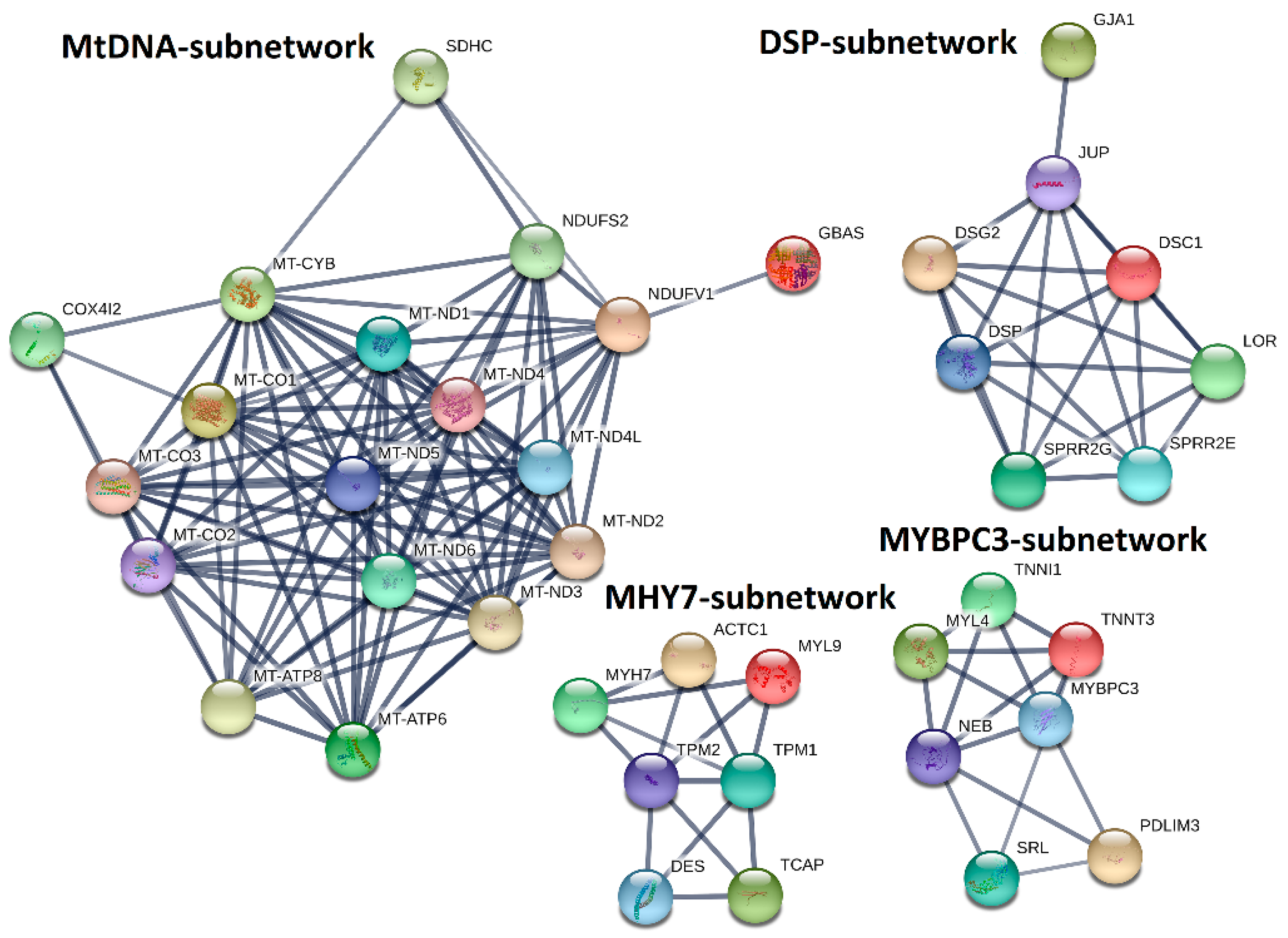
2.5. PPI-Subnetwork Analysis for MYH7 and MYBPC3 Modules
3. Discussion
3.1. Pathogenic Variant Analysis and Mechanisms of Variant Synergism
3.2. HCM Phenotypic Variability from Multi-Gene Modification and Differential Gene Expression
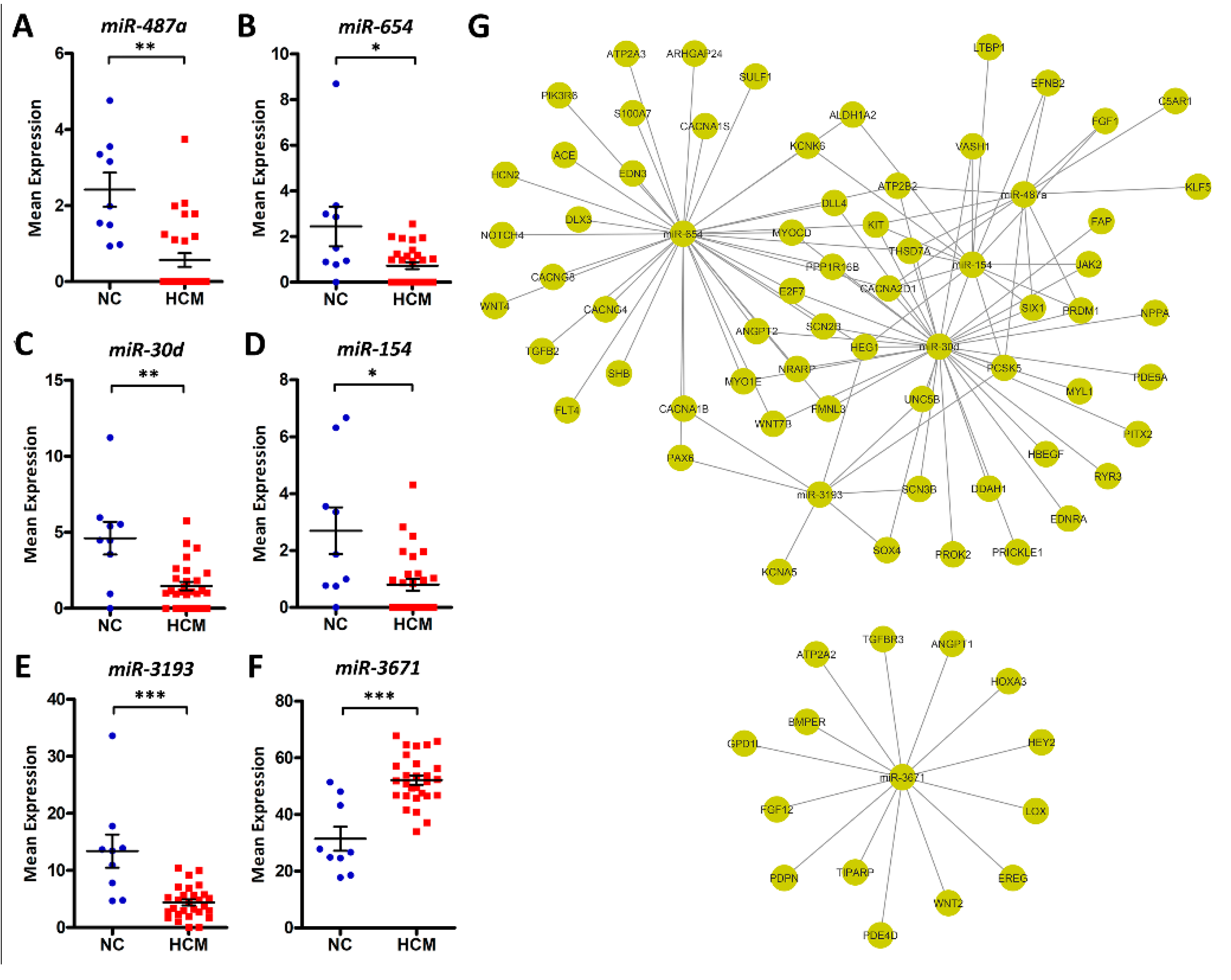
3.3. HCM Modulation with Noncoding RNAs
3.4. HCM Modulation with Gene Networks
3.5. Novelty and Limitations of This Study
4. Materials and Methods
4.1. RNA-Seq Data
4.2. Read Mapping and Variant Calling
4.3. Functional Variants and Gene Prioritization
4.4. Differential Expression Analysis
4.5. MiRNA Target Prediction
4.6. LncRNA Annotation
4.7. Co-Expression Network Analysis
4.8. PPI Network Analysis
4.9. Gene Over-Representation Analysis
4.10. Data Statement
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ARVC | arrhythmogenic right ventricular cardiomyopathy |
| DCM | dilated cardiomyopathy |
| DEG | differentially expressed gene |
| FDR | false discovery rate |
| GO | Gene Ontology |
| HCM | hypertrophic cardiomyopathy |
| Indel | insertion and deletion |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LncRNA | long noncoding RNA |
| MEs | module eigengenes |
| MiRNA | microRNA |
| NC | Normal control |
| PPI | protein-protein interaction |
| SNP | single nucleotide polymorphism |
| TPM | transcripts per million |
| WGCNA | weighted gene co-expression network analysis. |
References
- Maron, B.J.; Maron, M.S. Hypertrophic cardiomyopathy. Lancet 2013, 381, 242–255. [Google Scholar] [CrossRef]
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, M.S. Clinical utility of cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J. Cardiovasc. Magn. Reson. 2012, 14, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, L.R.; Rahman, M.S.; Elliott, P.M. A systematic review and meta-analysis of genotype–phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart 2013, 99, 1800–1811. [Google Scholar] [CrossRef]
- Jensen, M.K.; Havndrup, O.; Christiansen, M.; Andersen, P.S.; Diness, B.; Axelsson, A.; Skovby, F.; Køber, L.; Bundgaard, H. Penetrance of hypertrophic cardiomyopathy in children and adolescents: A 12-year follow-up study of clinical screening and predictive genetic testing. Circulation 2013, 127, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, M.; Elliott, P. The genetics of hypertrophic cardiomyopathy. Glob. Cardiol. Sci. Pract. 2018, 36, 1–9. [Google Scholar]
- Sabater-Molina, M.; Pérez-Sánchez, I.; Hernández del Rincón, J.; Gimeno, J. Genetics of hypertrophic cardiomyopathy: A review of current state. Clin. Genet. 2018, 93, 3–14. [Google Scholar] [CrossRef]
- Lopes, L.R.; Syrris, P.; Guttmann, O.P.; O’Mahony, C.; Tang, H.C.; Dalageorgou, C.; Jenkins, S.; Hubank, M.; Monserrat, L.; McKenna, W.J. Novel genotype–phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart 2015, 101, 294–301. [Google Scholar] [CrossRef]
- Forleo, C.; D’Erchia, A.M.; Sorrentino, S.; Manzari, C.; Chiara, M.; Iacoviello, M.; Guaricci, A.I.; De Santis, D.; Musci, R.L.; La Spada, A. Targeted next-generation sequencing detects novel gene–phenotype associations and expands the mutational spectrum in cardiomyopathies. PLoS ONE 2017, 12, e0181842. [Google Scholar] [CrossRef]
- Burns, C.; Bagnall, R.D.; Lam, L.; Semsarian, C.; Ingles, J. Multiple gene variants in hypertrophic cardiomyopathy in the era of next-generation sequencing. Circ. Cardiovasc. Genet. 2017, 10, e001666. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, D.; McKenna, W.J. Genetics of inherited cardiomyopathy. Eur. Heart J. 2012, 33, 296–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Ma, Y.; Yin, K.; Li, W.; Chen, W.; Zhang, Y.; Zhu, C.; Li, T.; Han, B.; Liu, X. Long non-coding and coding RNA profiling using strand-specific RNA-seq in human hypertrophic cardiomyopathy. Sci. Data 2019, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Ellims, A.H.; Moore, X.-l.; White, D.A.; Taylor, A.J.; Chin-Dusting, J.; Dart, A.M. Circulating microRNAs as biomarkers for diffuse myocardial fibrosis in patients with hypertrophic cardiomyopathy. J. Transl. Med. 2015, 13, 314. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Liu, W.; Wang, Z. MiR-154 promotes myocardial fibrosis through beta-catenin signaling pathway. Eur. Rev. Med Pharmacol. Sci. 2018, 22, 2052–2060. [Google Scholar] [PubMed]
