Shared Genomic and Proteomic Contribution of Amyloid and Tau Protein Characteristic of Alzheimer’s Disease to Brain Ischemia




Abstract
:1. Introduction
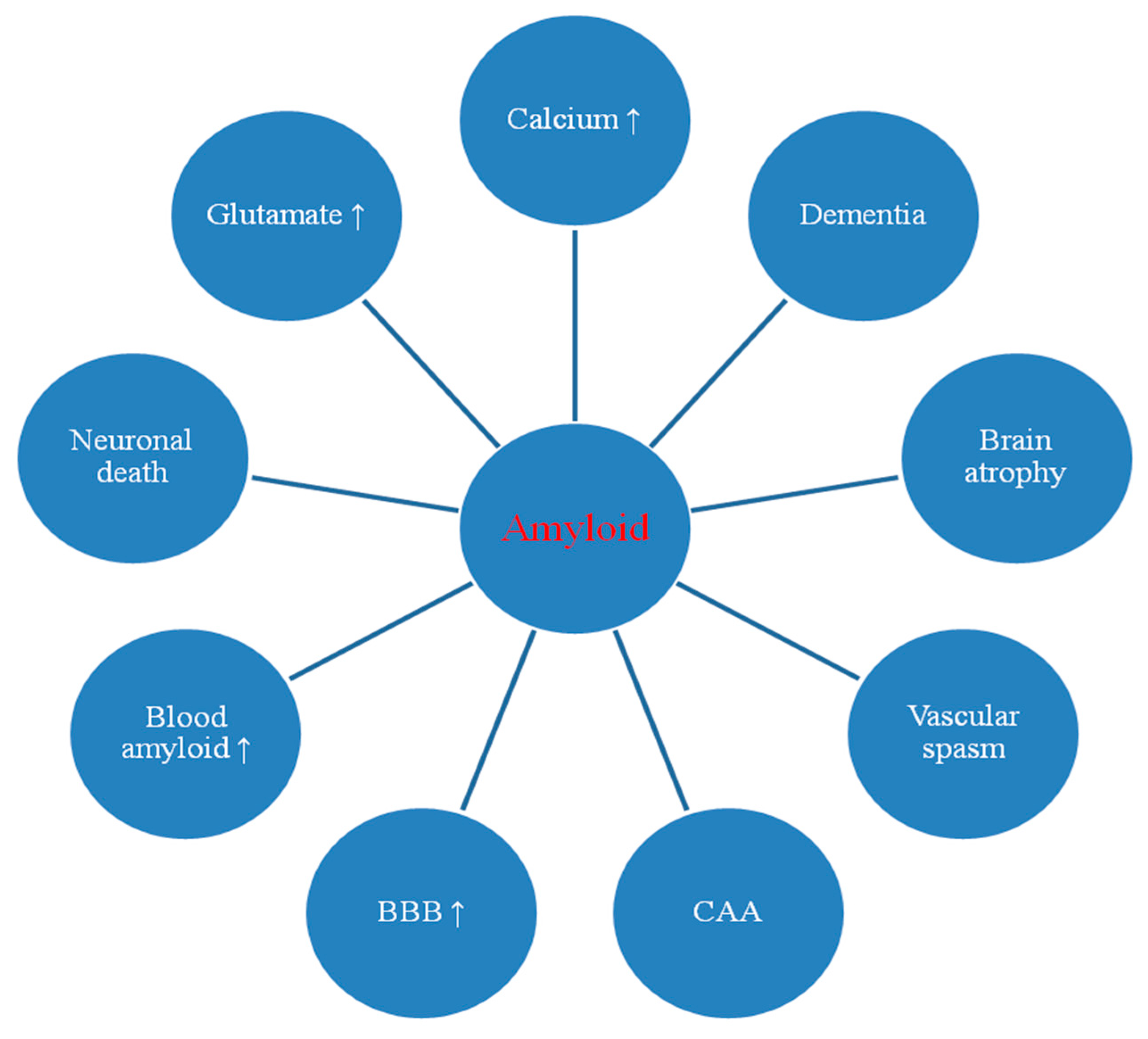
2. Amyloid in Post-Ischemic Brain
2.1. Dysregulation of Amyloid Associated Genes
2.2. Dysregulation of Amyloid Associated mRNAs
2.3. Changes in Amyloid Staining in Animal and Human Brain
2.4. Blood-Brain Barrier and Amyloid in the Blood
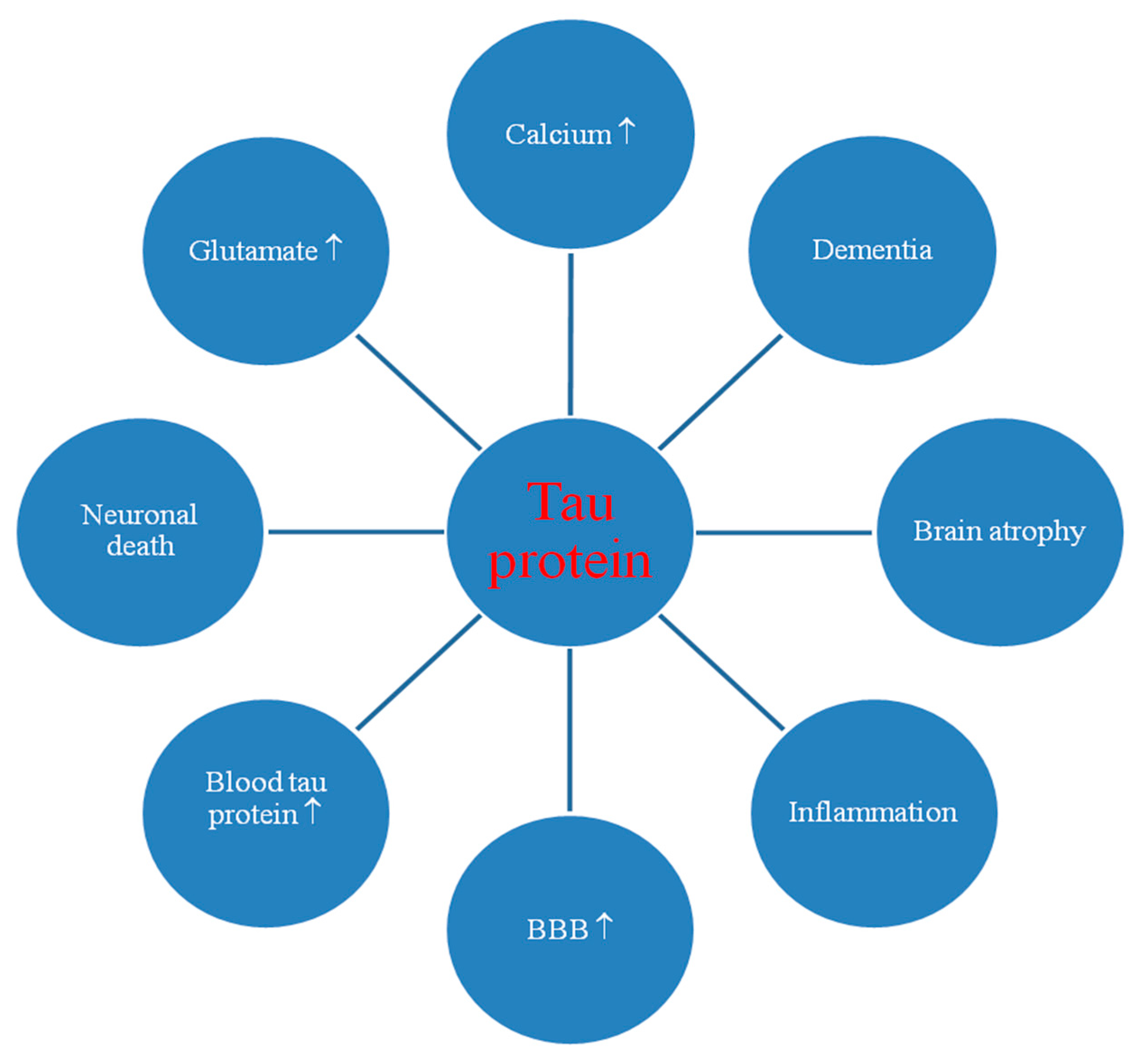
3. Tau Protein in Post-Ischemic Brain
3.1. Dysregulation of the Tau Protein Gene
3.2. Changes in Tau Protein Staining in Animal and Human Brain
3.3. Blood-Brain Barrier and Tau Protein in the Blood and Cerebrospinal Fluid
3.4. Tau Protein Hyperphosphorylation
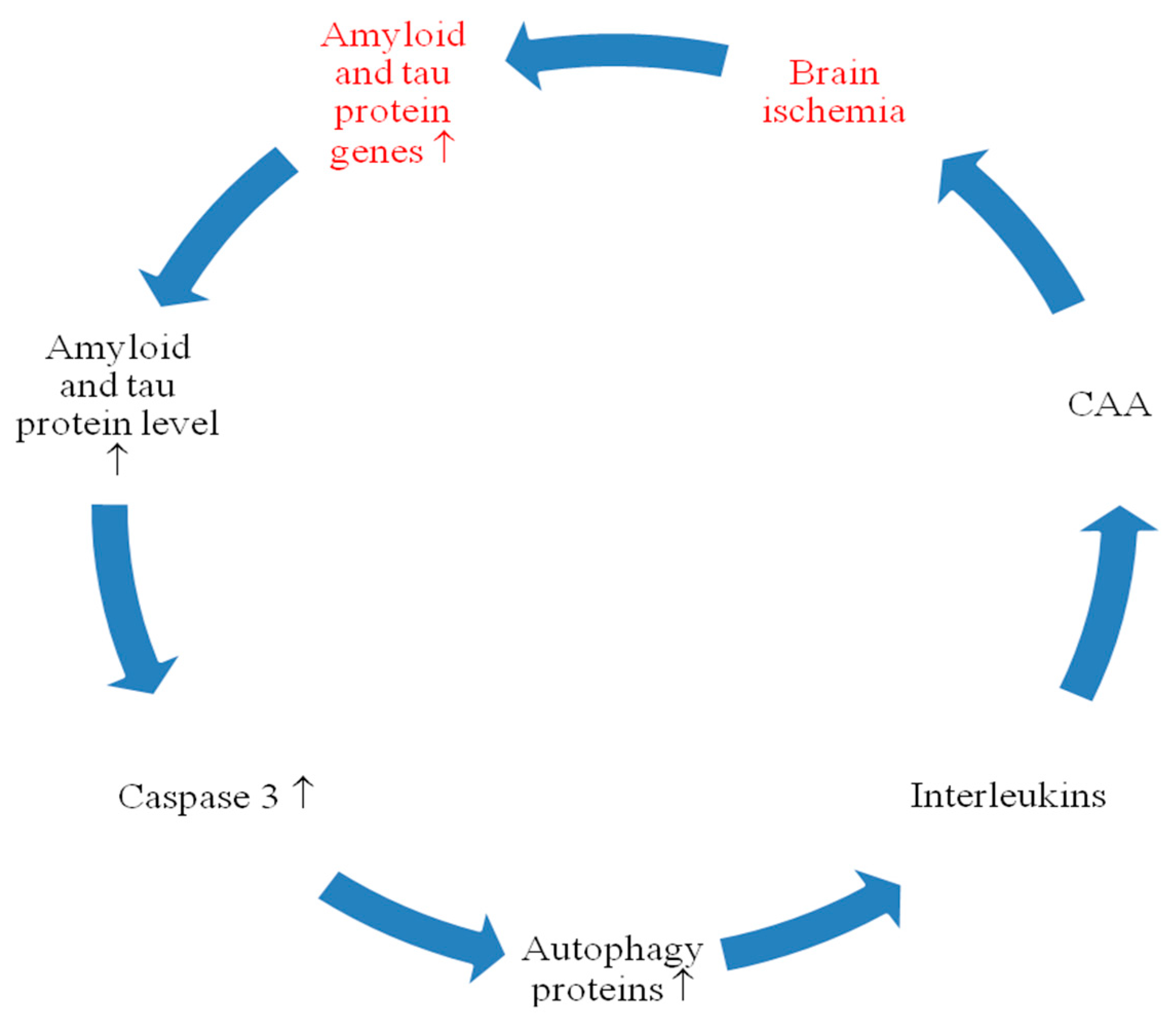
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pluta, R. The role of apolipoprotein E in the deposition of β-amyloid peptide during ischemia–reperfusion brain injury. A model of early Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2000, 903, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Ułamek, M.; Jabłoński, M. Alzheimer’s mechanisms in ischemic brain degeneration. Anat. Rec. 2009, 292, 1863–1881. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Tau protein dysfunction after brain ischemia. J. Alzheimer’s Dis. 2018, 66, 429–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Januszewski, S.; Pluta, R. Substantiation for the use of curcumin during the development of neurodegeneration after brain ischemia. Int. J. Mol. Sci. 2020, 21, 517. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R.; Salińska, E.; Puka, M.; Stafiej, A.; Łazarewicz, J.W. Early changes in extracellular amino acids and calcium concentrations in rabbit hippocampus following complete 15-min cerebral ischemia. Resuscitation 1988, 16, 193–210. [Google Scholar] [CrossRef]
- Ni, J.W.; Matsumoto, K.; Li, H.B.; Murakami, Y.; Watanabe, H. Neuronal damage and decrease of central acetylcholine level following permanent occlusion of bilateral common carotid arteries in rat. Brain Res. 1995, 673, 290–296. [Google Scholar] [CrossRef]
- Neumann, J.T.; Cohan, C.H.; Dave, K.R.; Wright, C.B.; Perez-Pinzon, M.A. Global cerebral ischemia: Synaptic and cognitive dysfunction. Curr. Drug Targets 2013, 14, 20–35. [Google Scholar] [CrossRef]
- Ruan, Y.W.; Han, X.J.; Shi, Z.S.; Lei, Z.G.; Xu, Z.C. Remodeling of synapses in the CA1 area of the hippocampus after transient global ischemia. Neuroscience 2012, 218, 268–277. [Google Scholar] [CrossRef]
- Zhao, Y.; Gu, J.H.; Dai, C.L.; Liu, Q.; Iqbal, K.; Liu, F.; Gong, C.X. Chronic cerebral hypoperfusion causes decrease of O-GlcNAcylation, hyperphosphorylation of tau and behavioral deficits in mice. Front. Aging Neurosci. 2014, 6, 10. [Google Scholar] [CrossRef]
- Hofmeijer, J.; van Putten, M.J. Ischemic cerebral damage: An appraisal of synaptic failure. Stroke 2012, 43, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Ułamek-Kozioł, M.; Kocki, J.; Bogucka-Kocka, A.; Januszewski, S.; Bogucki, J.; Czuczwar, S.J.; Pluta, R. Autophagy, mitophagy and apoptotic gene changes in the hippocampal CA1 area in a rat ischemic model of Alzheimer’s disease. Pharmacol. Rep. 2017, 69, 1289–1294. [Google Scholar] [CrossRef] [PubMed]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Kocki, J.; Januszewski, S.; Bogucki, J.; Bogucka-Kocka, A.; Pluta, R. Dysregulation of autophagy, mitophagy, and apoptosis genes in the CA3 region of the hippocampus in the ischemic model of Alzheimer’s disease in the rat. J. Alzheimers Dis. 2019, 72, 1279–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curcio, M.; Salazar, I.L.; Mele, M.; Canzoniero, L.M.; Duarte, C.B. Calpains and neuronal damage in the ischemic brain: The swiss knife in synaptic injury. Prog. Neurobiol. 2016, 143, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, W.D.; Kraydieh, S.; Prado, R.; Stagliano, N.E. White matter alterations following thromboembolic stroke: A β-amyloid precursor protein immunocytochemical study in rats. Acta Neuropathol. 1998, 95, 524–531. [Google Scholar] [CrossRef]
