Involvement of Senescence and Mitochondrial Fission in Endothelial Cell Pro-Inflammatory Phenotype Induced by Angiotensin II



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
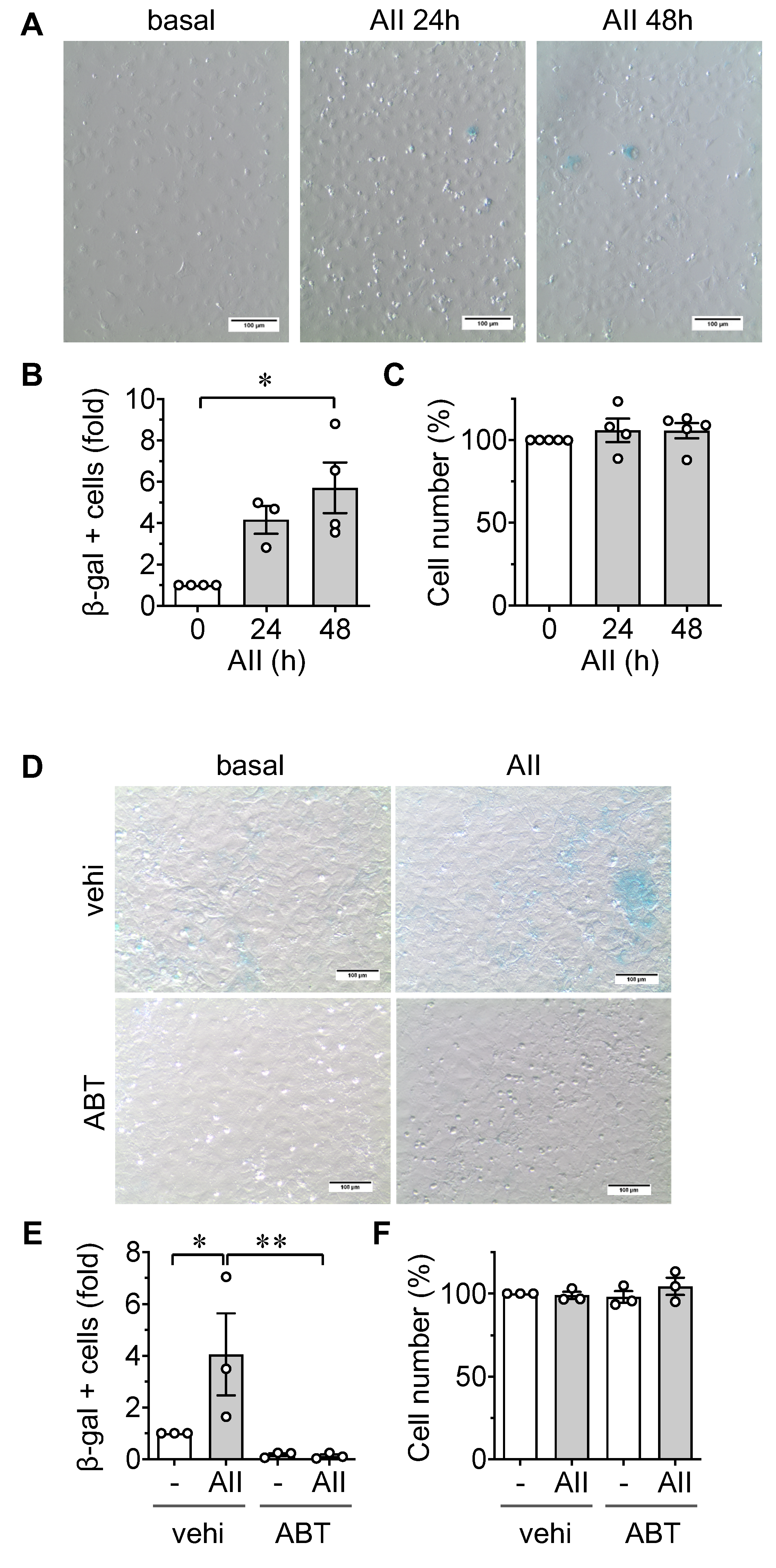
2.1. Induction of Senescence by AngII in ECs
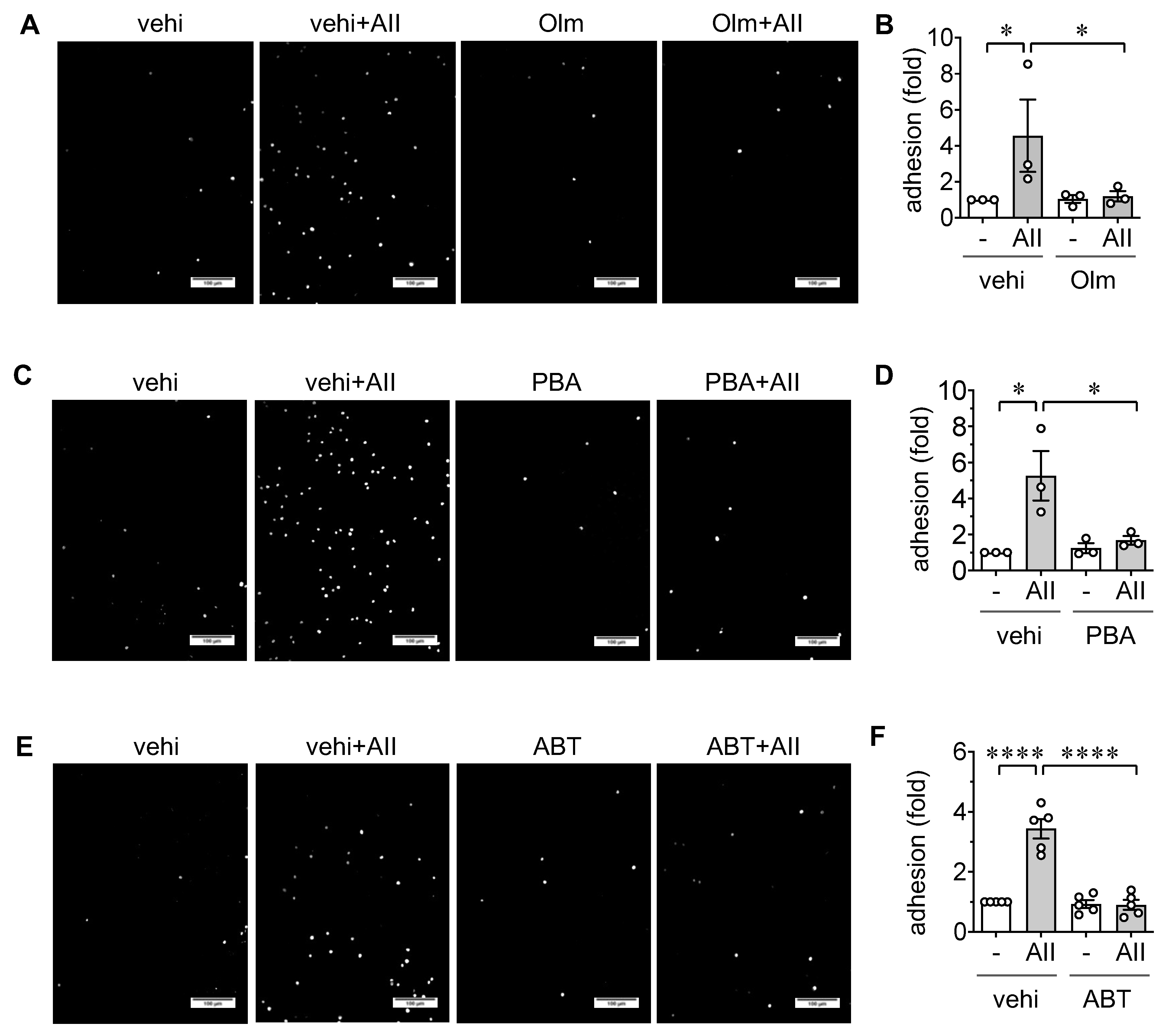
2.2. Induction of Leukocyte Adhesion via ER stress and Senescence in ECs
2.3. Role of Mitochondrial Fission in EC Senescence and Inflammation
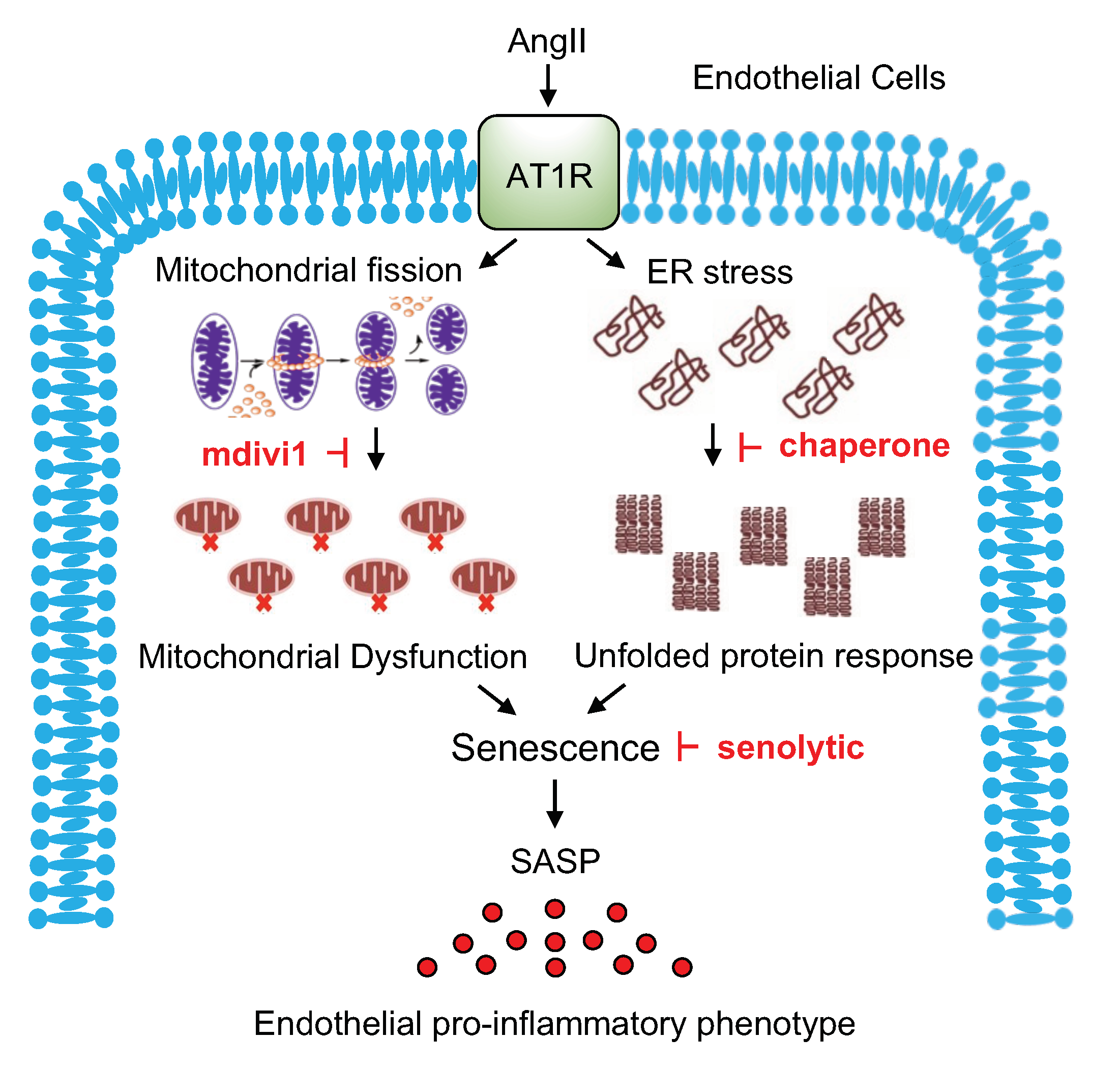
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Senescence-Associated β Galactosidase Assay
4.3. Leukocyte Adhesion Assay
4.4. Mitochondrial Morphology Analysis
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AngII | Angiotensin II |
| AT1R | Angiotensin II type 1 receptor |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| DMSO | Dimethyl sulfoxide |
| Drp1 | Dynamin-related protein 1 |
| ECs | Endothelial cells |
| ER | Endoplasmic reticulum |
| FBS | Fetal bovine serum |
| HPF | High power field |
| Mdivi1 | Mitochondrial division inhibitor-1 |
| MFC | Mitochondrial fragmentation count |
| PBS | Phosphate buffered saline |
| 4-PBA | 4-Phenylbutyrate |
| RAS | Renin angiotensin system |
| ROS | Reactive oxygen species |
| SASP | Senescence associated secretory phenotype |
| UPR | Unfolded protein response |
| VSMCs | Vascular smooth muscle cells |
References
- Brandes, R.P. Endothelial dysfunction and hypertension. Hypertension 2014, 64, 924–928. [Google Scholar] [CrossRef] [Green Version]
- Daiber, A.; Steven, S.; Weber, A.; Shuvaev, V.V.; Muzykantov, V.R.; Laher, I.; Li, H.; Lamas, S.; Munzel, T. Targeting vascular (endothelial) dysfunction. Br. J. Pharmacol. 2017, 174, 1591–1619. [Google Scholar] [CrossRef]