- Bos, J.M.; Poley, R.N.; Ny, M.; Tester, D.J.; Xu, X.; Vatta, M.; Towbin, J.A.; Gersh, B.J.; Ommen, S.R.; Ackerman, M.J. Genotype–phenotype relationships involving hypertrophic cardiomyopathy-associated mutations in titin, muscle LIM protein, and telethonin. Mol. Genet. Metab. 2006, 88, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, C.; Padron, R. Familial hypertrophic cardiomyopathy: Genes, mutations and animal models. A Review. Investig. Clin. 2004, 45, 69–99. [Google Scholar]
- Richard, P.; Charron, P.; Carrier, L.; Ledeuil, C.; Cheav, T.; Pichereau, C.; Benaiche, A.; Isnard, R.; Dubourg, O.; Burban, M. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003, 107, 2227–2232. [Google Scholar] [CrossRef]
- Morita, H.; Rehm, H.L.; Menesses, A.; McDonough, B.; Roberts, A.E.; Kucherlapati, R.; Towbin, J.A.; Seidman, J.; Seidman, C.E. Shared genetic causes of cardiac hypertrophy in children and adults. N. Engl. J. Med. 2008, 358, 1899–1908. [Google Scholar] [CrossRef] [Green Version]
- Gupte, T.M.; Haque, F.; Gangadharan, B.; Sunitha, M.S.; Mukherjee, S.; Anandhan, S.; Rani, D.S.; Mukundan, N.; Jambekar, A.; Thangaraj, K. Mechanistic heterogeneity in contractile properties of α-tropomyosin (TPM1) mutants associated with inherited cardiomyopathies. J. Biol. Chem. 2015, 290, 7003–7015. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Zhang, Z.; Hong, D.; Dai, Q.; Jiang, T. Myocardial fibrosis in desmin-related hypertrophic cardiomyopathy. J. Cardiovasc. Magn. Reson. 2010, 12, 68. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Arimura, T.; Itoh-Satoh, M.; Ueda, K.; Hohda, S.; Inagaki, N.; Takahashi, M.; Hori, H.; Yasunami, M.; Nishi, H. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 2192–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bortoli, M.; Calore, C.; Lorenzon, A.; Calore, M.; Poloni, G.; Mazzotti, E.; Rigato, I.; Marra, M.P.; Melacini, P.; Iliceto, S. Co-inheritance of mutations associated with arrhythmogenic cardiomyopathy and hypertrophic cardiomyopathy. Eur. J. Hum. Genet. 2017, 25, 1165–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Yang, Z.; Vatta, M.; Rampazzo, A.; Beffagna, G.; Pillichou, K.; Scherer, S.E.; Saffitz, J.; Kravitz, J.; Zareba, W. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Vite, A.; Gandjbakhch, E.; Prost, C.; Fressart, V.; Fouret, P.; Neyroud, N.; Gary, F.; Donal, E.; Varnous, S.; Fontaine, G. Desmosomal cadherins are decreased in explanted arrhythmogenic right ventricular dysplasia/cardiomyopathy patient hearts. PLoS ONE 2013, 8, e75082. [Google Scholar] [CrossRef]
- Alila-Fersi, O.; Chamkha, I.; Majdoub, I.; Gargouri, L.; Mkaouar-Rebai, E.; Tabebi, M.; Tlili, A.; Keskes, L.; Mahfoudh, A.; Fakhfakh, F. Co segregation of the m. 1555A> G mutation in the MT-RNR1 gene and mutations in MT-ATP6 gene in a family with dilated mitochondrial cardiomyopathy and hearing loss: A whole mitochondrial genome screening. Biochem. Biophys. Res. Commun. 2017, 484, 71–78. [Google Scholar] [CrossRef]
- Li, Y.Y.; Maisch, B.; Rose, M.L.; Hengstenberg, C. Point mutations in mitochondrial DNA of patients with dilated cardiomyopathy. J. Mol. Cell. Cardiol. 1997, 29, 2699–2709. [Google Scholar] [CrossRef]
- Pacheu-Grau, D.; Bareth, B.; Dudek, J.; Juris, L.; Vögtle, F.-N.; Wissel, M.; Leary, S.C.; Dennerlein, S.; Rehling, P.; Deckers, M. Cooperation between COA6 and SCO2 in COX2 maturation during cytochrome c oxidase assembly links two mitochondrial cardiomyopathies. Cell Metab. 2015, 21, 823–833. [Google Scholar] [CrossRef] [Green Version]