- Orzyłowska, O.; Oderfeld-Nowak, B.; Zaremba, M.; Januszewski, S.; Mossakowski, M.J. Prolonged and concomitant induction of astroglial immunoreactivity of interleukin-1 beta and interleukin-6 in the rat hippocampus after transient global ischemia. Neurosci. Lett. 1999, 263, 72–76. [Google Scholar] [CrossRef]
- Fernando, M.S.; Simpson, J.E.; Matthews, F.; Brayne, C.; Lewis, C.E.; Barber, R.; Kalaria, R.N.; Forster, G.; Esteves, F.; Wharton, S.B.; et al. White matter lesions in an unselected cohort of the elderly: Molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 2006, 37, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R.; Ułamek, M.; Januszewski, S. Micro-blood–brain barrier openings and cytotoxic fragments of amyloid precursor protein accumulation in white matter after ischemic brain injury in long-lived rats. Acta Neurochir. 2006, 96, 267–271. [Google Scholar]
- Pluta, R.; Januszewski, S.; Ułamek, M. Ischemic blood–brain barrier and amyloid in white matter as etiological factors in leukoaraiosis. Acta Neurochir. 2008, 102, 353–356. [Google Scholar]
- Scherr, M.; Trinka, E.; McCoy, M.; Krenn, Y.; Staffen, W.; Kirschner, M.; Bergmann, H.J.; Mutzenbach, J.S. Cerebral hypoperfusion during carotid artery stenosis can lead to cognitive deficits that may be independent of white matter lesion load. Curr. Neurovasc. Res. 2012, 9, 193–199. [Google Scholar] [CrossRef]
- Sekeljic, V.; Bataveljic, D.; Stamenkovic, S.; Ułamek, M.; Jabłoński, M.; Radenovic, L.; Pluta, R.; Andjus, P.R. Cellular markers of neuroinflammation and neurogenesis after ischemic brain injury in the long-term survival rat model. Brain Struct. Funct. 2012, 217, 411–420. [Google Scholar] [CrossRef]
- De Schotten, M.T.; Tomaiuolo, F.; Aiello, M.; Merola, S.; Silvetti, M.; Lecce, F.; Bartolomeo, P.; Doricchi, F. Damage to white matter pathways in subacute and chronic spatial neglect, a group study and 2 single-case studies with complete virtual “in, vivo” tractography dissection. Cereb. Cortex 2014, 24, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, S.; Andrzejewska, A.; Lukomska, B.; Jankowski, M. Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. J. Neuroinflamm. 2019, 16, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radenovic, L.; Nenadic, M.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J.; Andjus, P.R.; Pluta, R. Heterogeneity in brain distribution of activated microglia and astrocytes in a rat ischemic model of Alzheimer’s disease after 2 years of survival. Aging 2020, in press. [Google Scholar]
- Wakita, H.; Tomimoto, H.; Akiguchi, I.; Kimura, J. Glial activation and white matter changes in the rat brain induced by chronic cerebral hypoperfusion: An immunohistochemical study. Acta Neuropathol. 1994, 87, 484–492. [Google Scholar] [CrossRef]
- Yoshizaki, K.; Adachi, K.; Kataoka, S.; Watanabe, A.; Tabira, T.; Takahashi, K.; Wakita, H. Chronic cerebral hypoperfusion induced by right unilateral common carotid artery occlusion causes delayed white matter lesions and cognitive impairment in adult mice. Exp. Neurol. 2008, 210, 585–591. [Google Scholar]
