Structure–Activity Relationship of RGD-Containing Cyclic Octapeptide and αvβ3 Integrin Allows for Rapid Identification of a New Peptide Antagonist



Abstract
:
1. Introduction
2. Results
2.1. NMR Assignments of LXW64 and Verification of Disulfide Bond
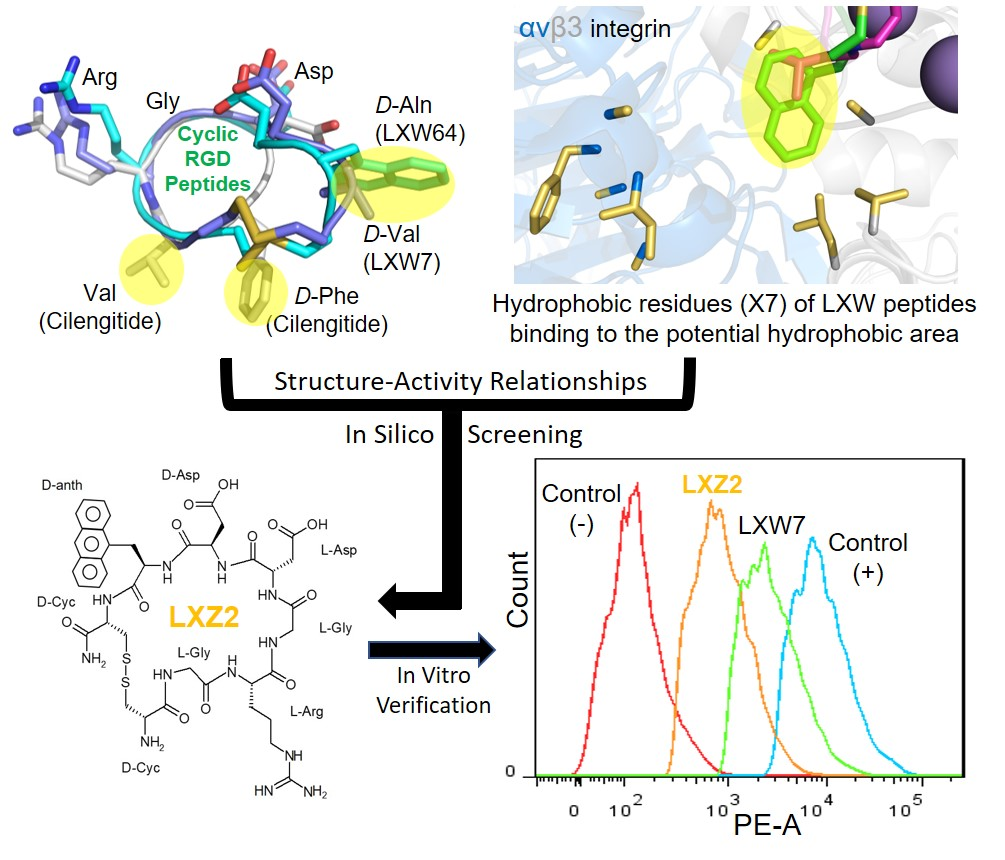
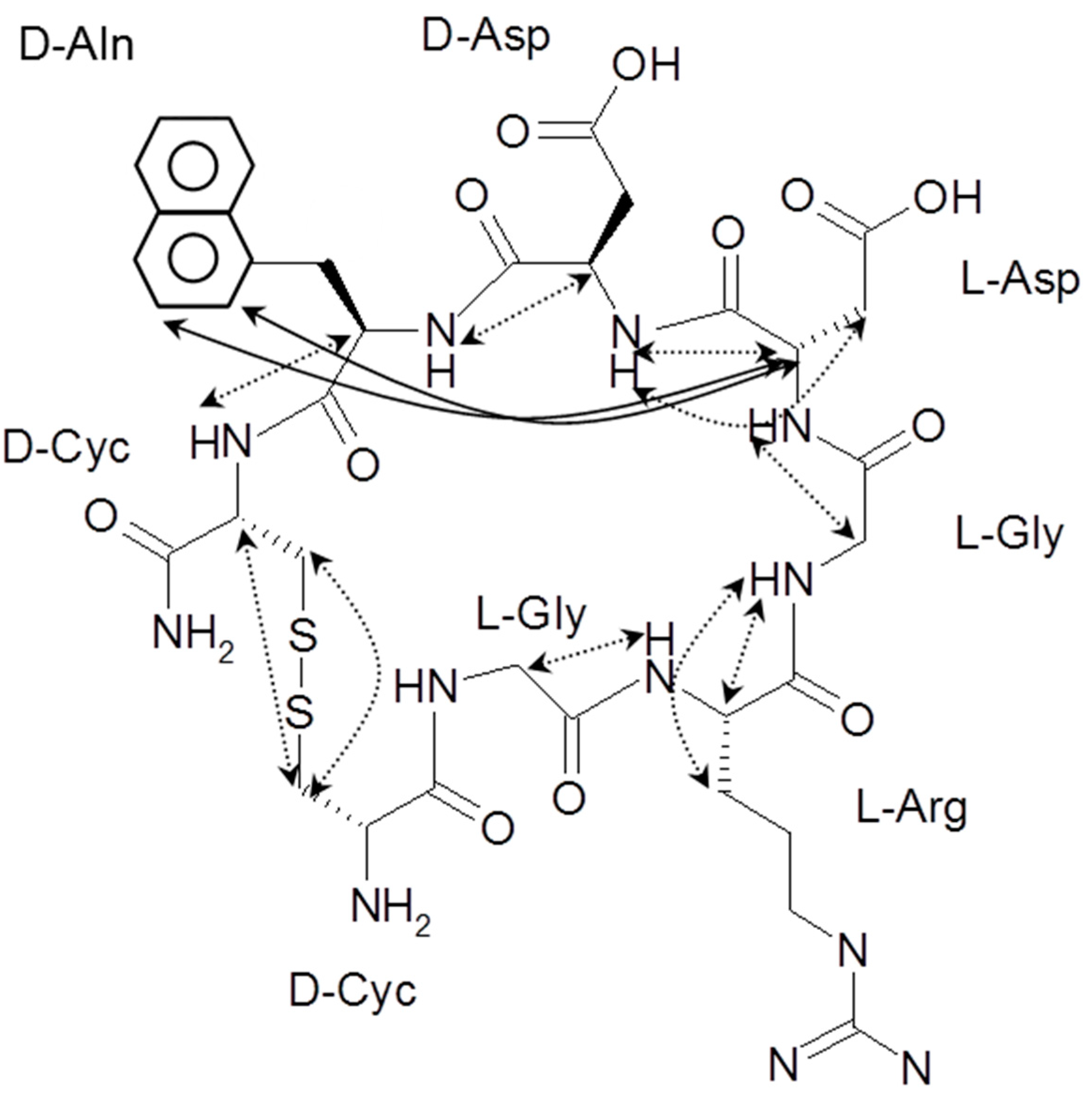
 ) contains 8 amino acids including non-proteinogenic amino acid—3-(1-naphthyl)-D-alanine (D-Nal1). The 1H-coupling spin system for each residue type was unique and easily distinguishable from 1H-1H TOCSY, for example, the residue, arginine, was unambiguously assigned based on its unique 1H resonances of Hβ and Hγ (1–2 ppm). The other three types of residues, two glycines, two aspartates, and two cysteines, were also identified and assigned with their HN, Hα, and Hβ. The sequential assignment was then completed through the connectivity of NOEs observed between the amide protons in 2D NOESY (Figure 1). Thus, all 1H resonances were unambiguously assigned for LXW64. The proton assignments allowed unambiguous assignments of all proton-attached 13C resonances using 1H-13C HMQC spectrum, while the chemical shifts of non-pronated 13C were assigned from 1H-13C HMBC spectrum due to their long-range couplings with other assigned protons. Native abundance gradient 15N HSQC spectrum was obtained for LXW64 on the 600 MHz Bruker spectrometer. The high-signal-to-noise quality of this spectrum enabled unambiguous assignments for all 15N chemical shifts. Chemical shifts of all 1H, 13C and 15N were fully assigned for LXW64 peptide and are available as Supporting Information in Table S1. The intramolecular disulfide bond of the peptide was confirmed via the NOEs between two cysteines (Cys1 and Cys8). Moreover, the disulfide bridge connectivity was identified by MS and 13Cβ chemical shifts of two cysteines [9].