- Mimaki, M.; Ikota, A.; Sato, A.; Komaki, H.; Akanuma, J.; Nonaka, I.; Goto, Y.-I. A double mutation (G11778A and G12192A) in mitochondrial DNA associated with Leber’s hereditary optic neuropathy and cardiomyopathy. J. Hum. Genet. 2003, 48, 0047–0050. [Google Scholar] [CrossRef] [Green Version]
- Ono, H.; Nakamura, H.; Matsuzaki, M. A NADH dehydrogenase ubiquinone flavoprotein is decreased in patients with dilated cardiomyopathy. Intern. Med. 2010, 49, 2039–2042. [Google Scholar] [CrossRef] [Green Version]
- Loeffen, J.; Elpeleg, O.; Smeitink, J.; Smeets, R.; Stöckler-Ipsiroglu, S.; Mandel, H.; Sengers, R.; Trijbels, F.; Van Den Heuvel, L. Mutations in the complex I NDUFS2 gene of patients with cardiomyopathy and encephalomyopathy. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2001, 49, 195–201. [Google Scholar]
- Antonicka, H.; Mattman, A.; Carlson, C.G.; Glerum, D.M.; Hoffbuhr, K.C.; Leary, S.C.; Kennaway, N.G.; Shoubridge, E.A. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2003, 72, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonne, G.; Carrier, L.; Richard, P.; Hainque, B.; Schwartz, K. Familial hypertrophic cardiomyopathy: From mutations to functional defects. Circ. Res. 1998, 83, 580–593. [Google Scholar] [CrossRef] [PubMed]
- Toepfer, C.N.; Wakimoto, H.; Garfinkel, A.C.; McDonough, B.; Liao, D.; Jiang, J.; Tai, A.C.; Gorham, J.M.; Lunde, I.G.; Lun, M. Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Sci. Transl. Med. 2019, 11, eaat1199. [Google Scholar] [CrossRef] [PubMed]
- Arimura, T.; Nakamura, T.; Hiroi, S.; Satoh, M.; Takahashi, M.; Ohbuchi, N.; Ueda, K.; Nouchi, T.; Yamaguchi, N.; Akai, J. Characterization of the human nebulette gene: A polymorphism in an actin-binding motif is associated with nonfamilial idiopathic dilated cardiomyopathy. Hum. Genet. 2000, 107, 440–451. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Parks, S.B.; Kushner, J.D.; Nauman, D.; Burgess, D.; Ludwigsen, S.; Partain, J.; Nixon, R.R.; Allen, C.N.; Irwin, R.P. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am. J. Hum. Genet. 2006, 79, 1030–1039. [Google Scholar] [CrossRef] [Green Version]
- Haapasalo, A.; Kovacs, D.M. The many substrates of presenilin/γ-secretase. J. Alzheimer’s Dis. 2011, 25, 3–28. [Google Scholar] [CrossRef]
- Bienengraeber, M.; Olson, T.M.; Selivanov, V.A.; Kathmann, E.C.; O’Cochlain, F.; Gao, F.; Karger, A.B.; Ballew, J.D.; Hodgson, D.M.; Zingman, L.V. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic K ATP channel gating. Nat. Genet. 2004, 36, 382–387. [Google Scholar] [CrossRef] [Green Version]
- Duggan, D.J.; Manchester, D.; Stears, K.P.; Mathews, D.J.; Hart, C.; Hoffman, E.P. Mutations in the δ-sarcoglycan gene are a rare cause of autosomal recessive limb-girdle muscular dystrophy (LGMD2). Neurogenetics 1997, 1, 49–58. [Google Scholar] [CrossRef]
- Tsubata, S.; Bowles, K.R.; Vatta, M.; Zintz, C.; Titus, J.; Muhonen, L.; Bowles, N.E.; Towbin, J.A. Mutations in the human δ-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J. Clin. Investig. 2000, 106, 655–662. [Google Scholar] [CrossRef] [Green Version]
- Rader, E.P.; Turk, R.; Willer, T.; Beltrán, D.; Inamori, K.-i.; Peterson, T.A.; Engle, J.; Prouty, S.; Matsumura, K.; Saito, F. Role of dystroglycan in limiting contraction-induced injury to the sarcomeric cytoskeleton of mature skeletal muscle. Proc. Natl. Acad. Sci. USA 2016, 113, 10992–10997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh-Satoh, M.; Hayashi, T.; Nishi, H.; Koga, Y.; Arimura, T.; Koyanagi, T.; Takahashi, M.; Hohda, S.; Ueda, K.; Nouchi, T. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2002, 291, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohapatra, B.; Jimenez, S.; Lin, J.H.; Bowles, K.R.; Coveler, K.J.; Marx, J.G.; Chrisco, M.A.; Murphy, R.T.; Lurie, P.R.; Schwartz, R.J. Mutations in the muscle LIM protein and α-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol. Genet. Metab. 2003, 80, 207–215. [Google Scholar] [CrossRef]