- Jabłoński, M.; Maciejewski, R.; Januszewski, S.; Ułamek, M.; Pluta, R. One year follow up in ischemic brain injury and the role of Alzheimer factors. Physiol. Res. 2011, 60 (Suppl. 1), 113–119. [Google Scholar]
- Hossmann, K.A.; Schmidt-Kastner, R.; Ophoff, B.G. Recovery of integrative central nervous function after one hour global cerebro-circulatory arrest in normothermic cat. J. Neurol. Sci. 1987, 77, 305–320. [Google Scholar] [CrossRef]
- De la Tremblaye, P.B.; Plamondon, H. Impaired conditioned emotional response and object recognition are concomitant to neuronal damage in the amygdale and perirhinal cortex in middle-aged ischemic rats. Behav. Brain Res. 2011, 219, 227–233. [Google Scholar] [CrossRef]
- Kiryk, A.; Pluta, R.; Figiel, I.; Mikosz, M.; Ułamek, M.; Niewiadomska, G.; Jabłoński, M.; Kaczmarek, L. Transient brain ischemia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav. Brain Res. 2011, 219, 1–7. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.J.; Zhang, M.; Fang, C.Q.; Zhou, H.D. Cerebral ischemia aggravates cognitive impairment in a rat model of Alzheimer’s disease. Life Sci. 2011, 89, 86–92. [Google Scholar] [CrossRef]
- Cohan, C.H.; Neumann, J.T.; Dave, K.R.; Alekseyenko, A.; Binkert, M.; Stransky, K.; Lin, H.W.; Barnes, C.A.; Wright, C.B.; Perez-Pinzon, M.A. Effect of cardiac arrest on cognitive impairment and hippocampal plasticity in middle-aged rats. PLoS ONE 2015, 10, e0124918. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wong, A.; Au, L.; Yang, J.; Wang, Z.; Leung, E.Y.; Chen, S.; Ho, C.L.; Mok, V.C. Influence of amyloid-beta on cognitive decline after stroke/transient ischemic attack: Three-year longitudinal study. Stroke 2015, 46, 3074–3080. [Google Scholar] [PubMed] [Green Version]
- Pluta, R. Ischemia-Reperfusion Pathways in Alzheimer’s Disease; Nova, Science Publishers, Inc.: New York, NY, USA, 2007. [Google Scholar]
- Pluta, R.; Furmaga-Jabłońska, W.; Maciejewski, R.; Ułamek-Kozioł, M.; Jabłoński, M. Brain ischemia activates β- and γ-secretase cleavage of amyloid precursor protein: Significance in sporadic Alzheimer’s disease. Mol. Neurobiol. 2013, 47, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluta, R.; Jabłoński, M.; Ułamek-Kozioł, M.; Kocki, J.; Brzozowska, J.; Januszewski, S.; Furmaga-Jabłońska, W.; Bogucka-Kocka, A.; Maciejewski, R.; Czuczwar, S.J. Sporadic, Alzheimer’s disease begins as episodes of brain ischemia and ischemically dysregulated Alzheimer’s disease genes. Mol. Neurobiol. 2013, 48, 500–515. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Hypoxia/ischemia activate processing of amyloid precursor protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2017, 140, 536–549. [Google Scholar]
- Pluta, R. Brain Ischemia: Alzheimer’s Disease Mechanisms; Nova, Science Publishers, Inc.: New York, NY, USA, 2019; p. 311. [Google Scholar]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Common proteomic and genomic contribution to ischemic brain damage and Alzheimer’s disease. In Alzheimer’s Disease; Wisniewski, T., Ed.; Codon Publications: Brisbane, Australia, 2019; pp. 53–68. [Google Scholar]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S. Amyloid pathology in the brain after ischemia. Folia Neuropathol. 2019, 57, 220–226. [Google Scholar]
- Pluta, R.; Ułamek-Kozioł, M. The role of degenerative pathways in the development of irreversible consequences after brain ischemia. Neural Regen. Res. 2019, 14, 982–983. [Google Scholar] [CrossRef]
