Investigating Programmed Cell Death and Tumor Invasion in a Three-Dimensional (3D) Microfluidic Model of Glioblastoma




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
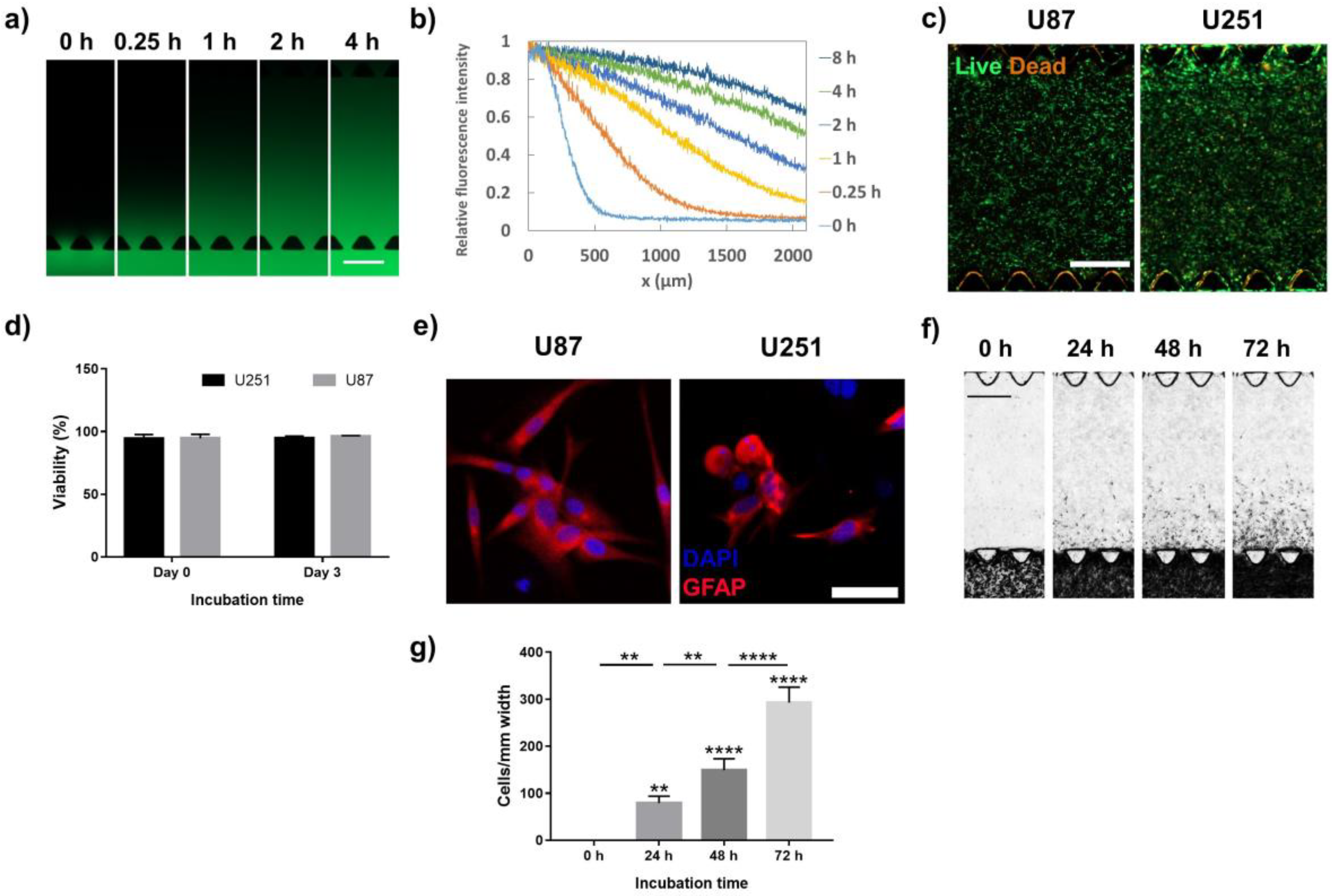
2.1. Development and Analysis of Glioblastoma-on-a-Chip (GoC) Model
2.2. Chemotherapy Treatment: Analysis of Programmed Cell Death and Invasion
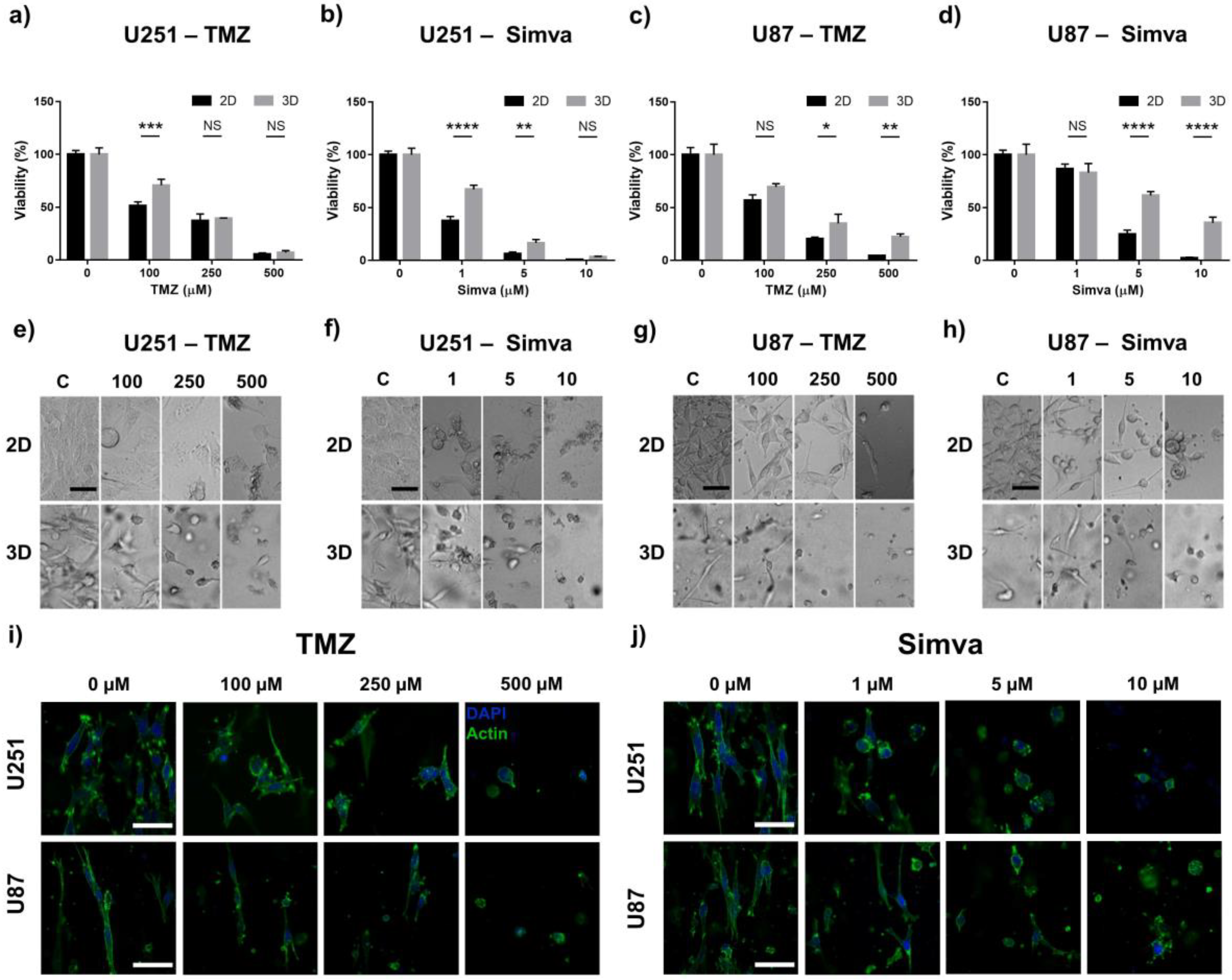
2.3. Drug Sensitivity Analysis of Cells in 3D GoC Model and 2D Cultures
2.4. Drug-Induced Apoptosis in 2D Culture System and GoC
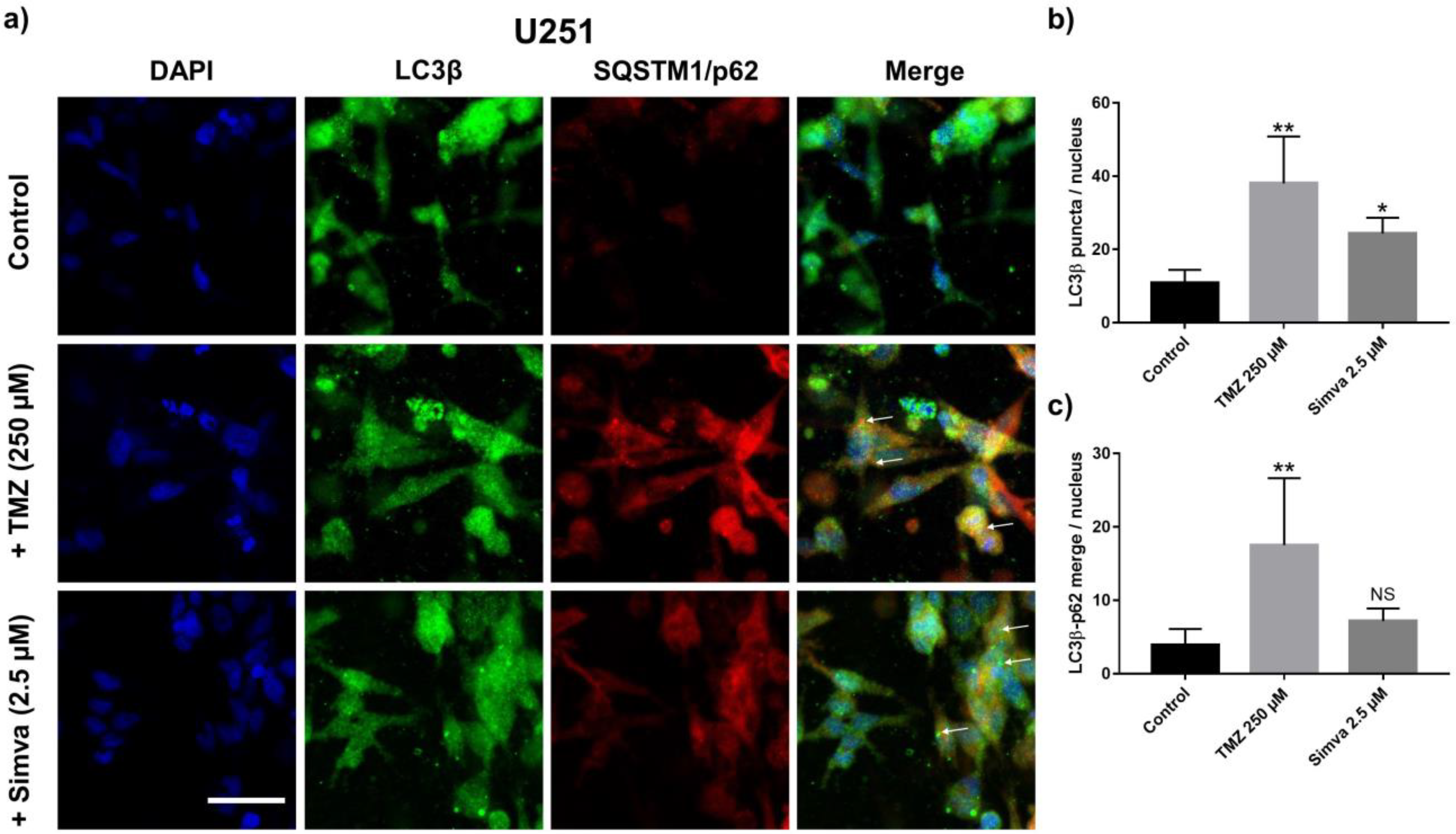
2.5. Effect of TMZ and Simva in the Autophagy Pathway and Its Effect in Their Apoptosis Induction
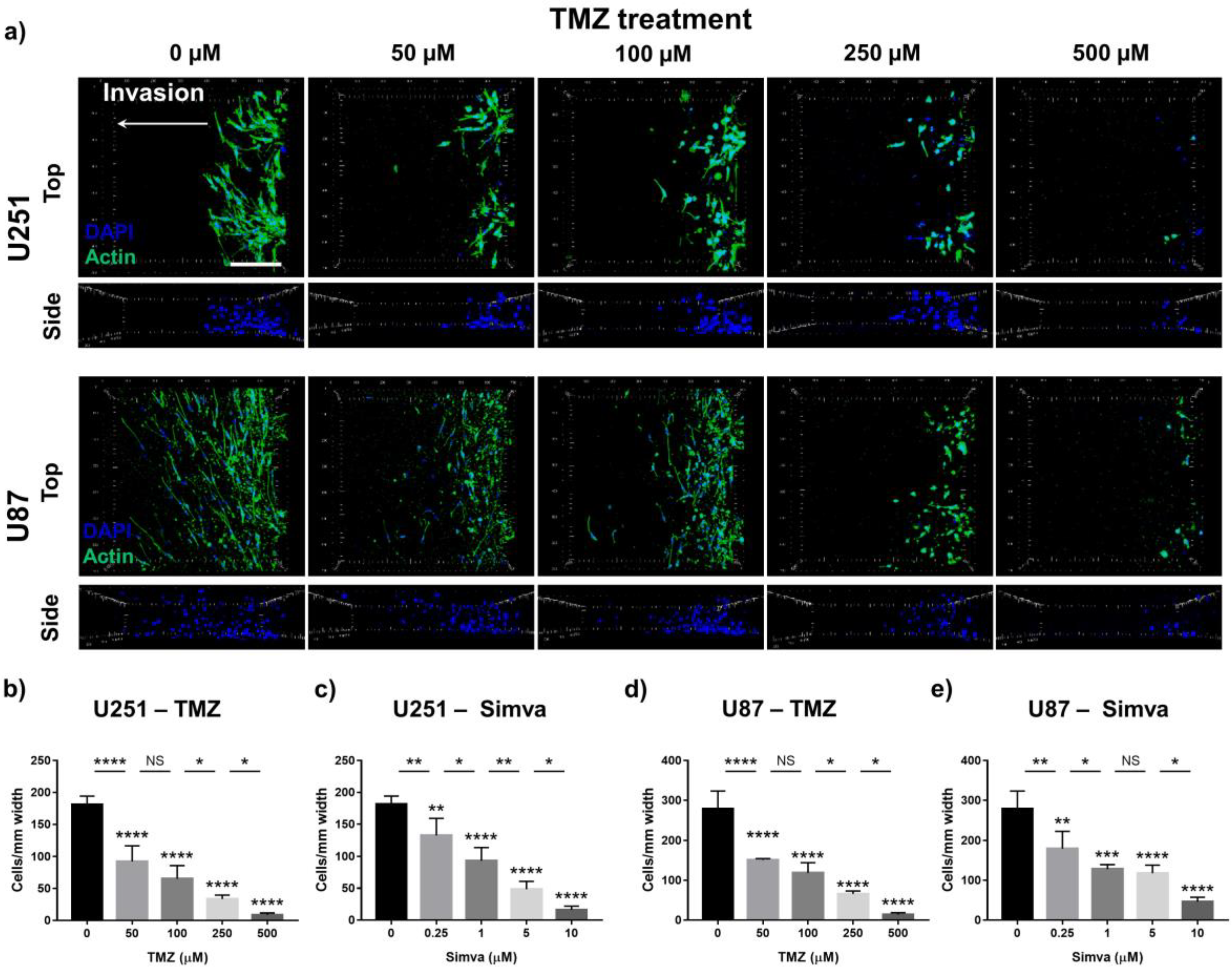
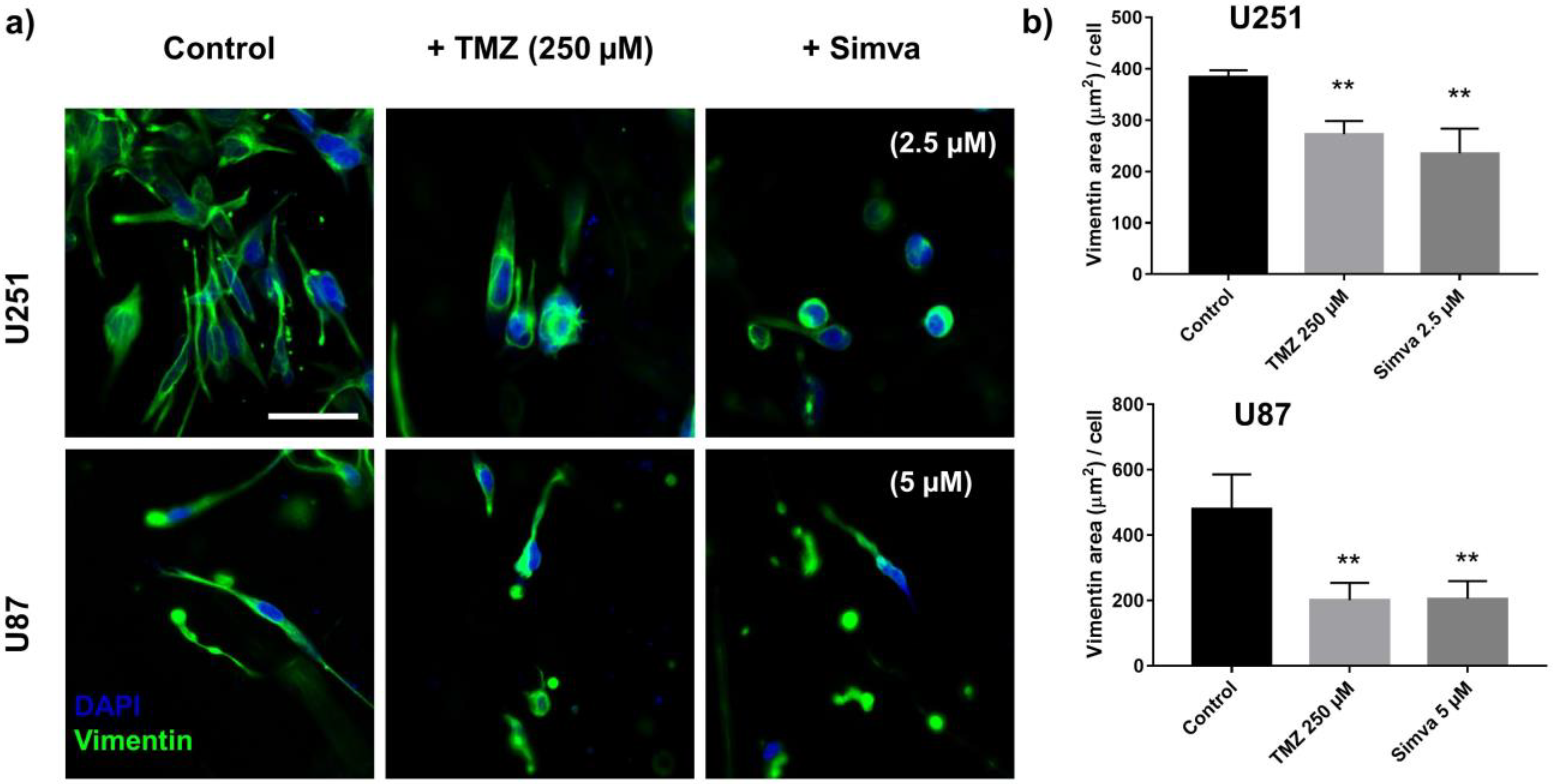
2.6. Effect of TMZ and Simva on Cell Invasion in GoC
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Microfluidic Device Fabrication
4.3. Monolayer Cell Culture
4.4. 3D Cell Culture
4.5. Cell Viability Analysis
4.6. Immunofluorescence Staining
4.7. Imaging
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2009–2013. Neuro Oncol. 2016, 18, v1–v75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, E.C. Glioblastoma multiforme: The terminator. Proc. Natl. Acad. Sci. USA 2000, 97, 6242–6244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Pitz, M.; Hamai, A.; Siemianowicz, K.; Likus, W.; Wiechec, E.; Toyota, B.D.; et al. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol. Ther. 2018, 184, 13–41. [Google Scholar] [CrossRef]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.J.; Yang, I.; Aghi, M.K.; Ohgaki, H.; Hau, P.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. Neuropathology 2005, 10, S26–S29. [Google Scholar]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin Use and Reduced Cancer-Related Mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassif, C.A.; Kratz, L.; Sparks, S.E.; Wheeler, C.; Bianconi, S.; Gropman, A.; Calis, K.A.; Kelley, R.I.; Tierney, E.; Porter, F.D. A placebo-controlled trial of simvastatin