- Geier, C.; Gehmlich, K.; Ehler, E.; Hassfeld, S.; Perrot, A.; Hayess, K.; Cardim, N.; Wenzel, K.; Erdmann, B.; Krackhardt, F. Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum. Mol. Genet. 2008, 17, 2753–2765. [Google Scholar] [CrossRef] [Green Version]
- Fujii, J.; Zarain-Herzberg, A.; Willard, H.F.; Tada, M.; MacLennan, D. Structure of the rabbit phospholamban gene, cloning of the human cDNA, and assignment of the gene to human chromosome 6. J. Biol. Chem. 1991, 266, 11669–11675. [Google Scholar]
- Ha, K.N.; Masterson, L.R.; Hou, Z.; Verardi, R.; Walsh, N.; Veglia, G.; Robia, S.L. Lethal Arg9Cys phospholamban mutation hinders Ca2+-ATPase regulation and phosphorylation by protein kinase A. Proc. Natl. Acad. Sci. USA 2011, 108, 2735–2740. [Google Scholar] [CrossRef] [Green Version]
- Minamisawa, S.; Sato, Y.; Tatsuguchi, Y.; Fujino, T.; Imamura, S.-i.; Uetsuka, Y.; Nakazawa, M.; Matsuoka, R. Mutation of the phospholamban promoter associated with hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 2003, 304, 1–4. [Google Scholar] [CrossRef]
- Carniel, E.; Taylor, M.R.; Sinagra, G.; Di Lenarda, A.; Ku, L.; Fain, P.R.; Boucek, M.M.; Cavanaugh, J.; Miocic, S.; Slavov, D. α-Myosin heavy chain: A sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation 2005, 112, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.S.; Hassanzadeh, S.; Winitsky, S.; Lin, H.; Satorius, C.; Vemuri, R.; Aletras, A.H.; Wen, H.; Epstein, N.D. The overall pattern of cardiac contraction depends on a spatial gradient of myosin regulatory light chain phosphorylation. Cell 2001, 107, 631–641. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, F.W.; Reischmann, S.; Schwalm, A.; Unger, A.; Ramanujam, D.; Münch, J.; Müller, O.J.; Hengstenberg, C.; Galve, E.; Charron, P. FHL2 expression and variants in hypertrophic cardiomyopathy. Basic Res. Cardiol. 2014, 109, 451. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Xu, J.; Li, Y.; Jia, C.; Ma, X.; Zhang, L.; Xie, X.; Zhang, Y.; Gao, X.; Zhang, Y. Cardiac ankyrin repeat protein attenuates cardiac hypertrophy by inhibition of ERK1/2 and TGF-β signaling pathways. PLoS ONE 2012, 7, e50436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandary, B.; Meng, Q.; James, J.; Osinska, H.; Gulick, J.; Valiente-Alandi, I.; Sargent, M.A.; Bhuiyan, M.S.; Blaxall, B.C.; Molkentin, J.D. Cardiac fibrosis in proteotoxic cardiac disease is dependent upon myofibroblast TGF-β signaling. J. Am. Heart Assoc. 2018, 7, e010013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A. Regulatory mutations in transforming growth factor-β3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005, 65, 366–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Shen, X.J.; Zou, Q.; Wang, S.P.; Tang, S.M.; Zhang, G.Z. Biological functions of microRNAs: A review. J. Physiol. Biochem. 2011, 67, 129–139. [Google Scholar] [CrossRef]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell. Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Gao, R.; Bei, Y.; Zhou, Q.; Zhou, Y.; Zhang, H.; Jin, M.; Wei, S.; Wang, K.; Xu, X. Circulating miR-30d predicts survival in patients with acute heart failure. Cell. Physiol. Biochem. 2017, 41, 865–874. [Google Scholar] [CrossRef]
- Bukauskas, T.; Mickus, R.; Cereskevicius, D.; Macas, A. Value of serum miR-23a, miR-30d, and miR-146a biomarkers in ST-elevation myocardial infarction. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 3925. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Nguyen, S.S.; Gao, X.-M.; Tham, Y.K.; Ooi, J.Y.; Patterson, N.L.; Kiriazis, H.; Su, Y.; Thomas, C.J.; Lin, R.C. Inhibition of miR-154 protects against cardiac dysfunction and fibrosis in a mouse model of pressure overload. Sci. Rep. 2016, 6, 22442. [Google Scholar] [CrossRef] [Green Version]