- Pluta, R.; Lossinsky, A.S.; Walski, M.; Wiśniewski, H.M.; Mossakowski, M.J. Platelet occlusion phenomenon after short- and long-term survival following complete cerebral ischemia in rats produced by cardiac arrest. J. Brain Res. 1994, 35, 463–471. [Google Scholar]
- Pluta, R.; Lossinsky, A.S.; Wiśniewski, H.M.; Mossakowski, M.J. Early blood–brain barrier changes in the rat following transient complete cerebral ischemia induced by cardiac arrest. Brain Res. 1994, 633, 41–52. [Google Scholar] [CrossRef]
- Pluta, R.; Kida, E.; Lossinsky, A.S.; Golabek, A.A.; Mossakowski, M.J.; Wisniewski, H.M. Complete cerebral ischemia with short-term survival in rats induced by cardiac arrest. I. Extracellular accumulation of Alzheimer’s β-amyloid protein precursor in the brain. Brain Res. 1994, 649, 323–328. [Google Scholar]
- Wisniewski, H.M.; Pluta, R.; Lossinsky, A.S.; Mossakowski, M.J. Ultrastructural studies of cerebral vascular spasm after cardiac arrest-related global cerebral ischemia in rats. Acta Neuropathol. 1995, 90, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Barcikowska, M.; Januszewski, S.; Misicka, A.; Lipkowski, A.W. Evidence of blood–brain barrier permeability/leakage for circulating human Alzheimer’s β-amyloid-(1–42)-peptide. Neuroreport 1996, 7, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Barcikowska, M.; Misicka, A.; Lipkowski, A.W.; Spisacka, S.; Januszewski, S. Ischemic rats as a model in the study of the neurobiological role of human β-amyloid peptide. Time-dependent disappearing diffuse amyloid plaques in brain. Neuroreport 1999, 10, 3615–3619. [Google Scholar] [PubMed]
- Pluta, R.; Misicka, A.; Barcikowska, M.; Spisacka, S.; Lipkowski, A.W.; Januszewski, S. Possible reverse transport of β-amyloid peptide across the blood-brain barrier. Acta Neurochir. 2000, 76, 73–77. [Google Scholar]
- Pluta, R. Blood–brain barrier dysfunction and amyloid precursor protein accumulation in microvascular compartment following ischemia–reperfusion brain injury with 1-year survival. Acta Neurochir. 2003, 86, 117–122. [Google Scholar]
- Anfuso, C.D.; Assero, G.; Lupo, G.; Nicota, A.; Cannavo, G.; Strosznajder, R.P.; Rapisarda, P.; Pluta, R.; Alberghia, M. Amyloid beta(1-42) and its beta(25-35) fragment induce activation and membrane translocation of cytosolic phospholipase A(2) in bovine retina capillary pericytes. Biochim. Biophys. Acta 2004, 1686, 125–138. [Google Scholar]
- Lee, P.H.; Bang, O.Y.; Hwang, E.M.; Lee, J.S.; Joo, U.S.; Mook-Jung, I.; Huh, K. Circulating beta amyloid protein is elevated in patients with acute ischemic stroke. J. Neural Transm. 2005, 112, 1371–1379. [Google Scholar]
- Pluta, R. Pathological opening of the blood–brain barrier to horseradish peroxidase and amyloid precursor protein following ischemia–reperfusion brain injury. Chemotherapy 2005, 51, 223–226. [Google Scholar] [CrossRef]
- Pluta, R.; Januszewski, S.; Jabłoński, M.; Ułamek, M. Factors in creepy delayed neuronal death in hippocampus following brain ischemia-reperfusion injury with long-term survival. Acta Neurochir. 2010, 106, 37–41. [Google Scholar]
- Mörtberg, E.; Zetterberg, H.; Nordmark, J.; Blennow, K.; Catry, C.; Decraemer, H.; Vanmechelen, E.; Rubertsson, S. Plasma tau protein in comatose patients after cardiac arrest treated with therapeutic hypothermia. Acta Anaesthesiol. Scand. 2011, 55, 1132–1138. [Google Scholar]