therapy in Smith-Lemli-Opitz syndrome. Genet. Med. 2017, 19, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Patel, Y.T.; Jacus, M.O.; Davis, A.D.; Boulos, N.; Turner, D.C.; Vuppala, P.K.; Freeman, B.B.; Gilbertson, R.J.; Stewart, C.F. Simvastatin Hydroxy Acid Fails to Attain Sufficient Central Nervous System Tumor Exposure to Achieve a Cytotoxic Effect: Results of a Preclinical Cerebral Microdialysis Study. Drug Metab. Dispos. 2016, 44, 591–594. [Google Scholar] [CrossRef] [Green Version]
- International, T.; Epidemiology, C.; Gaist, D. Statin use and survival following glioblastoma multiforme. Cancer Epidemiol. 2014, 38, 722–727. [Google Scholar]
- Ricci, M.S.; Zong, W. Chemotherapeutic Approaches for Targeting Cell Death Pathways. Oncologist 2006, 11, 342–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaitanya, G.V.; Alexander, J.S.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workman, P.; Collins, I. Corrigendum: New approaches to molecular cancer therapeutics. Nat. Chem. Biol. 2007, 3, 126. [Google Scholar]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef]
- William, W.N.; Heymach, J.V.; Kim, E.S.; Lippman, S.M. Molecular targets for cancer chemoprevention. Nat. Rev. Drug Discov. 2009, 8, 213–225. [Google Scholar] [CrossRef]
- Ferté, C.; André, F.; Soria, J.C. Molecular circuits of solid tumors: Prognostic and predictive tools for bedside use. Nat. Rev. Clin. Oncol. 2010, 7, 367–380. [Google Scholar] [CrossRef]
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef]
- Wistuba, I.I.; Gelovani, J.G.; Jacoby, J.J.; Davis, S.E.; Herbst, R.S. Methodological and practical challenges for personalized cancer therapies. Nat. Rev. Clin. Oncol. 2011, 8, 135–141. [Google Scholar] [CrossRef]