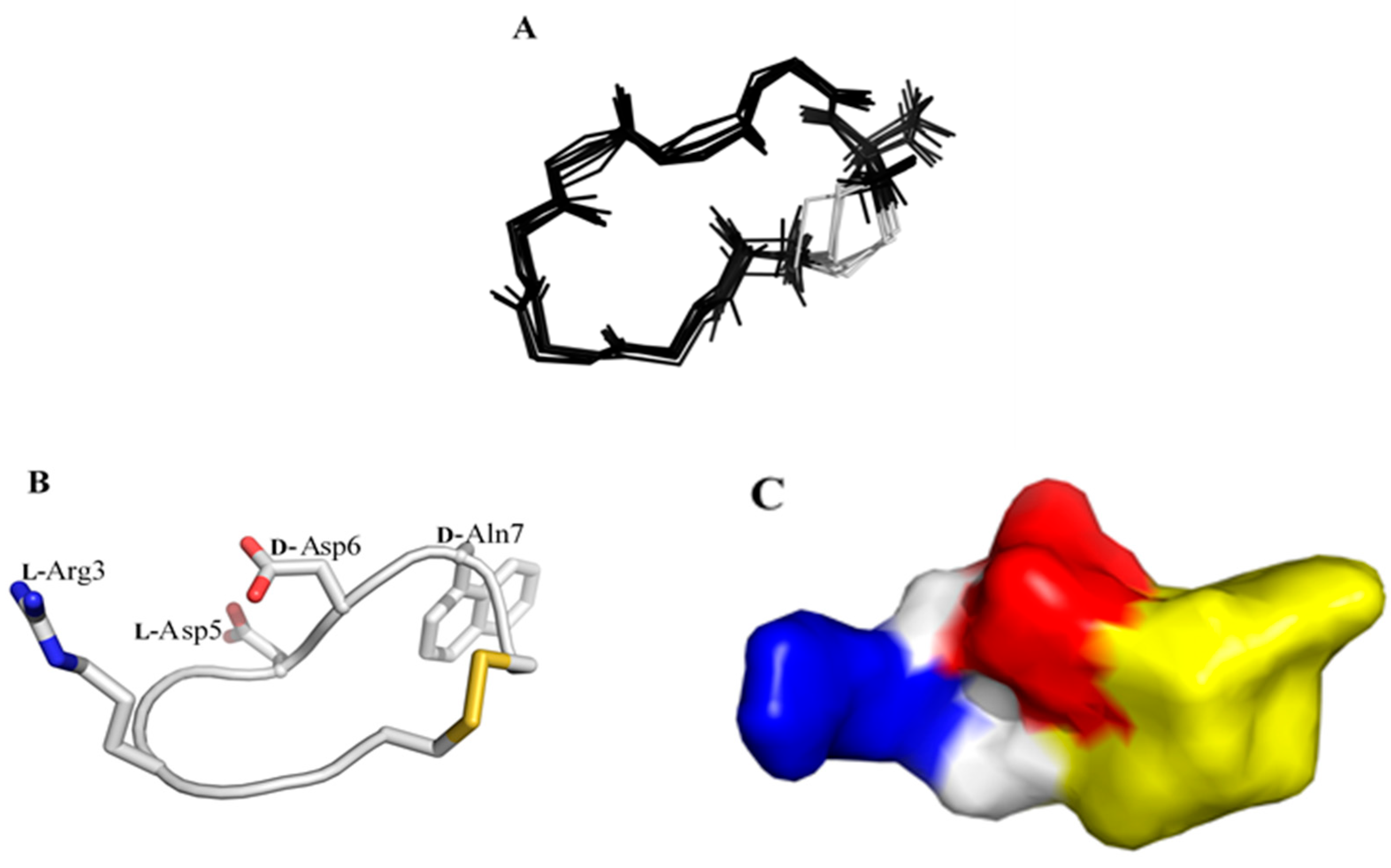
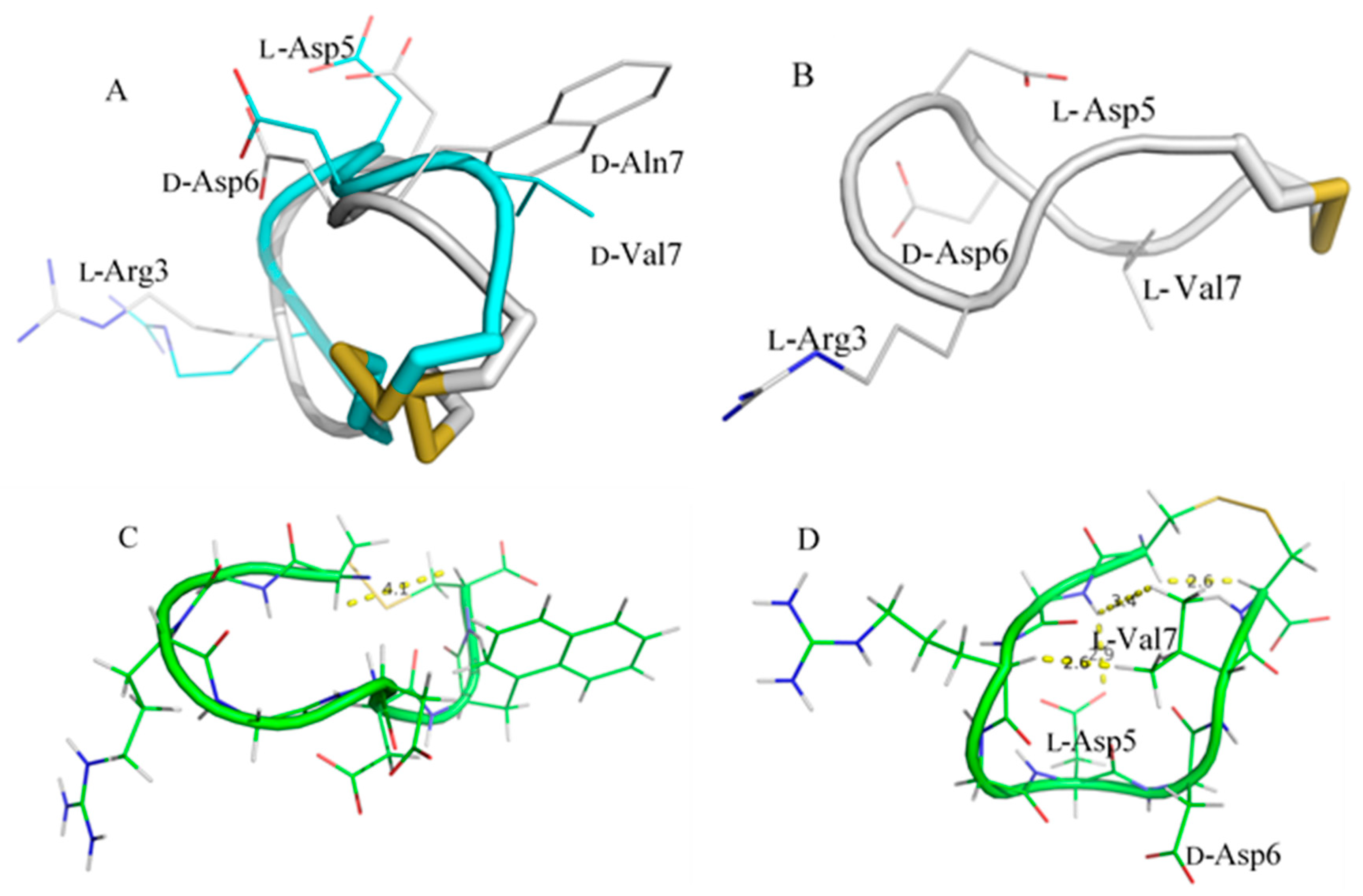
) contains 8 amino acids including non-proteinogenic amino acid—3-(1-naphthyl)-D-alanine (D-Nal1). The 1H-coupling spin system for each residue type was unique and easily distinguishable from 1H-1H TOCSY, for example, the residue, arginine, was unambiguously assigned based on its unique 1H resonances of Hβ and Hγ (1–2 ppm). The other three types of residues, two glycines, two aspartates, and two cysteines, were also identified and assigned with their HN, Hα, and Hβ. The sequential assignment was then completed through the connectivity of NOEs observed between the amide protons in 2D NOESY (Figure 1). Thus, all 1H resonances