- Zetterberg, H.; Mörtberg, E.; Song, L.; Chang, L.; Provuncher, G.K.; Patel, P.P.; Ferrell, E.; Fournier, D.R.; Kan, C.W.; Campbell, T.G.; et al. Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid β levels in humans. PLoS ONE 2011, 6, e28263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randall, J.; Mörtberg, E.; Provuncher, G.K.; Fournier, D.R.; Duffy, D.C.; Rubertsson, S.; Blennow, K.; Zetterberg, H.; Wilson, D.H. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: Results of a pilot study. Resuscitation 2013, 84, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Cao, H.Y.; Wang, Y.R.; Jiao, S.S.; Bu, X.L.; Zeng, F.; Wang, Q.H.; Li, J.; Deng, J.; Zhou, H.D.; et al. Aβ is predictive for short-term neurological deficits after acute ischemic stroke. Neurotox. Res. 2015, 27, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Koistinaho, M.; Kettunen, M.I.; Goldsteins, G.; Keinanen, R.; Salminen, A.; Ort, M.; Bures, J.; Liu, D.; Kauppinen, R.A.; Higgins, L.S.; et al. β-amyloid precursor protein transgenic mice that harbor diffuse Aβ deposits but do not form plaques show increased ischemic vulnerability: Role of inflammation. Proc. Natl. Acad. Sci. USA 2002, 99, 1610–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluta, R.; Kida, E.; Golabek, A.A.; Mossakowski, M.J. Possible involvement of patomechanism(s) operating in brain ischemia, in Alzheimer’s disease pathology. J. Cereb. Blood Flow Metab. 1995, 15 (Suppl. 1), S803. [Google Scholar]
- Pluta, R. Proteins associated with Alzheimer’s disease in conditions predisposing to Alzheimer’s-type neurodegeneration. J. Cereb. Blood Flow Metab. 2001, 21 (Suppl. 1), S424. [Google Scholar]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Dysregulation of Alzheimer’s disease-related genes and proteins following cardiac arrest. Folia Neuropathol. 2017, 55, 283–288. [Google Scholar] [CrossRef]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Januszewski, S.; Pluta, R. Proteomic and genomic changes in tau protein, which are associated with Alzheimer’s disease after ischemia-reperfusion brain injury. Int. J. Mol. Sci. 2020, 21, 892. [Google Scholar] [CrossRef] [Green Version]
- Kocki, J.; Ułamek-Kozioł, M.; Bogucka-Kocka, A.; Januszewski, S.; Jabłoński, M.; Gil-Kulik, P.; Brzozowska, J.; Petniak, A.; Furmaga-Jabłońska, W.; Bogucki, J.; et al. Dysregulation of amyloid precursor protein, β-secretase, presenilin 1 and 2 genes in the rat selectively vulnerable CA1 subfield of hippocampus following transient global brain ischemia. J. Alzheimers Dis. 2015, 47, 1047–1056. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R.; Ułamek-Kozioł, M.; Kocki, J.; Bogucki, J.; Januszewski, S.; Bogucka-Kocka, A.; Czuczwar, S.J. Expression of the tau protein and amyloid protein precursor processing genes in the CA3 area of the hippocampus in the ischemic model of Alzheimer’s disease in the rat. Mol. Neurobiol. 2020, 57, 1281–1290. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R.; Kocki, J.; Ułamek-Kozioł, M.; Petniak, A.; Gil-Kulik, P.; Januszewski, S.; Bogucki, J.; Jabłoński, M.; Brzozowska, J.; Furmaga-Jabłońska, W.; et al. Discrepancy in expression of β-secretase and amyloid-β protein precursor in Alzheimer-related genes in the rat medial temporal lobe cortex following transient global brain ischemia. J. Alzheimers Dis. 2016, 51, 1023–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluta, R.; Kocki, J.; Ułamek-Kozioł, M.; Bogucka-Kocka, A.; Gil-Kulik, P.; Januszewski, S.; Jabłoński, M.; Petniak, A.; Brzozowska, J.; Bogucki, J.; et al. Alzheimer-associated presenilin 2 gene is dysregulated in rat medial temporal lobe cortex after complete brain ischemia due to cardiac arrest. Pharmacol. Rep. 2016, 68, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Panickar, K.S.; Yang, S.H.; Rabbani, O.; Day, A.L.; Simpkins, J.W. Estrogen attenuates over-expression of beta-amyloid precursor protein messenger RNA in an animal model of focal ischemia. Brain Res. 1998, 810, 87–92. [Google Scholar] [CrossRef]