- Junttila, M.R.; De Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Bartlett, R.; Everett, W.; Lim, S.; Natasha, G.; Loizidou, M.; Jell, G.; Tan, A.; Seifalian, A.M. Personalized in vitro cancer modeling—Fantasy or reality? Transl. Oncol. 2014, 7, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Nyga, A.; Cheema, U.; Loizidou, M. 3D tumour models: Novel in vitro approaches to cancer studies. J. Cell Commun. Signal. 2011, 5, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Valente, K.P.; Khetani, S.; Kolahchi, A.R.; Sanati-Nezhad, A.; Suleman, A.; Akbari, M. Microfluidic technologies for anticancer drug studies. Drug Discov. Today 2017, 22, 1654–1670. [Google Scholar] [CrossRef]
- Annabi, N.; Tamayol, A.; Uquillas, J.A.; Akbari, M.; Bertassoni, L.E.; Cha, C.; Camci-Unal, G.; Dokmeci, M.R.; Peppas, N.A.; Khademhosseini, A. 25th anniversary article: Rational design and applications of hydrogels in regenerative medicine. Adv. Mater. 2014, 26, 85–124. [Google Scholar] [CrossRef] [PubMed]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslin, S.; O’Driscoll, L. Three-dimensional cell culture: The missing link in drug discovery. Drug Discov. Today 2013, 18, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Tekin, H.; Simmons, S.; Cummings, B.; Gao, L.; Adiconis, X.; Hession, C.C.; Ghoshal, A.; Dionne, D.; Choudhury, S.R.; Yesilyurt, V.; et al. Effects of 3D culturing conditions on the transcriptomic profile of stem-cell-derived neurons. Nat. Biomed. Eng. 2018, 2, 540–554. [Google Scholar] [CrossRef]
- Seyfoori, A.; Samiei, E.; Godau, B.; Jalili, N.; Rahmanian, M.; Farahmand, L.; Majidzadeh, K.; Akbari, M. Self-Filling Microwell Arrays (SFMAs) for Tumor Spheroid Formation. Lab Chip 2018, 18, 3516–3528. [Google Scholar] [CrossRef]
- Murphy, S.V.; Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 2014, 32, 773–785. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Bischel, L.L.; Young, E.W.K.; Mader, B.R.; Beebe, D.J. Tubeless microfluidic angiogenesis assay with three-dimensional endothelial-lined microvessels. Biomaterials 2013, 34, 1471–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, D.; Fiorelli, R.; Barrientos, E.S.; Melendez, E.L.; Sanai, N.; Mehta, S.; Nikkhah, M. A three-dimensional (3D) organotypic microfluidic model for glioma