- Widmer, R.J.; Lerman, A. Endothelial dysfunction and cardiovascular disease. Glob. Cardiol. Sci. Pract. 2014, 2014, 291–308. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Mohammadmoradi, S.; Chen, J.Z.; Sawada, H.; Daugherty, A.; Lu, H.S. Renin-Angiotensin System and Cardiovascular Functions. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e108–e116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, Y.; Guddeti, R.R.; Kwon, T.G.; Lerman, L.O.; Lerman, A. Treating coronary disease and the impact of endothelial dysfunction. Prog. Cardiovasc. Dis. 2015, 57, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Brasier, A.R.; Recinos, A.; Eledrisi, M.S. Vascular inflammation and the renin-angiotensin system. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1257–1266. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, S.; Ohtsu, H.; Suzuki, H.; Shirai, H.; Frank, G.D.; Eguchi, S. Angiotensin II signal transduction through the AT1 receptor: Novel insights into mechanisms and pathophysiology. Clin. Sci. 2007, 112, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-induced production of mitochondrial reactive oxygen species: Potential mechanisms and relevance for cardiovascular disease. Antioxid. Redox Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef] [PubMed]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diebold, I.; Hennigs, J.K.; Miyagawa, K.; Li, C.G.; Nickel, N.P.; Kaschwich, M.; Cao, A.; Wang, L.; Reddy, S.; Chen, P.I.; et al. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab. 2015, 21, 596–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliuso, A.; Cossart, P.; Stavru, F. The ever-growing complexity of the mitochondrial fission machinery. Cell. Mol. Life Sci. 2018, 75, 355–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Lee, S.Y.; Seo, H.H.; Ham, O.; Lee, C.; Park, J.H.; Lee, J.; Seung, M.; Yun, I.; Han, S.M.; et al. Regulation of mitochondrial morphology by positive feedback interaction between PKCdelta and Drp1 in vascular smooth muscle cell. J. Cell. Biochem. 2015, 116, 648–660. [Google Scholar] [CrossRef]