- Shi, J.; Yang, S.H.; Stubley, L.; Day, A.L.; Simpkins, J.W. Hypoperfusion induces overexpression of β-amyloid precursor protein mRNA in a focal ischemic rodent model. Brain Res. 2000, 853, 1–4. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, S.H.; Kim, S.S.; Kim, Y.K.; Jeong, S.J.; Ma, J.; Han, D.H.; Cho, B.K.; Suh, Y.H. Post-ischemic changes in the expression of Alzheimer’s APP isoforms in rat cerebral cortex. Neuroreport 1998, 9, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Tanzi, R.E.; Kogure, K. Selective induction of Kunitz-type protease inhibitor domain-containing amyloid precursor protein mRNA after persistent focal ischemia in rat cerebral cortex. Neurosci. Lett. 1991, 125, 172–174. [Google Scholar] [CrossRef]
- Koistinaho, J.; Pyykonen, I.; Keinanen, R.; Hokfelt, T. Expression of β-amyloid precursor protein mRNAs following transient focal ischaemia. Neuroreport 1996, 7, 2727–2731. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Fisk, L.; Kochkina, E.G.; Plesneva, S.A.; Zhuravin, I.A.; Babusikova, E.; Dobrota, D.; Turner, A.J. Effect of hypoxia/ischemia and hypoxic preconditioning/reperfusion on expression of some amyloid-degrading enzymes. Ann. N. Y. Acad. Sci. 2004, 1035, 21–33. [Google Scholar] [CrossRef]
- Yan, F.L.; Zhang, J.; Guan, X.N.; Hong, Z. mRNA expression and activity of ADAM17 in hippocampus after chronic cerebral hypoperfusion: Experiment with aged rats. Zhonghua Yi Xue Za Zhi 2007, 87, 2515–2517. [Google Scholar]
- Blasko, I.; Beer, R.; Bigl, M.; Apelt, J.; Franz, G.; Rudzki, D.; Ransmayr, G.; Kampfl, A.; Schliebs, R. Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer’s disease β-secretase (BACE-1). J. Neural Transm. 2004, 111, 523–536. [Google Scholar] [CrossRef]
- Chen, X.H.; Siman, R.; Iwata, A.; Meaney, D.F.; Trojanowski, J.Q.; Smith, D.H. Long-term accumulation of amyloid-β, β-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am. J. Pathol. 2004, 165, 357–371. [Google Scholar] [CrossRef]
- Wen, Y.; Onyewuchi, O.; Yang, S.; Liu, R.; Simpkins, J.W. Increased beta-secretase activity and expression in rats following transient cerebral ischemia. Brain Res. 2004, 1009, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.M.; Hsieh, C.L.; Lin, H.Y.; Lin, J.G. Panax, Notoginseng Burk attenuates impairment of learning and memory functions and increases ED1, BDNF and beta-secretase immunoreactive cells in chronic stage ischemia-reperfusion injured rats. Am. J. Chin. Med. 2008, 36, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Pi, R.; Mao, X.; Chen, X.; Qin, J.; Xu, S.; Liu, P. Alterations in mRNA expression of BACE1, cathepsin B.; and glutaminyl cyclase in mice ischemic brain. Neuroreport 2009, 20, 1456–1460. [Google Scholar] [CrossRef]
- Tanimukai, H.; Imaizumi, K.; Kudo, T.; Katayama, T.; Tsuda, M.; Takagi, T.; Tohyama, M.; Takeda, M. Alzheimer-associated presenilin-1 gene is induced in gerbil hippocampus after transient ischemia. Mol. Brain Res. 1998, 54, 212–218. [Google Scholar] [CrossRef]
- Pennypacker, K.R.; Hernandez, H.; Benkovic, S.; Morgan, D.G.; Willing, A.E.; Sanberg, P.R. Induction of presenilins in the rat brain after middle cerebral arterial occlusion. Brain Res. Bull. 1999, 48, 539–543. [Google Scholar] [CrossRef]
- Hall, E.D.; Oostveen, J.A.; Dunn, E.; Carter, D.B. Increased amyloid protein precursor and apolipoprotein E immunoreactivity in the selectively vulnerable hippocampus following transient forebrain ischemia in gerbils. Exp. Neurol. 1995, 135, 17–27. [Google Scholar] [CrossRef]