stem cells—Vascular interactions. Biomaterials 2019, 198, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.; Puleo, J.; Llave, A.; Mouneimne, G.; Kamm, R.D.; Nikkhah, M. Breast cancer cell invasion into a three dimensional tumor-stroma microenvironment. Sci. Rep. 2016, 6, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zervantonakisa, I.K.; Hughes-Alfor, S.K.; Charest, J.L.; Condeelis, J.S.; Gertler, F.B.; Kamm, R.D. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. USA 2012, 109, 13515–13520. [Google Scholar] [CrossRef] [Green Version]
- Ayuso, J.M.; Monge, R.; Martínez-González, A.; Virumbrales-Muñoz, M.; Llamazares, G.A.; Berganzo, J.; Hernández-Laín, A.; Santolaria, J.; Doblaré, M.; Hubert, C.; et al. Glioblastoma on a microfluidic chip: Generating pseudopalisades and enhancing aggressiveness through blood vessel obstruction events. Neuro Oncol. 2017, 19, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211.e5. [Google Scholar] [CrossRef] [Green Version]
- Kenig, S.; Alonso, M.B.D.; Mueller, M.M.; Lah, T.T. Glioblastoma and endothelial cells cross-talk, mediated by SDF-1, enhances tumour invasion and endothelial proliferation by increasing expression of cathepsins B, S, and MMP-9. Cancer Lett. 2010, 289, 53–61. [Google Scholar] [CrossRef]
- Demuth, T.; Berens, M.E. Molecular mechanisms of glioma cell migration and invasion. J. Neurooncol. 2004, 70, 217–228. [Google Scholar] [CrossRef]
- Shin, Y.; Han, S.; Jeon, J.S.; Yamamoto, K.; Zervantonakis, I.K.; Sudo, R.; Kamm, R.D.; Chung, S. Microfluidic assay for simultaneous culture of multiple cell types on surfaces or within hydrogels. Nat. Protoc. 2012, 7, 1247–1259. [Google Scholar] [CrossRef] [Green Version]
- Velpula, K.K.; Dasari, V.R.; Tsung, A.J.; Dzung, H.; Rao, J.S. Cord blood stem cells revert glioma stem cell EMT by down regulating transcriptional activation of Sox2 and Twist1. Oncotarget 2011, 2, 1028–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, J.H.; Park, S.J.; Jo, A.; Kang, J.L.; Jou, I. Caveolin-1 