- Seals, D.R.; Jablonski, K.L.; Donato, A.J. Aging and vascular endothelial function in humans. Clin. Sci. 2011, 120, 357–375. [Google Scholar] [CrossRef] [Green Version]
- Laurent, S.; Boutouyrie, P.; Cunha, P.G.; Lacolley, P.; Nilsson, P.M. Concept of Extremes in Vascular Aging. Hypertension 2019, 74, 218–228. [Google Scholar] [CrossRef]
- Kunieda, T.; Minamino, T.; Nishi, J.; Tateno, K.; Oyama, T.; Katsuno, T.; Miyauchi, H.; Orimo, M.; Okada, S.; Takamura, M.; et al. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation 2006, 114, 953–960. [Google Scholar] [CrossRef]
- Xiong, S.; Salazar, G.; Patrushev, N.; Ma, M.; Forouzandeh, F.; Hilenski, L.; Alexander, R.W. Peroxisome proliferator-activated receptor gamma coactivator-1alpha is a central negative regulator of vascular senescence. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 988–998. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Z.; Wang, F.; Gao, P.; Pei, J.F.; Liu, Y.; Xu, T.T.; Tang, X.; Fu, W.Y.; Lu, J.; Yan, Y.F.; et al. Age-Associated Sirtuin 1 Reduction in Vascular Smooth Muscle Links Vascular Senescence and Inflammation to Abdominal Aortic Aneurysm. Circ. Res. 2016, 119, 1076–1088. [Google Scholar] [CrossRef]
- Liang, B.; Wang, S.; Wang, Q.; Zhang, W.; Viollet, B.; Zhu, Y.; Zou, M.H. Aberrant endoplasmic reticulum stress in vascular smooth muscle increases vascular contractility and blood pressure in mice deficient of AMP-activated protein kinase-α2 in vivo. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 595–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitler, K.M.; Webb, R.C. Endoplasmic reticulum stress contributes to aortic stiffening via proapoptotic and fibrotic signaling mechanisms. Hypertension 2014, 63, e40–e45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayanagi, T.; Kawai, T.; Forrester, S.J.; Obama, T.; Tsuji, T.; Fukuda, Y.; Elliott, K.J.; Tilley, D.G.; Davisson, R.L.; Park, J.Y.; et al. Role of epidermal growth factor receptor and endoplasmic reticulum stress in vascular remodeling induced by angiotensin II. Hypertension 2015, 65, 1349–1355. [Google Scholar] [CrossRef]