- Tomimoto, H.; Akiguchi, I.; Wakita, H.; Nakamura, S.; Kimura, J. Ultrastructural localization of amyloid protein precursor in the normal and postischemic gerbil brain. Brain Res. 1995, 672, 187–195. [Google Scholar] [CrossRef]
- Ishimaru, H.; Ishikawa, K.; Haga, S.; Shoji, M.; Ohe, Y.; Haga, C.; Sasaki, A.; Takashashi, A.; Maruyama, Y. Accumulation of apolipoprotein E and β-amyloid-like protein in a trace of the hippocampal CA1 pyramidal cell layer after ischaemic delayed neuronal death. Neuroreport 1996, 7, 3063–3067. [Google Scholar] [CrossRef]
- Yokota, M.; Saido, T.C.; Tani, E.; Yamaura, I.; Minami, N. Cytotoxic fragment of amyloid precursor protein accumulates in hippocampus after global forebrain ischemia. J. Cereb. Blood Flow Metab. 1996, 16, 1219–1223. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R. Experimental model of neuropathological changes characteristic for Alzheimer’s disease. Folia Neuropathol. 1997, 35, 94–98. [Google Scholar] [PubMed]
- Pluta, R.; Barcikowska, M.; Dębicki, G.; Ryba, M.; Januszewski, S. Changes in amyloid precursor protein and apolipoprotein E immunoreactivity following ischemic brain injury in rat with long-term survival: Influence of idebenone treatment. Neurosci. Lett. 1997, 232, 95–98. [Google Scholar] [CrossRef]
- Pluta, R.; Misicka, A.; Januszewski, J.; Barcikowska, M.; Lipkowski, A.W. Transport of human β-amyloid peptide through the rat blood-brain barrier after global cerebral ischemia. Acta Neurochir. 1997, 70, 247–249. [Google Scholar]
- Pluta, R.; Barcikowska, M.; Mossakowski, M.J.; Zelman, I. Cerebral accumulation of beta-amyloid following ischemic brain injury with long-term survival. Acta Neurochir. 1998, 71, 206–208. [Google Scholar]
- Lin, B.; Schmidt-Kastner, R.; Busto, R.; Ginsberg, M.D. Progressive parenchymal deposition of β-amyloid precursor protein in rat brain following global cerebral ischemia. Acta Neuropathol. 1999, 97, 359–368. [Google Scholar] [CrossRef]
- Pluta, R. No effect of anti-oxidative therapy on cerebral amyloidosis following ischemia–reperfusion brain injury. Folia Neuropathol. 2000, 38, 188–190. [Google Scholar]
- Lin, B.; Ginsberg, M.D.; Busto, R. Hyperglycemic but not normoglycemic global ischemia induces marked early intraneuronal expression of β-amyloid precursor protein. Brain Res. 2001, 888, 107–116. [Google Scholar] [CrossRef]
- Sinigaglia-Coimbra, R.; Cavalheiro, E.A.; Coimbra, C.G. Postischemic hypertermia induces Alzheimer-like pathology in the rat brain. Acta Neuropathol. 2002, 103, 444–452. [Google Scholar]
- Fujioka, M.; Taoka, T.; Matsuo, Y.; Mishima, K.; Ogoshi, K.; Kondo, Y.; Isuda, M.; Fujiwara, M.; Asano, T.; Sakaki, T.; et al. Magnetic resonance imaging shows delayed ischemic striatal neurodegeneration. Ann. Neurol. 2003, 54, 732–747. [Google Scholar] [CrossRef]
- Pluta, R.; Jabłoński, M. Alzheimer’s factors in ischemic brain injury. In Brain Injury, Pathogenesis, Monitoring, Recovery and Management; Agrawal, A., Ed.; InTech, Open Book: Rjeka, Croatia, 2012; pp. 97–138. [Google Scholar]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Ściślewska, M.; Bogucka-Kocka, A.; Kocki, J. Alzheimer’s factors in postischemic dementia. Rom. J. Morphol. Embryol. 2012, 53, 461–466. [Google Scholar]