is involved in reactive oxygen species-induced SHP-2 activation in astrocytes. Exp. Mol. Med. 2011, 43, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, A.R.; da Silva Rosa, S.C.; Samiei, E.; Alizadeh, J.; Field, J.; Kawalec, P.; Thliveris, J.; Akbari, M.; Ghavami, S.; Gordon, J.W. Autophagy modulates temozolomide-induced cell death in alveolar Rhabdomyosarcoma cells. Cell Death Discov. 2018, 4, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emami, A.; Shojaei, S.; da Silva Rosa, S.C.; Aghaei, M.; Samiei, E.; Vosoughi, A.R.; Kalantari, F.; Kawalec, P.; Thliveris, J.; Sharma, P.; et al. Mechanisms of simvastatin myotoxicity: The role of autophagy flux inhibition. Eur. J. Pharmacol. 2019, 862, 172616. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Hashemi, M.; Ande, S.R.; Yeganeh, B.; Xiao, W.; Eshraghi, M.; Bus, C.J.; Kadkhoda, K.; Wiechec, E.; Halayko, A.J. Apoptosis and cancer: Mutations within caspase genes. J. Med. Genet. 2009, 46, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.J.; Lee, C.C.; Shih, Y.L.; Lin, T.Y.; Wang, S.H.; Lin, Y.F.; Shih, C.M. Resveratrol enhances the therapeutic effect of temozolomide against malignant glioma in vitro and in vivo by inhibiting autophagy. Free Radic. Biol. Med. 2012, 52, 377–391. [Google Scholar] [CrossRef]
- Yanae, M.; Tsubaki, M.; Satou, T.; Itoh, T.; Imano, M.; Yamazoe, Y.; Nishida, S. Statin-induced apoptosis via the suppression of ERK1/2 and Akt activation by inhibition of the geranylgeranyl-pyrophosphate biosynthesis in glioblastoma. J. Exp. Clin. Cancer Res. 2011, 30, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Proskuryakov, S.Y.; Konoplyannikov, A.G.; Gabai, V.L. Necrosis: A specific form of programmed cell death? Exp. Cell Res. 2003, 283, 1–16. [Google Scholar] [CrossRef]
- Porter, A.G.; Ja, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Lin, L.; Baehrecke, E.H. Autophagy, cell death, and cancer. Mol. Cell. Oncol. 2015, 2, e985913. [Google Scholar] [CrossRef] [Green Version]