were unambiguously assigned for LXW64. The proton assignments allowed unambiguous assignments of all proton-attached 13C resonances using 1H-13C HMQC spectrum, while the chemical shifts of non-pronated 13C were assigned from 1H-13C HMBC spectrum due to their long-range couplings with other assigned protons. Native abundance gradient 15N HSQC spectrum was obtained for LXW64 on the 600 MHz Bruker spectrometer. The high-signal-to-noise quality of this spectrum enabled unambiguous assignments for all 15N chemical shifts. Chemical shifts of all 1H, 13C and 15N were fully assigned for LXW64 peptide and are available as Supporting Information in Table S1. The intramolecular disulfide bond of the peptide was confirmed via the NOEs between two cysteines (Cys1 and Cys8). Moreover, the disulfide bridge connectivity was identified by MS and 13Cβ chemical shifts of two cysteines [9].2.2. Structure Determination of LXW64 and Structural Comparison with Other LXW Peptides
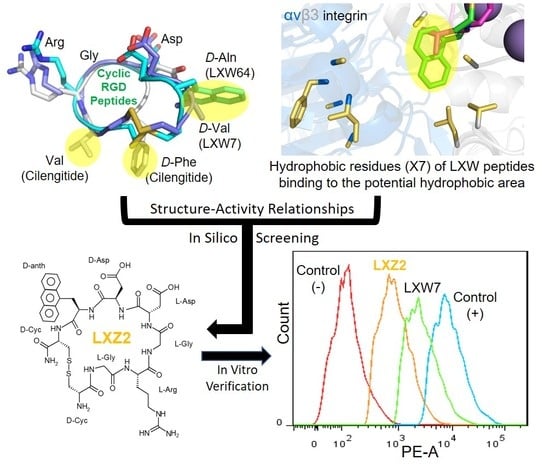
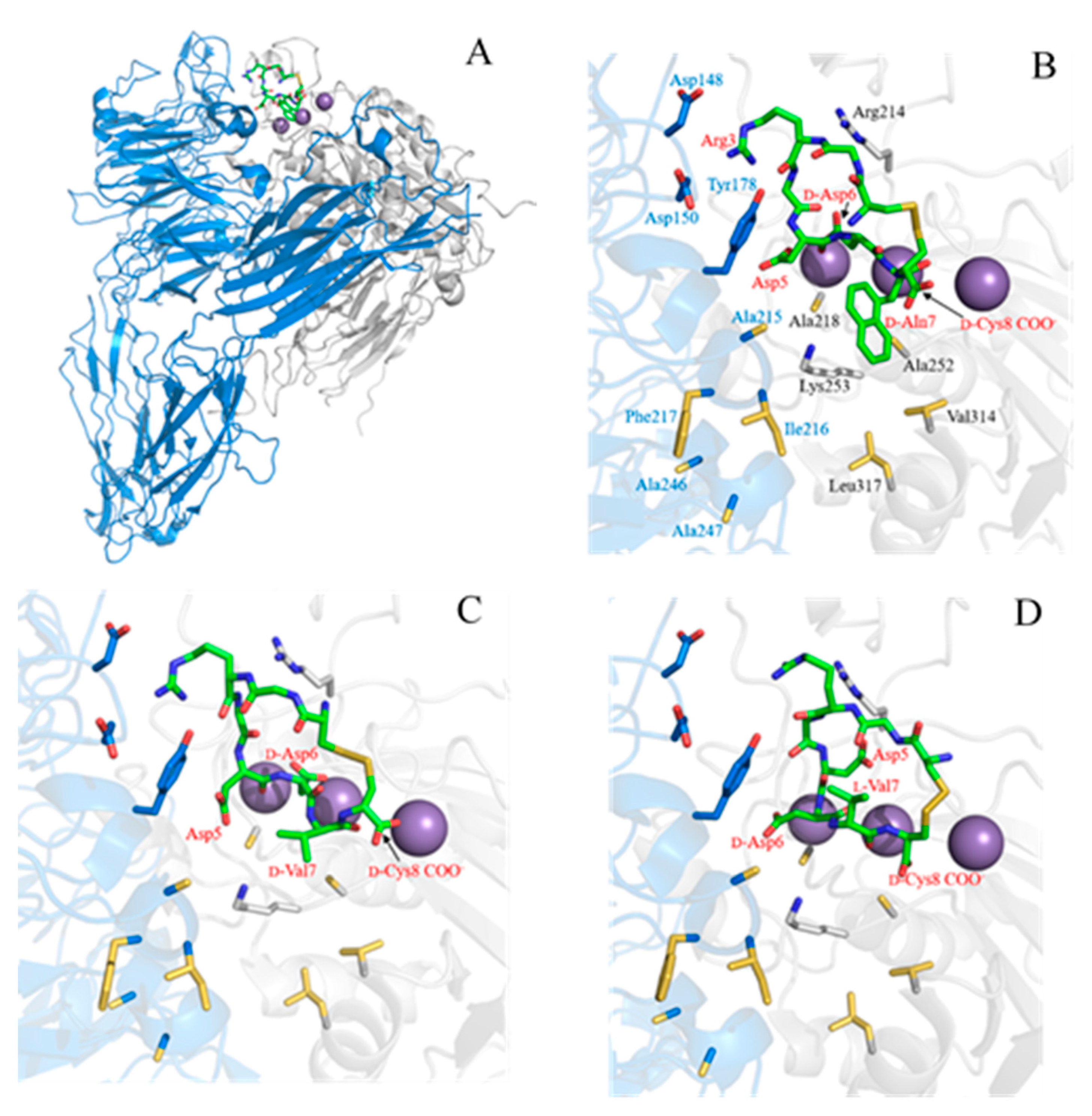
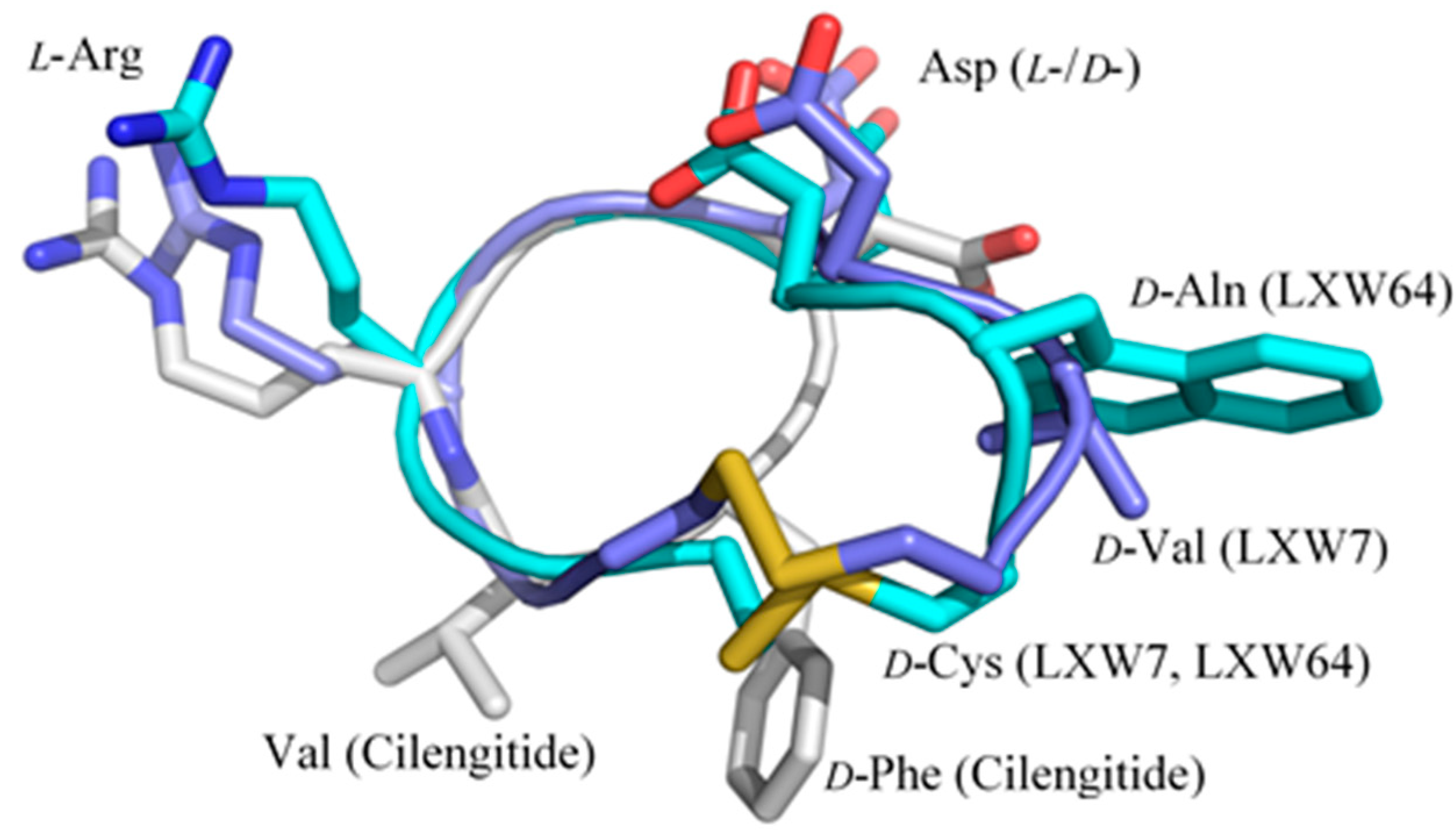
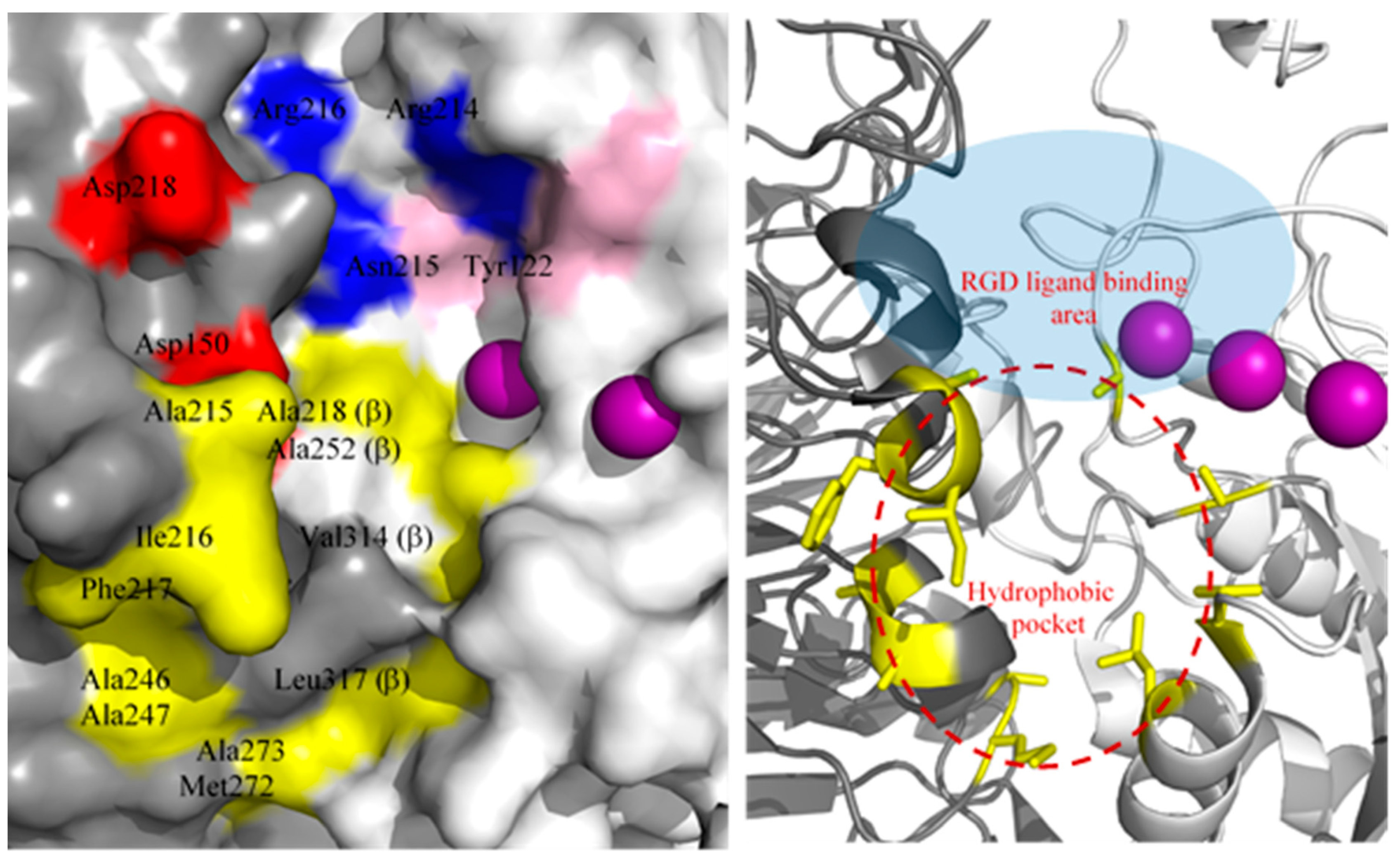
2.3. Complex Structure Models of LXW Peptides and αvβ3 Integrin
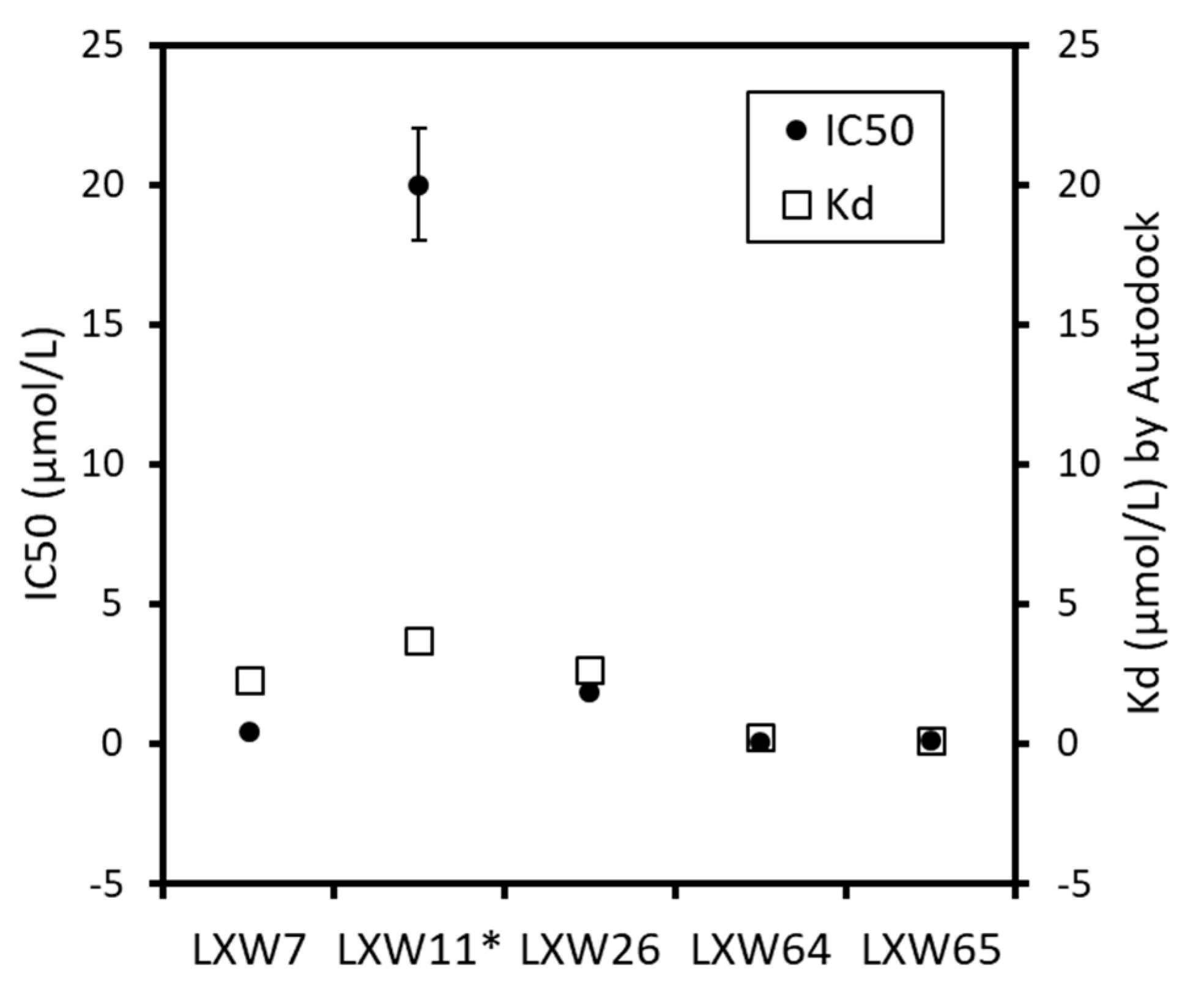
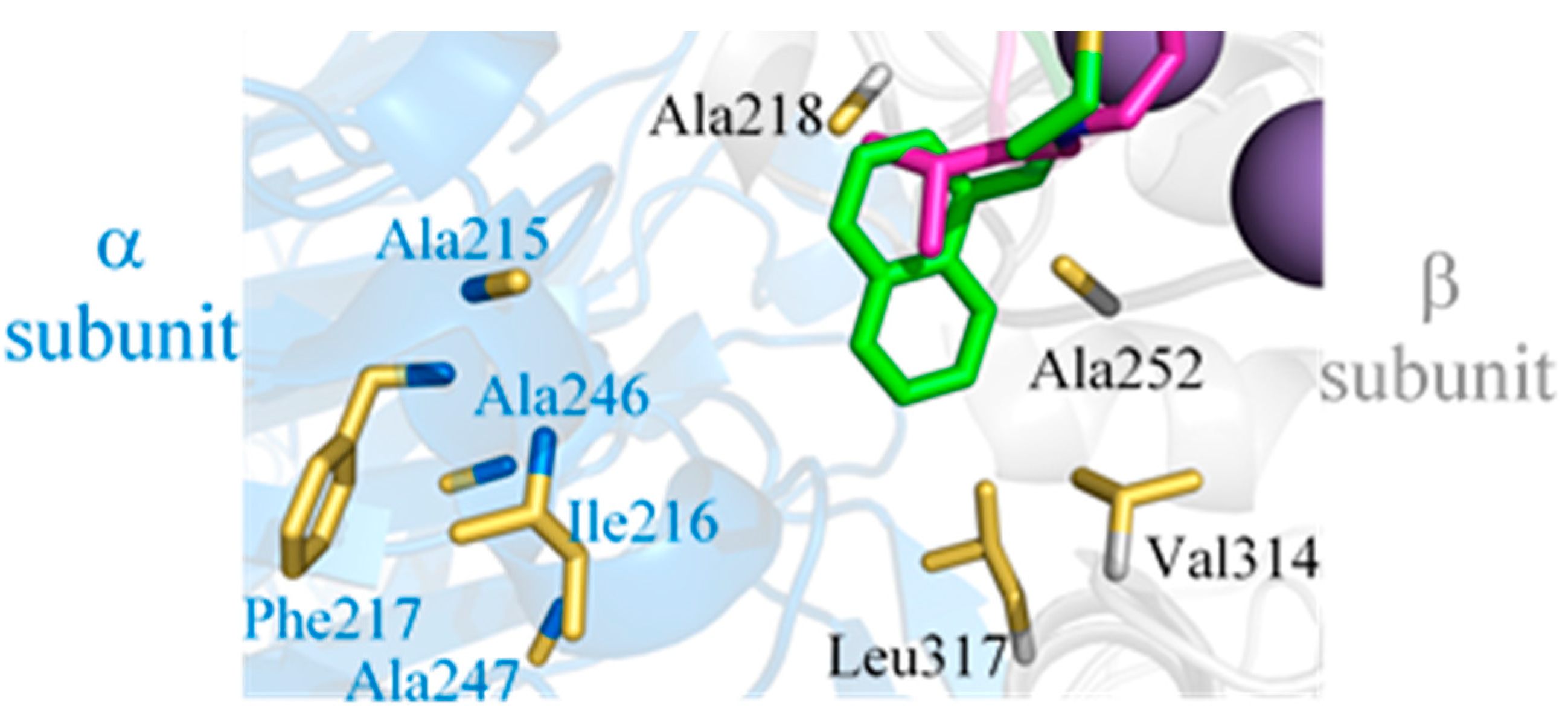
2.4. New LXW-Analogous Peptide Screening by Autodock
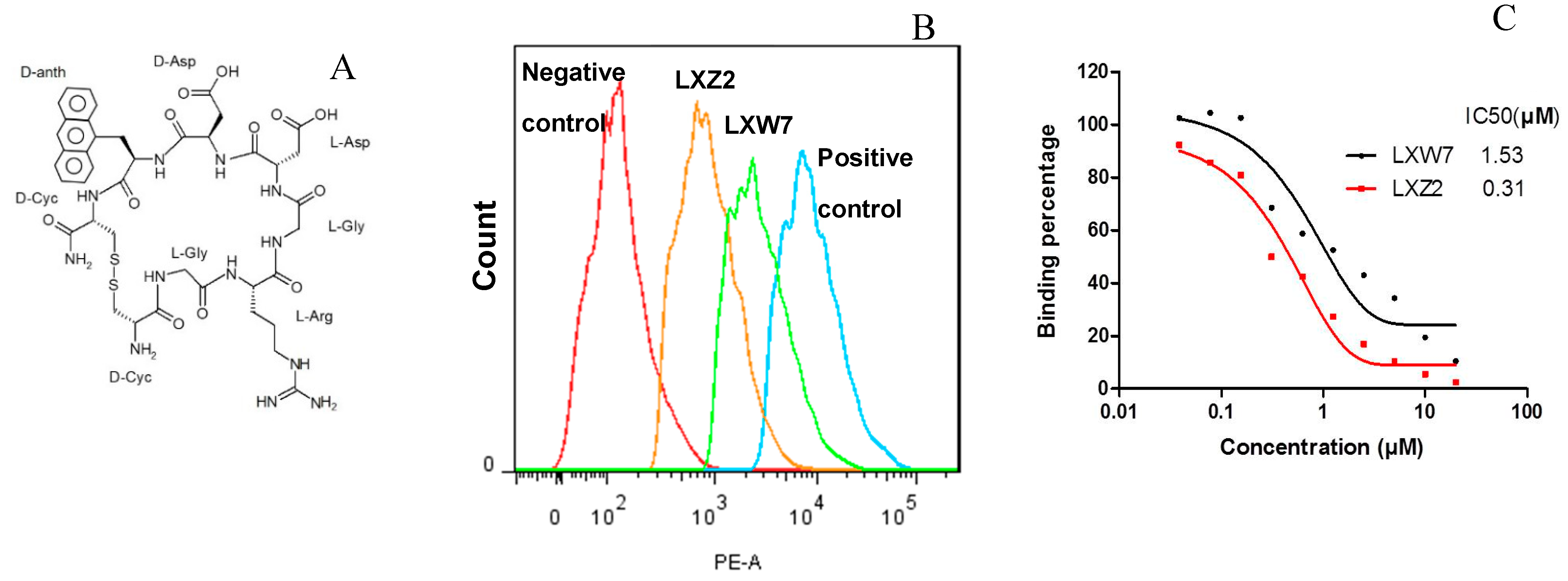
2.5. In Vitro Examination of New LXW-Analogous Peptides