- Tomita, Y.; Kusama, Y.; Seino, Y.; Munakata, K.; Kishida, H.; Hayakawa, H. Increased accumulation of acidic fibroblast growth factor in left ventricular myocytes of patients with idiopathic cardiomyopathy. Am. Heart J. 1997, 134, 779–786. [Google Scholar] [CrossRef]
- Templin, C.; Ghadri, J.-R.; Rougier, J.-S.; Baumer, A.; Kaplan, V.; Albesa, M.; Sticht, H.; Rauch, A.; Puleo, C.; Hu, D. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur. Heart J. 2011, 32, 1077–1088. [Google Scholar] [CrossRef]
- Uchida, S.; Dimmeler, S. Long noncoding RNAs in cardiovascular diseases. Circ. Res. 2015, 116, 737–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobuß, L.; Bär, C.; Thum, T. Long non-coding RNAs: At the heart of cardiac dysfunction? Front. Physiol. 2019, 10, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdonschot, J.A.; Hazebroek, M.R.; Derks, K.W.; Barandiarán Aizpurua, A.; Merken, J.J.; Wang, P.; Bierau, J.; van den Wijngaard, A.; Schalla, S.M.; Abdul Hamid, M.A. Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. Eur. Heart J. 2018, 39, 864–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol. ) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wahid, F.; Shehzad, A.; Khan, T.; Kim, Y.Y. MicroRNAs: Synthesis, mechanism, function, and recent clinical trials. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2010, 1803, 1231–1243. [Google Scholar] [CrossRef] [Green Version]
- Rajewsky, N. microRNA target predictions in animals. Nat. Genet. 2006, 38, S8–S13. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Sticht, C.; Pandey, P.; Gretz, N. miRWalk–database: Prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 2011, 44, 839–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchese, F.P.; Raimondi, I.; Huarte, M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017, 18, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, J.D.; Wei, Y.; Khavari, P.A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 2018, 19, 143. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Vasaikar, S.; Shi, Z.; Greer, M.; Zhang, B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017, 45, W130–W137. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Group# | Sex | Age | Smoking | LAD (mm)a | LVST (mm)b | LVEDD (mm)c | LVEF (%)d | Maxi LVWT (mm)e | Maxi LVOTG (mmHg)f |
|---|---|---|---|---|---|---|---|---|---|---|
| HCM420 | GENETUN | male | 32 | NA | 41 | 14 | 44 | 76 | 18 | 55 |
| HCM405 | GENETUN | female | 31 | NO | 42 | 14 | 47 | 78 | 16 | 126 |
| HCM541 | GENETUN | male | 38 | NA | 47 | 22 | 46 | 71 | 25 | 96 |
| HCM493 | GENETUN | male | 40 | NA | 40 | 15 | 45 | 75 | 17 | 95 |
| HCM273 | GENETUN | male | 30 | NO | 38 | 15 | 33 | 50 | 21 | 30 |
| HCM269 | GENETUN | male | 25 | NO | 53 | 29 | 51 | 63 | 38 | 56 |
| HCM395 | GENETUN | male | 20 | NO | 49 | 19 | 43 | 60 | 22 | 78 |
| HCM282 | GENETUN | male | 48 | YES | 49 | 21 | 49 | 65.3 | 23 | 75 |
| HCM591 | GENETUN | male | 42 | NA | 39 | 21 | 45 | 70 | 32 | 53 |
| HCM552 | GENETUN | male | 32 | NA | NA | NA | NA | 75 | 21 | 100 |
| HCM515 | MYBPC3 | male | 21 | YES | 39 | 18 | 50 | 75 | 25 | 84 |
| HCM504 | MYBPC3 | male | 25 | YES | 39 | 25 | 39 | 75 | 28 | 66 |
| HCM498 | MYBPC3 | male | 39 | YES | 38 | 20 | 42 | 72 | 21 | 55 |
| HCM486 | MYBPC3 | female | 36 | NA | 45 | 16 | 35 | 75 | 20 | 100 |
| HCM460 | MYBPC3 | male | 43 | YES | 45 | 16 | 45 | 65 | 19 | 61 |
| HCM439 | MYBPC3 | female | 47 | NA | 36 | 15 | 38 | 80 | 18 | 103 |
| HCM437 | MYBPC3 | male | 30 | YES | 54 | 19 | 47 | 78 | 26 | 54 |
| HCM429 | MYBPC3 | female | 36 | NA | 48 | 17 | 42 | 78 | 18 | 70 |
| HCM533 | MYBPC3 | male | 47 | YES | 62 | 24 | 50 | 75 | 29 | 118 |