- Ghavami, S.; Eshragi, M.; Ande, S.R.; Chazin, W.J.; Klonisch, T.; Halayko, A.J.; McNeill, K.D.; Hashemi, M.; Kerkhoff, C.; Los, M. S100A8/A9 induces autophagy and apoptosis via ROS-mediated cross-talk between mitochondria and lysosomes that involves BNIP3. Cell Res. 2010, 20, 314–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Tan, J.; Miao, Y.; Li, M.; Zhang, Q. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J. Cell. Physiol. 2017, 232, 2977–2984. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, M.I.; Vosoughi, A.R.; Albokashy, M.; Butterfield, Y.; et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, J.; Glogowska, A.; Thliveris, J.; Kalantari, F.; Shojaei, S.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 749–768. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- McGonigle, P.; Ruggeri, B. Animal models of human disease: Challenges in enabling translation. Biochem. Pharmacol. 2014, 87, 162–171. [Google Scholar] [CrossRef]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T.; et al. Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 2015, 33, 1837–1843. [Google Scholar] [CrossRef] [Green Version]
- Taubenberger, A.V.; Bray, L.J.; Haller, B.; Shaposhnykov, A.; Binner, M.; Freudenberg, U.; Guck, J.; Werner, C. 3D extracellular matrix interactions modulate tumour cell growth, invasion and angiogenesis in engineered tumour microenvironments. Acta Biomater. 2016, 36, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Wen, P.Y.; Reardon, D.A. Progress in glioma diagnosis, classification and treatment. Nat. Rev. Neurol. 2016, 12, 69–70. [Google Scholar] [CrossRef]
- Alizadeh, J.; Zeki, A.A.; Mirzaei, N.; Tewary, S.; Moghadam, A.R.; Glogowska, A.; Nagakannan, P.; Eftekharpour, E.; Wiechec, E.; Gordon, J.W.; et al. Mevalonate Cascade Inhibition by Simvastatin Induces the Intrinsic Apoptosis Pathway via Depletion of Isoprenoids in Tumor Cells. Sci. Rep. 2017, 7, 44841. [Google Scholar] [CrossRef]