3. Discussion
Identification of a New LXW Analog—LXZ2
4. Materials and Methods
4.1. Synthesis of Peptides
 where a lower case letter represents D-amino acid (x is a variable amino acid) and a disulfide bridge is formed between two D-cysteines (D-Cys1 and D-Cys8), were chemically synthesized and purified by preparative RP-HPLC) from C S Bio Co (Menlo Park, CA, USA). For synthesis of LXW64 and LXZ2, the non-natural amino acids—Fmoc-3-(1-naphthyl)-D-alanine and Fmoc-3-(9-anthryl)-D-alanine—were purchased from Chem Impex (Wood Dale, IL, USA). HPLC purification was performed using an Agilent 1200 instrument and a Phenomenex Luna 5 μm C18(2) 100A 250 × 4.6 mm column. The peptides were eluted using a gradient of buffer A (0.1% TFA in water) and B (0.1% TFA in acetonitrile) with a flow rate of 1 mL/min. Each peptide eluted as a single peak via HPLC with > 97% purity, was verified by MS. The theoretical mass of LXW64 (918.98) was very close to the experimental value (918.73). For LXZ2, the experimentally measured mass of 968.72 was also similar to the expected 969.09. For NMR sample preparation, 20 mg peptides were dissolved in 0.6 mL DMSO-d6 solvent (Cambridge Isotope Laboratories, Inc., Tewksbury, MA, USA). The peptide solution was carefully transferred into an NMR tube after centrifugation. The repeated NMR analysis showed that the peptide samples were stable over several months in the chosen DMSO solvent.