| HCM518 | MYBPC3 | female | 31 | NA | 37 | 22 | 38 | 68 | 26 | 105 |
| HCM506 | MYH7 | male | 19 | YES | 48 | 20 | 43 | 75 | 21 | 59 |
| HCM562 | MYH7 | female | 36 | YES | 36 | 0 | 36 | 66 | 21 | 70 |
| HCM490 | MYH7 | female | 41 | NA | 44 | 15 | 39 | 58 | 21 | 63 |
| HCM491 | MYH7 | female | 30 | NA | 44 | 23 | 32 | 75 | 28 | 64 |
| HCM456 | MYH7 | male | 28 | NA | 51 | 15 | 45 | 69 | 20 | 90 |
| HCM483 | MYH7 | male | 37 | YES | 57 | 23 | 40 | 72 | 26 | 70 |
| HCM431 | MYH7 | male | 24 | NA | 57 | 18 | 49 | 80 | 21 | 80 |
| HCM443 | MYH7 | female | 28 | NA | 46 | 17 | 44 | 69 | 18 | 68 |
| sc5-LV | NORMAL | female | NA | NA | NA | NA | NA | NA | NA | NA |
| sc2-LV | NORMAL | male | NA | NA | NA | NA | NA | NA | NA | NA |
| sc6-LV | NORMAL | male | NA | NA | NA | NA | NA | NA | NA | NA |
| N105-LV | NORMAL | male | NA | NA | NA | NA | NA | NA | NA | NA |
| N104-LV | NORMAL | male | NA | NA | NA | NA | NA | NA | NA | NA |
| ND2 | NORMAL | male | NA | NA | NA | NA | NA | NA | NA | NA |
| ND1-LV | NORMAL | male | NA | NA | NA | NA | NA | NA | NA | NA |
| N102-LV | NORMAL | male | NA | NA | NA | NA | NA | NA | NA | NA |
| N103-LV | NORMAL | male | NA | NA | NA | NA | NA | NA | NA | NA |
| Sample ID | Raw Reads | HQ Reads | HQ Reads (%) | Mapping Rate (%) # | Total Variant | Exonic Variant |
|---|---|---|---|---|---|---|
| HCM420 | 117,254,002 | 110,409,284 | 94.16 | 96.87 | 742,007 | 34,208 |
| HCM405 | 96,660,588 | 90,958,152 | 94.10 | 96.72 | 697,916 | 32,501 |
| HCM541 | 118,364,902 | 112,199,122 | 94.79 | 96.56 | 748,918 | 34,438 |
| HCM493 | 135,595,138 | 127,235,398 | 93.83 | 96.67 | 802,057 | 35,285 |
| HCM273 | 100,383,614 | 94,562,944 | 94.20 | 96.10 | 674,997 | 32,320 |
| HCM269 | 104,573,612 | 98,483,828 | 94.18 | 96.69 | 657,403 | 30,937 |
| HCM395 | 101,463,898 | 94,778,784 | 93.41 | 96.50 | 718,773 | 32,792 |
| HCM282 | 135,385,378 | 128,496,976 | 94.91 | 96.41 | 835,560 | 36,065 |
| HCM591 | 117,087,250 | 110,633,048 | 94.49 | 96.33 | 742,357 | 33,694 |
| HCM552 | 110,633,048 | 97,644,508 | 88.26 | 96.57 | 714,914 | 32,157 |
| HCM515 | 126,385,206 | 120,132,036 | 95.05 | 96.53 | 797,070 | 35,707 |
| HCM504 | 126,614,812 | 120,041,188 | 94.81 | 96.46 | 747,293 | 34,481 |
| HCM498 | 118,114,640 | 110,661,262 | 93.69 | 96.18 | 745,414 | 33,633 |
| HCM486 | 102,005,260 | 95,821,344 | 93.94 | 96.76 | 688,159 | 32,517 |
| HCM460 | 83,321,826 | 78,346,916 | 94.03 | 97.01 | 641,381 | 30,658 |
| HCM439 | 115,966,430 | 108,531,416 | 93.59 | 96.48 | 777,052 | 34,950 |
| HCM437 | 114,409,150 | 107,630,394 | 94.07 | 96.59 | 718,353 | 33,216 |
| HCM429 | 107,327,856 | 100,460,638 | 93.60 | 96.51 | 781,013 | 34,514 |
| HCM533 | 113,801,296 | 107,068,190 | 94.08 | 96.47 | 765,001 | 34,584 |
| HCM518 | 123,140,430 | 116,174,372 | 94.34 | 96.54 | 821,103 | 36,229 |
| HCM506 | 104,464,396 | 98,442,104 | 94.24 | 96.88 | 721,500 | 33,453 |
| HCM562 | 119,176,332 | 113,518,840 | 95.25 | 96.63 | 782,848 | 34,382 |
| HCM490 | 109,178,274 | 102,325,212 | 93.72 | 96.79 | 728,218 | 33,873 |
| HCM491 | 123,213,328 | 115,698,990 | 93.90 | 95.97 | 756,623 | 34,040 |
| HCM456 | 124,188,740 | 116,826,920 | 94.07 | 96.65 | 793,088 | 35,368 |
| HCM483 | 132,375,212 | 125,845,538 | 95.07 | 96.52 | 838,862 | 35,806 |
| HCM431 | 95,467,544 | 91,611,504 | 95.96 | 96.60 | 701,266 | 33,263 |
| HCM443 | 103,385,170 | 96,969,874 | 93.79 | 96.18 | 707,524 | 33,016 |
| sc5-LV | 115,206,460 | 109,427,776 | 94.98 | 94.80 | 731,586 | 35,144 |
| sc2-LV | 119,628,518 | 113,668,538 | 95.02 | 96.52 | 747,079 | 34,629 |
| sc6-LV | 133,156,688 | 126,807,678 | 95.23 | 95.59 | 789,274 | 37,289 |
| N105-LV | 125,064,692 | 118,533,240 | 94.78 | 96.39 | 830,057 | 35,975 |
| N104-LV | 151,765,118 | 143,413,882 | 94.50 | 96.50 | 930,969 | 37,875 |
| ND2 | 115,981,584 | 110,192,388 | 95.01 | 94.90 | 649,196 | 33,173 |