- Pan, Q.; Wang, X.Y.H. Chemoresistance to Temozolomide in Human Glioma Cell Line U251 is Associated with Increased Activity of O 6 -methylguanine- DNA Methyltransferase and Can be Overcome by Metronomic Temozolomide Regimen. Cell Biochem. Biophys. 2012, 62, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Kubelt, C.; Hattermann, K.; Sebens, S.; Mehdorn, H.M.; Held-feindt, J. Epithelial-to-mesenchymal transition in paired human primary and recurrent glioblastomas. Int. J. Oncol. 2015, 46, 2515–2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.M.; Cukierman, E. Modeling Tissue Morphogenesis and Cancer in 3D. Cell 2007, 130, 601–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, W.P.; Batista, L.F.Z.; Naumann, S.C.; Wick, W.; Weller, M.; Menck, C.F.M.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O 6 -methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlak, E.; Damasceno, R.; Arnold, H.; Terzis, A.J.; Allee, R. Temozolomide induces apoptosis and senescence in glioma cells cultured as multicellular spheroids. Br. J. Cancer 2003, 88, 463–469. [Google Scholar]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Würstle, S.; Schneider, F.; Ringel, F.; Gempt, J.; Lämmer, F.; Delbridge, C.; Wu, W.E.I.; Schlegel, J.; Neuropathology, D.; München, T.U.; et al. Temozolomide induces autophagy in primary and established glioblastoma cells in an EGFR independent manner. Oncol. Lett. 2017, 14, 322–328. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Verity, M.A.; Reue, K. Lipin-1 regulates autophagy clearance and intersects with statin drug effects in skeletal muscle. Cell Metab. 2014, 20, 267–279. [Google Scholar] [CrossRef] [Green Version]
- Su, F.; Shi, M.; Zhang, J.; Zheng, Q.; Zhang, D.; Zhang, W.; Wang, H.; Li, X. Simvastatin Protects Heart from Pressure Overload Injury by Inhibiting Excessive Autophagy. Int. J. Med. Sci. 2018, 15, 1508–1516. [Google Scholar] [CrossRef] [Green Version]