where a lower case letter represents D-amino acid (x is a variable amino acid) and a disulfide bridge is formed between two D-cysteines (D-Cys1 and D-Cys8), were chemically synthesized and purified by preparative RP-HPLC) from C S Bio Co (Menlo Park, CA, USA). For synthesis of LXW64 and LXZ2, the non-natural amino acids—Fmoc-3-(1-naphthyl)-D-alanine and Fmoc-3-(9-anthryl)-D-alanine—were purchased from Chem Impex (Wood Dale, IL, USA). HPLC purification was performed using an Agilent 1200 instrument and a Phenomenex Luna 5 μm C18(2) 100A 250 × 4.6 mm column. The peptides were eluted using a gradient of buffer A (0.1% TFA in water) and B (0.1% TFA in acetonitrile) with a flow rate of 1 mL/min. Each peptide eluted as a single peak via HPLC with > 97% purity, was verified by MS. The theoretical mass of LXW64 (918.98) was very close to the experimental value (918.73). For LXZ2, the experimentally measured mass of 968.72 was also similar to the expected 969.09. For NMR sample preparation, 20 mg peptides were dissolved in 0.6 mL DMSO-d6 solvent (Cambridge Isotope Laboratories, Inc., Tewksbury, MA, USA). The peptide solution was carefully transferred into an NMR tube after centrifugation. The repeated NMR analysis showed that the peptide samples were stable over several months in the chosen DMSO solvent.4.2. NMR Spectroscopy
4.3. Experimental Constraints and Structure Calculation
4.4. Complex Modeling and Autodock Screening
4.5. Flow Cytometry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SAR | Structure–Activity Relationship |
| RGD | Arginylglycylaspartic acid |
| NMR | Nuclear Magnetic Resonance |
| NOE | Nuclear Overhauser Effect |
| HSQC | Heteronuclear Single Quantum Correlation |
| TOCSY | TOtal Correlated SpectroscopY |
| NOESY | Nuclear Overhauser Effect Spectroscopy |
| HMQC | Heteronuclear Multiple-Quantum Correlation |
| HMBC | Heteronuclear Multiple Bond Correlation |
| RMSD | Root-Mean-Squared Derivation |
| RP-HPLC | Reversed Phase-High Performance Liquid Chromatography |
| MS | Mass spectrometry |
| DMSO | Dimethyl sulfoxide |
| DSS | 2,2-Dimethyl-2-silapentane-5-sulfonate |
| PBS | Phosphate-Buffered Saline |
| FBS | Fetal Calf Serum |
| IC50 | Half-Maximal Inhibitory Concentration |
References
- Horton, M.A. The αvβ3 integrin “vitronectin receptor”. Int. J. Biochem. Cell Biol. 1997, 29, 721–725. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, F.; Chen, X. Integrin αvβ3-target cancer therapy. Drug Dev. Res. 2008, 69, 329–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, P.C.; Strömblad, S.; Klemke, R.; Visscher, D.; Sarkar, F.H.; Cheresh, D.A. Antiintegrin alpha v beta 3 blocks human breast cancer growth and angiogenesis in human skin. J. Clin. Invest. 1995, 96, 1815–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, K.A.; Bicknell, R. Anti-angiogenic alternatives to VEGF blockade. Clin. Exp. Metastasis 2016, 33, 197–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin αvβ3 in complex with an arg-gly-asp ligand. Science 2002, 296, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Le Breton, A.; Préat, V. RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol. Pharm. 2012, 9, 2961–2973. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Wang, Y.; Lau, E.Y.; Luo, J.; Yao, N.; Shi, C.; Meza, L.; Tseng, H.; Maeda, Y.; Kumaresan, P.; et al. The use of one-bead one-compound combinatorial library technology to discover high-affinity αvβ3 integrin and cancer targeting arginine-glycine-aspartic acid ligands with a built-in handle. Mol. Cancer Ther. 2010, 9, 2714–2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xiao, W.; Zhang, Y.; Meza, L.; Tseng, H.; Takada, Y.; Ames, J.B.; Lam, K.S. Optimization of RGD-Containing Cyclic Peptides against αvβ3 Integrin. Mol. Cancer Ther. 2016, 15, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Rajarathnam, K. 13C NMR chemical shifts can predict disulfide bond formation. J. Biomol. NMR 2000, 18, 165–171. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Gfeller, D.; Michielin, O.; Zoete, V. SwissSidechain: A molecular and structural database of non-natural sidechains. Nucleic Acids Res. 2013, 41, D327–D332. [Google Scholar] [CrossRef] [PubMed]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate. Design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, M.A.; Paolillo, M.; Sanchez-Hernandez, Y.; Curti, D.; Ciusani, E.; Serra, M.; Colombo, L.; Schinelli, S. A small-molecule RGD-integrin antagonist inhibits cell adhesion, cell migration and induces anoikis in glioblastoma cells. Int. J. Oncol. 2013, 42, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Pan, Y.; Xu, Y. Binding investigation of integrin alphavbeta3 with its inhibitors by SPR technology and molecular docking simulation. J. Biomol. Screen. 2010, 15, 131–137. [Google Scholar] [CrossRef]
- Ma, Y.; Ai, G.; Zhang, C.; Zhao, M.; Dong, X.; Han, Z.; Wang, Z.; Zhang, M.; Liu, Y.; Gao, W.; et al. Novel Linear Peptides with High Affinity to αvβ3 Integrin for Precise Tumor Identification. Theranostics 2017, 7, 1511–1523. [Google Scholar] [CrossRef]
- Joo, S.H. Cyclic Peptides as Therapeutic Agents and Biochemical Tools. Biomol. Ther. (Seoul) 2012, 20, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Rezai, T.; Yu, B.; Millhauser, G.L.; Jacobson, M.P.; Lokey, R.S. Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers. J. Am. Chem. Soc. 2006, 128, 2510–2511. [Google Scholar] [CrossRef]
- Reardon, D.A.; Neyns, B.; Weller, M.; Tonn, J.C.; Nabors, L.B.; Stupp, R. Cilengitide: An RGD pentapeptide ανβ3 and ανβ5 integrin inhibitor in development for glioblastoma and other malignancies. Future Oncol. 2011, 7, 339–354. [Google Scholar] [CrossRef] [Green Version]
- Koshland, D.E. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Sykoutri, D.; Geetha, N.; Hayer, S.; Mandl, P.; Smolen, S.S.; Prager, G.; Redlich, K. αvβ3 Integrin Inhibition with Cilengitide both Prevents and Treats Collagen Induced Arthritis. Ann. Rheum Dis. 2013, 72 (Suppl. 1), A1–A88. [Google Scholar] [CrossRef] [Green Version]
- Collin, G.; Hӧke, H.; Talbiersky, J. Anthracene. Ullmann’s Encyclopedia of Industrial Chemistry, 7th ed.; John Wiley & Sons: New York, NY, USA, 2006. [Google Scholar]
- Toxicology and carcinogenesis studies of naphthalene (cas no. 91-20-3) in F344/N rats (inhalation studies). Natl. Toxicol. Program Tech. Rep. Ser. 2000, 500, 1–173.