| ND1-LV | 111,029,746 | 105,848,092 | 95.33 | 95.66 | 715,284 | 33,920 |
| N102-LV | 111,141,904 | 102,446,288 | 92.18 | 96.59 | 754,272 | 33,343 |
| N103-LV | 146,673,012 | 138,682,818 | 94.55 | 96.50 | 864,176 | 35,856 |
| Group | Sample ID | Gene | dbSNP | Variant Type | AAChange# |
|---|---|---|---|---|---|
| GENETUN | HCM541 | ABCC9 | rs763968252 | nonsynonymous | NM_005691:exon24:c.T2935C:p.W979R |
| HCM493 | SGCD | rs397516338 | nonsynonymous | NM_001128209:exon8:c.A845G:p.Q282R | |
| TTN | rs146400809 | nonsynonymous | NM_133378:exon126:c.C28730T:p.P9577L | ||
| HCM395 | CSRP3 | NA | stop loss | NM_003476:exon7:c.A585C:p.X195C | |
| HCM282 | NEXN | rs146245480 | nonsynonymous | NM_001172309:exon7:c.C643T:p.R215C | |
| HCM591 | DSP | rs116888866 | nonsynonymous | NM_001008844:exon24:c.G6119A:p.R2040Q | |
| PSEN2 | rs574125890 | nonsynonymous | NM_000447:exon8:c.G640T:p.V214L | ||
| HCM552 | NEBL | rs75301590 | nonsynonymous | NM_006393:exon6:c.G561C:p.Q187H | |
| MYBPC3 | HCM515 | MYBPC3 | rs869025465 | frameshift deletion | NM_000256:exon13:c.1153_1168del:p.V385Mfs*15 |
| NEXN | rs146245480 | nonsynonymous | NM_001172309:exon7:c.C643T:p.R215C | ||
| LDB3 | rs566463138 | nonsynonymous | NM_001080114:exon10:c.T1367G:p.M456R | ||
| TNNT2 | rs141121678 | nonsynonymous | NM_001001432:exon14:c.G839A:p.R280H | ||
| HCM504 | MYBPC3 | NA | frameshift deletion | NM_000256:exon28:c.3018delC:p.W1007Gfs*12 | |
| TTN | rs118161093 | nonsynonymous | NM_003319:exon27:c.G5602A:p.A1868T | ||
| TTN | rs139517732 | nonsynonymous | NM_001256850:exon3:c.G160A:p.V54M | ||
| HCM498 | MYBPC3 | NA | stop gain | NM_000256:exon5:c.G587A:p.W196X | |
| ACTN2 | rs376144003 | nonsynonymous | NM_001103:exon11:c.T1162A:p.W388R | ||
| PSEN2 | NA | nonsynonymous | NM_000447:exon10:c.C902A:p.T301K | ||
| HCM486 | PLN | NA | nonsynonymous | NM_002667:exon2:c.T106C:p.C36R | |
| HCM460 | MYBPC3 | rs730880576 | stop gain | NM_000256:exon26:c.G2748A:p.W916X | |
| LDB3 | rs397517221 | nonsynonymous | NM_001080114:exon2:c.C236T:p.T79I | ||
| TTN | rs199932621 | nonsynonymous | NM_003319:exon186:c.G75632A:p.R25211Q | ||
| HCM439 | MYBPC3 | rs397516073 | splicing | NA | |
| LAMA4 | rs3752579 | nonsynonymous | NM_001105206:exon12:c.T1475A:p.L492H | ||
| PSEN2 | NA | nonsynonymous | NM_000447:exon13:c.G1234A:p.A412T | ||
| HCM437 | MYBPC3 | NA | frameshift insertion | NM_000256:exon13:c.1201dupC:p.Q401Pfs*12 | |
| MYH7 | rs727503278 | nonsynonymous | NM_000257:exon5:c.C427T:p.R143W | ||
| COX15 | rs769275933 | nonsynonymous | NM_001320976:exon9:c.C584T:p.T195M | ||
| HCM429 | MYBPC3 | NA | frameshift deletion | NM_000256:exon22:c.2237delA:p.E746Gfs*6 | |
| NEXN | rs146245480 | nonsynonymous | NM_001172309:exon7:c.C643T:p.R215C | ||
| HCM533 | MYBPC3 | NA | stop gain | NM_000256:exon24:c.C2526G:p.Y842X | |
| TTN | rs549841864 | nonsynonymous | NM_003319:exon167:c.C66059T:p.P22020L | ||
| HCM518 | MYBPC3 | NA | frameshift deletion | NM_000256:exon4:c.480delG:p.P161Hfs*5 | |
| MYH7 | HCM506 | MYH7 | rs121913627 | nonsynonymous | NM_000257:exon16:c.G1816A:p.V606M |
| RBM20 | rs372923744 | nonsynonymous | NM_001134363:exon9:c.G2201A:p.R734Q | ||
| HCM562 | MYH7 | rs727503278 | nonsynonymous | NM_000257:exon5:c.C427T:p.R143W | |
| DSP | rs116888866 | nonsynonymous | NM_001008844:exon24:c.G6119A:p.R2040Q | ||
| HCM490 | MYH7 | rs397516201 | nonsynonymous | NM_000257:exon30:c.C4130T:p.T1377M | |
| TTN | rs368057764 | nonsynonymous | NM_003319:exon79:c.C19652T:p.T6551M | ||
| TTN | NA | nonsynonymous | NM_003319:exon154:c.G56641A:p.D18881N | ||
| TTN | rs567446185 | nonsynonymous | NM_003319:exon154:c.G46322A:p.G15441D | ||
| HCM491 | MYH7 | rs397516127 | nonsynonymous | NM_000257:exon18:c.C1987T:p.R663C | |
| MYH6 | NA | splicing | NA | ||
| HCM456 | MYH7 | rs3218714 | nonsynonymous | NM_000257:exon13:c.C1207T:p.R403W | |