- Ayyadevara, S.; Mercanti, F.; Wang, X.; Mackintosh, S.G.; Tackett, A.J.; Prayaga, S.V.; Romeo, F.; Shmookler Reis, R.J.; Mehta, J.L. Age- and Hypertension-Associated Protein Aggregates in Mouse Heart Have Similar Proteomic Profiles. Hypertension 2016, 67, 1006–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Guo, X.; Lei, P.; Shi, S.; Luo, S.; Cheng, X. PI3K/Akt/uncoupling protein 2 signaling pathway may be involved in cell senescence and apoptosis induced by angiotensin II in human vascular endothelial cells. Mol. Biol. Rep. 2014, 41, 6931–6937. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Mistriotis, P.; Andreadis, S.T. Vascular aging: Molecular mechanisms and potential treatments for vascular rejuvenation. Ageing Res. Rev. 2017, 37, 94–116. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Cooper, H.A.; Scalia, R.; Rizzo, V.; Eguchi, S. Angiotensin II- and Alzheimer-Type Cardiovascular Aging. Circ. Res. 2018, 123, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Youn, S.W.; Sudhahar, V.; Das, A.; Chandhri, R.; Cuervo Grajal, H.; Kweon, J.; Leanhart, S.; He, L.; Toth, P.T.; et al. Redox Regulation of Mitochondrial Fission Protein Drp1 by Protein Disulfide Isomerase Limits Endothelial Senescence. Cell Rep. 2018, 23, 3565–3578. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.; Gallo, G. To mdivi-1 or not to mdivi-1: Is that the question? Dev. Neurobiol. 2017, 77, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Salazar, G. NADPH Oxidases and Mitochondria in Vascular Senescence. Int. J. Mol. Sci. 2018, 19, 1327. [Google Scholar] [CrossRef] [Green Version]