- Loos, B.; Du Toit, A.; Hofmeyr, J.H.S. Defining and measuring autophagosome flux—Concept and reality. Autophagy 2014, 10, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in Mammalian Autophagy Research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Xu, Z.; Dai, S.; Qian, L.; Sun, L.; Gong, Z. Targeting autophagy to sensitive glioma to temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguemgo Kouam, P.; Rezniczek, G.A.; Kochanneck, A.; Priesch-Grzeszkowiak, B.; Hero, T.; Adamietz, I.A.; Bühler, H. Robo1 and vimentin regulate radiation- induced motility of human glioblastoma cells. PLoS ONE 2018, 13, e0198508. [Google Scholar] [CrossRef] [Green Version]
- Ghavami, S.; Yeganeh, B.; Stelmack, G.L.; Kashani, H.H.; Sharma, P.; Cunnington, R.; Rattan, S.; Bathe, K.; Klonisch, T.; Dixon, I.M.C.; et al. Apoptosis, autophagy and ER stress in mevalonate cascade inhibition-induced cell death of human atrial fibroblasts. Cell Death Dis. 2012, 3, e330. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samiei, E.; Seyfoori, A.; Toyota, B.; Ghavami, S.; Akbari, M. Investigating Programmed Cell Death and Tumor Invasion in a Three-Dimensional (3D) Microfluidic Model of Glioblastoma. Int. J. Mol. Sci. 2020, 21, 3162. https://doi.org/10.3390/ijms21093162
Samiei E, Seyfoori A, Toyota B, Ghavami S, Akbari M. Investigating Programmed Cell Death and Tumor Invasion in a Three-Dimensional (3D) Microfluidic Model of Glioblastoma. International Journal of Molecular Sciences. 2020; 21(9):3162. https://doi.org/10.3390/ijms21093162
Chicago/Turabian StyleSamiei, Ehsan, Amir Seyfoori, Brian Toyota, Saeid Ghavami, and Mohsen Akbari. 2020. "Investigating Programmed Cell Death and Tumor Invasion in a Three-Dimensional (3D) Microfluidic Model of Glioblastoma" International Journal of Molecular Sciences 21, no. 9: 3162. https://doi.org/10.3390/ijms21093162
APA StyleSamiei, E., Seyfoori, A., Toyota, B., Ghavami, S., & Akbari, M. (2020). Investigating Programmed Cell Death and Tumor Invasion in a Three-Dimensional (3D) Microfluidic Model of Glioblastoma. International Journal of Molecular Sciences, 21(9), 3162. https://doi.org/10.3390/ijms21093162