| HCM483 | MYH7 | rs121913627 | nonsynonymous | NM_000257:exon16:c.G1816A:p.V606M | |
| HCM431 | MYH7 | rs727503246 | nonsynonymous | NM_000257:exon30:c.G4066A:p.E1356K | |
| PSEN1 | NA | splicing | NA | ||
| HCM443 | MYH7 | rs121913632 | nonsynonymous | NM_000257:exon20:c.G2221T:p.G741W | |
| NEBL | rs75301590 | nonsynonymous | NM_006393:exon6:c.G561C:p.Q187H |
| GO Term | Description | Gene Count | p-value | FDR |
|---|---|---|---|---|
| GO:0072358 | cardiovascular system development | 95 | 6.21 × 10−14 | 8.38 × 10−11 |
| GO:0007507 | heart development | 75 | 2.71 × 10−10 | 1.10 × 10−7 |
| GO:0003007 | heart morphogenesis | 42 | 7.29 × 10−9 | 1.89 × 10−6 |
| GO:0003231 | cardiac ventricle development | 26 | 1.18 × 10−7 | 1.61 × 10−5 |
| GO:0003206 | cardiac chamber morphogenesis | 25 | 6.39 × 10−7 | 6.43 × 10−5 |
| GO:0003205 | cardiac chamber development | 29 | 1.09 × 10−6 | 9.88 × 10−5 |
| GO:0048738 | cardiac muscle tissue development | 31 | 4.73 × 10−6 | 3.00 × 10−4 |
| GO:0060411 | cardiac septum morphogenesis | 16 | 5.09 × 10−6 | 3.11 × 10−4 |
| GO:0003208 | cardiac ventricle morphogenesis | 17 | 7.56 × 10−6 | 4.43 × 10−4 |
| GO:0035051 | cardiocyte differentiation | 22 | 2.22 × 10−5 | 1.12 × 10−3 |
| GO:0003279 | cardiac septum development | 19 | 3.35 × 10−5 | 1.59 × 10−3 |
| GO:0003215 | cardiac right ventricle morphogenesis | 8 | 3.44 × 10−5 | 1.62 × 10−3 |
| GO:0003197 | endocardial cushion development | 11 | 8.84 × 10−5 | 3.62 × 10−3 |
| GO:1905207 | regulation of cardiocyte differentiation | 10 | 2.19 × 10−4 | 7.33 × 10−3 |
| GO:0003203 | endocardial cushion morphogenesis | 9 | 2.62 × 10−4 | 8.54 × 10−3 |
| GO:0055007 | cardiac muscle cell differentiation | 17 | 3.08 × 10−4 | 9.57 × 10−3 |
| GO:0055008 | cardiac muscle tissue morphogenesis | 13 | 3.15 × 10−4 | 9.72 × 10−3 |
| GO:0060047 | heart contraction | 33 | 5.04 × 10−4 | 1.46 × 10−2 |
| GO:0003015 | heart process | 33 | 6.21 × 10−4 | 1.72 × 10−2 |
| GO:0035050 | embryonic heart tube development | 14 | 6.58 × 10 −4 | 1.80 × 10−2 |
| GO:0055017 | cardiac muscle tissue growth | 13 | 6.92 × 10−4 | 1.88 × 10−2 |
| GO:0061323 | cell proliferation involved in heart morphogenesis | 6 | 7.55 × 10−4 | 1.98 × 10−2 |
| GO:0055012 | ventricular cardiac muscle cell differentiation | 6 | 7.55 × 10−4 | 1.98 × 10−2 |
| GO:1905209 | positive regulation of cardiocyte differentiation | 7 | 1.00 × 10−3 | 2.44 × 10−2 |
| GO:0003170 | heart valve development | 9 | 1.27 × 10−3 | 2.97 × 10−2 |
| GO:0003272 | endocardial cushion formation | 7 | 1.31 × 10−3 | 3.02 × 10−2 |
| GO:0010002 | cardioblast differentiation | 6 | 1.46 × 10−3 | 3.32 × 10−2 |
| GO:0060419 | heart growth | 13 | 1.59 × 10−3 | 3.52 × 10−2 |
| GO:0003143 | embryonic heart tube morphogenesis | 12 | 1.73 × 10−3 | 3.78 × 10−2 |
| GO:2000725 | regulation of cardiac muscle cell differentiation | 7 | 2.14 × 10−3 | 4.40 × 10−2 |
| GO:0001947 | heart looping | 11 | 2.44 × 10−3 | 4.85 × 10−2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Collyer, J.; Wang, M.; Sun, F.; Xu, F. Genetic Dissection of Hypertrophic Cardiomyopathy with Myocardial RNA-Seq. Int. J. Mol. Sci. 2020, 21, 3040. https://doi.org/10.3390/ijms21093040
Gao J, Collyer J, Wang M, Sun F, Xu F. Genetic Dissection of Hypertrophic Cardiomyopathy with Myocardial RNA-Seq. International Journal of Molecular Sciences. 2020; 21(9):3040. https://doi.org/10.3390/ijms21093040
Chicago/Turabian StyleGao, Jun, John Collyer, Maochun Wang, Fengping Sun, and Fuyi Xu. 2020. "Genetic Dissection of Hypertrophic Cardiomyopathy with Myocardial RNA-Seq" International Journal of Molecular Sciences 21, no. 9: 3040. https://doi.org/10.3390/ijms21093040
APA StyleGao, J., Collyer, J., Wang, M., Sun, F., & Xu, F. (2020). Genetic Dissection of Hypertrophic Cardiomyopathy with Myocardial RNA-Seq. International Journal of Molecular Sciences, 21(9), 3040. https://doi.org/10.3390/ijms21093040