- Prattichizzo, F.; Bonafe, M.; Ceka, A.; Giuliani, A.; Rippo, M.R.; Re, M.; Antonicelli, R.; Procopio, A.D.; Olivieri, F. Endothelial Cell Senescence and Inflammaging: MicroRNAs as Biomarkers and Innovative Therapeutic Tools. Curr. Drug Targets 2016, 17, 388–397. [Google Scholar] [CrossRef]
- Kotla, S.; Le, N.T.; Vu, H.T.; Ko, K.A.; Gi, Y.J.; Thomas, T.N.; Giancursio, C.; Lusis, A.J.; Cooke, J.P.; Fujiwara, K.; et al. Endothelial senescence-associated secretory phenotype (SASP) is regulated by Makorin-1 ubiquitin E3 ligase. Metabolism 2019, 100, 153962. [Google Scholar] [CrossRef]
- Civelek, M.; Manduchi, E.; Riley, R.J.; Stoeckert, C.J., Jr.; Davies, P.F. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ. Res. 2009, 105, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Chen, B.; Xiao, F.Q.; Wu, Y.T.; Wang, R.H.; Sun, Z.W.; Fu, G.S.; Mou, Y.; Tao, W.; Hu, X.S.; et al. Autophagy protects against senescence and apoptosis via the RAS-mitochondria in high-glucose-induced endothelial cells. Cell. Physiol. Biochem. 2014, 33, 1058–1074. [Google Scholar] [CrossRef]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, A.; Shimauchi, T.; Tanaka, T.; Shimoda, K.; Toyama, T.; Kitajima, N.; Ishikawa, T.; Shindo, N.; Numaga-Tomita, T.; Yasuda, S.; et al. Hypoxia-induced interaction of filamin with Drp1 causes mitochondrial hyperfission-associated myocardial senescence. Sci. Signal. 2018, 11, eaat5185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Xiao, C.; Long, F.; Wu, W.; Huang, M.; Qu, L.; Liu, X.; Zhu, Y. Fra-1 plays a critical role in angiotensin II-induced vascular senescence. FASEB J. 2019, 33, 7603–7614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, S.J.; Preston, K.J.; Cooper, H.A.; Boyer, M.J.; Escoto, K.M.; Poltronetti, A.J.; Elliott, K.J.; Kuroda, R.; Miyao, M.; Sesaki, H.; et al. Mitochondrial fission mediates endothelial inflammation. Hypertension 2020, in press. [Google Scholar]