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Kneller, D.G. SPARKY 3; University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Wuthrich, K.; Billeter, M.; Braun, W. Pseudo-structures for the 20 common amino acids for use in studies of protein conformations by measurements of intramolecular proton-proton distance constraints with nuclear magnetic resonance. J. Mol. Biol. 1983, 169, 949–961. [Google Scholar] [CrossRef]
- Wang, A.C.; Bax, A. Determination of the Backbone Dihedral Angles f in Human Ubiquitin from Reparametrized Empirical Karlpus Equations. J. Am. Chem. Soc. 1996, 118, 2483–2494. [Google Scholar] [CrossRef]
- Assa-Munt, N.; Jia, X.; Laakkonen, P.; Ruoslahti, E. Solution structures and integrin binding activities of an RGD peptide with two isomers. Biochemistry 2001, 40, 2373–2378. [Google Scholar] [CrossRef]
- Kessler, H.; Seip, S. NMR of Peptides. In Two-Dimensional NMR Spectroscopy: Applications for Chemists and Biochemists, 2nd ed.; Croasmun, W.R., Carlson, R.M.K., Eds.; VCH Publishers: New York, NY, USA, 1994; pp. 642–643. [Google Scholar]
- Brunger, A.T. X-PLOR, Version 3.1: A System for X-Ray Crystallography and NMR; Yale University Press: New Haven, CT, USA, 1992. [Google Scholar]
- Badger, J.; Kumar, R.A.; Yip, P.; Szalma, S. New features and enhancements in the X-PLOR computer program. Proteins 1999, 35, 25–33. [Google Scholar] [CrossRef]
- Bagby, S.; Harvey, T.S.; Eagle, S.G.; Inouye, S.; Ikura, M. NMR-derived three-dimensional solution structure of protein S complexed with calcium. Structure 1994, 2, 107–122. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Mohd, S.; Rizvi, D.; Shakil, S.; Mohd, H. A simple click by click protocol to perform docking: Autodock 4.2 made easy for non-bioinformaticians. EXCLI J. 2013, 12, 831–857. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Feinstein, W.P.; Brylinski, M. Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets. J. Cheminform. 2015, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Amino Acid Sequence # | IC50 (μmoL/L) |
|---|---|---|
| LXW7 | cGRGDdvc-NH2 | 0.46 |
| LXW11 | CGRGDdvC-NH2 | >20 |
| LXW64 | cGRGDd-DNal1-c-NH2 | 0.07 |
| NOE Restraints (Total) | 58 |
|---|---|
| dihedral angle restraints | 14 |
| RMSD from ideal geometry | |
| bond length (Å) | 0.0094 ± 0.00041 |
| bond angles (degree) | 2.27 ± 0.04 |
| Ramachandran plot | |
| allowed region (%) | 100 |
| disallowed region (%) | 0 |
| RMSD of atom position from average structure | |
| main chain (Å) | 0.34 ± 0.072 |
| non-hydrogen (Å) | 1.47 ± 0.16 |
| RGD Peptide | X Amino Acids in LXW Analogs (CGRGDdXc-NH2) | IC50 (µmoL/L) | Binding Free Energy (kcaL/moL) | Kd (µmoL/L) by Autodock # |
|---|---|---|---|---|
| LXW7 | D-Val | 0.46 | −7.7 | 2.27 |
| LXW11 * | D-Val and L-Cys | >20 | −7.4 | 3.70 |
| LXW26 | D-Ile | 1.84 | −7.6 | 2.67 |
| LXW64 | D-Nal1 | 0.07 | −9.0 | 0.24 |
| LXW65 | D-Nal2 | 0.13 | −9.3 | 0.15 |
| Peptide/X7 Residue | Kd (µmoL/L) | Peptide/X7 Residue | Kd (µmoL/L) | Peptide/X7 Residue | Kd (µmoL/L) |
|---|---|---|---|---|---|
DNAL1 (LXW64) | 0.25 | DCPE | 0.70 | DNLE | 1.63 |
DNTL (LXZ2) | 0.29 | DHL1 | 1.38 | DNVA | 1.16 |
DTRP | 0.30 | DALC | 0.50 | DQ33 | 0.50 |
DPZ4 | 0.59 | DLVG | 0.83 | DQ36 | 0.30 |
DLEU | 1.16 | D5MW | 0.30 | DQX3 | 0.38 |
DPHE | 0.36 | D6MW | 0.42 | DTH9 | 0.59 |
DMET | 0.83 | D2TH | 0.98 | DAHP | 1.63 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, A.; Xiao, W.; Wang, Y.; Wang, W.; Chang, H.W.; Ames, J.B.; Lam, K.S.; Zhang, Y. Structure–Activity Relationship of RGD-Containing Cyclic Octapeptide and αvβ3 Integrin Allows for Rapid Identification of a New Peptide Antagonist. Int. J. Mol. Sci. 2020, 21, 3076. https://doi.org/10.3390/ijms21093076
Silva A, Xiao W, Wang Y, Wang W, Chang HW, Ames JB, Lam KS, Zhang Y. Structure–Activity Relationship of RGD-Containing Cyclic Octapeptide and αvβ3 Integrin Allows for Rapid Identification of a New Peptide Antagonist. International Journal of Molecular Sciences. 2020; 21(9):3076. https://doi.org/10.3390/ijms21093076
Chicago/Turabian StyleSilva, Aaron, Wenwu Xiao, Yan Wang, Wei Wang, Heng Wei Chang, James B. Ames, Kit S. Lam, and Yonghong Zhang. 2020. "Structure–Activity Relationship of RGD-Containing Cyclic Octapeptide and αvβ3 Integrin Allows for Rapid Identification of a New Peptide Antagonist" International Journal of Molecular Sciences 21, no. 9: 3076. https://doi.org/10.3390/ijms21093076
APA StyleSilva, A., Xiao, W., Wang, Y., Wang, W., Chang, H. W., Ames, J. B., Lam, K. S., & Zhang, Y. (2020). Structure–Activity Relationship of RGD-Containing Cyclic Octapeptide and αvβ3 Integrin Allows for Rapid Identification of a New Peptide Antagonist. International Journal of Molecular Sciences, 21(9), 3076. https://doi.org/10.3390/ijms21093076