- Cooper, H.A.; Cicalese, S.; Preston, K.J.; Kawai, T.; Okuno, K.; Choi, E.T.; Kasahara, K.; Uchida, H.A.; Otaka, N.; Scalia, R.; et al. Targeting mitochondrial fission as a potential therapeutic for abdominal aortic aneurysm. Cardiovasc. Res. 2020, in press. [Google Scholar]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyao, M.; Cicalese, S.; Kawai, T.; Cooper, H.A.; Boyer, M.J.; Elliott, K.J.; Forrester, S.J.; Kuroda, R.; Rizzo, V.; Hashimoto, T.; et al. Involvement of Senescence and Mitochondrial Fission in Endothelial Cell Pro-Inflammatory Phenotype Induced by Angiotensin II. Int. J. Mol. Sci. 2020, 21, 3112. https://doi.org/10.3390/ijms21093112
Miyao M, Cicalese S, Kawai T, Cooper HA, Boyer MJ, Elliott KJ, Forrester SJ, Kuroda R, Rizzo V, Hashimoto T, et al. Involvement of Senescence and Mitochondrial Fission in Endothelial Cell Pro-Inflammatory Phenotype Induced by Angiotensin II. International Journal of Molecular Sciences. 2020; 21(9):3112. https://doi.org/10.3390/ijms21093112
Chicago/Turabian StyleMiyao, Masashi, Stephanie Cicalese, Tatsuo Kawai, Hannah A. Cooper, Michael J. Boyer, Katherine J. Elliott, Steven J. Forrester, Ryohei Kuroda, Victor Rizzo, Tomoki Hashimoto, and et al. 2020. "Involvement of Senescence and Mitochondrial Fission in Endothelial Cell Pro-Inflammatory Phenotype Induced by Angiotensin II" International Journal of Molecular Sciences 21, no. 9: 3112. https://doi.org/10.3390/ijms21093112
APA StyleMiyao, M., Cicalese, S., Kawai, T., Cooper, H. A., Boyer, M. J., Elliott, K. J., Forrester, S. J., Kuroda, R., Rizzo, V., Hashimoto, T., Scalia, R., & Eguchi, S. (2020). Involvement of Senescence and Mitochondrial Fission in Endothelial Cell Pro-Inflammatory Phenotype Induced by Angiotensin II. International Journal of Molecular Sciences, 21(9), 3112. https://doi.org/10.3390/ijms21093112