- Banati, R.B.; Gehrmann, J.; Wießner, C.; Hossmann, K.A.; Kreutzberg, G.W. Glial expression of the β-amyloid precursor protein (APP) in global ischemia. J. Cereb. Blood Flow Metab. 1995, 15, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; Mengod, G.; Tortosa, A.; Ferrer, I.; Palacios, J.M. Increased β-amyloid precursor protein expression in astrocytes in the gerbil hippocampus following ischaemia: Association with proliferation of astrocytes. Eur. J. Neurosci. 1995, 7, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Nihashi, T.; Inao, S.; Kajita, Y.; Kawai, T.; Sugimoto, T.; Niwa, M.; Kabeya, R.; Hata, N.; Hayashi, S.; Yoshida, J. Expression and distribution of beta amyloid precursor protein and beta amyloid peptide in reactive astrocytes after transient middle cerebral artery occlusion. Acta Neurochir. 2001, 143, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Badan, I.; Platt, D.; Kessler, C.; Popa-Wagner, A. Temporal dynamics of degenerative and regenerative events associated with cerebral ischemia in aged rats. Gerontology 2003, 49, 356–365. [Google Scholar] [CrossRef]
- Badan, I.; Dinca, I.; Buchhold, B.; Suofu, Y.; Walker, L.; Gratz, M.; Platt, D.; Kessler, C.H.; Popa-Wagner, A. Accelerated accumulation of N- and C-terminal beta APP fragments and delayed recovery of microtubule-associated protein 1B expression following stroke in aged rats. Eur. J. Neurosci. 2004, 19, 2270–2280. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef]
- Takuma, K.; Baba, A.; Matsuda, T. Astrocyte apoptosis: Implications for neuroprotection. Prog. Neurobiol. 2004, 72, 111–127. [Google Scholar] [CrossRef]
- Yam, P.S.; Takasago, T.; Dewar, D.; Graham, D.I.; McCulloch, J. Amyloid precursor protein accumulates in white matter at the margin of a focal ischaemic lesion. Brain Res. 1997, 760, 150–157. [Google Scholar] [CrossRef]
- Pluta, R. Glial expression of the β-amyloid peptide in cardiac arrest. J. Neurol. Sci. 2002, 203–204, 277–280. [Google Scholar] [CrossRef]
- Pluta, R. Astroglial expression of the beta-amyloid in ischemia-reperfusion brain injury. Ann. N. Y. Acad. Sci. 2002, 977, 102–108. [Google Scholar] [CrossRef]
- Pluta, R. Role of ischemic blood–brain barrier on amyloid plaques development in Alzheimer’s disease brain. Curr. Neurovasc. Res. 2007, 4, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Van Groen, T.; Puurunen, K.; Maki, H.M.; Sivenius, J.; Jolkkonen, J. Transformation of diffuse beta-amyloid precursor protein and beta-amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke 2005, 36, 1551–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluta, R.; Jolkkonen, J.; Cuzzocrea, S.; Pedata, F.; Cechetto, D.; PopaWagner, A. Cognitive impairment with vascular impairment and degeneration. Curr. Neurovasc. Res. 2011, 8, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Kocki, J.; Maciejewski, R.; Ułamek-Kozioł, M.; Jabłoński, M.; Bogucka-Kocka, A.; Czuczwar, S.J. Ischemia signaling to Alzheimer-related genes. Folia Neuropathol. 2012, 50, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R.; Jabłoński, M.; Czuczwar, S.J. Postischemic dementia with Alzheimer phenotype: Selectively vulnerable versus resistant areas of the brain and neurodegeneration versus β-amyloid peptide. Folia Neuropathol. 2012, 50, 101–109. [Google Scholar]
- Cotter, R.L.; Burke, W.J.; Thomas, V.S.; Potter, J.F.; Zheng, J.; Gendelman, H.E. Insights into the neurodegenerative process of Alzheimer’s disease: A role for mononuclear phagocyte associated inflammation and neurotoxicity. J. Leukoc. Biol. 1999, 65, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Giulian, D.; Haverkamp, L.J.; Li, J.; Karshin, W.L.; Yu, J.; Tom, D.; Li, X.; Kirkpatrick, J.B. Senile plaques stimulate microglia to release a neurotoxin found in Alzheimer brain. Neurochem. Int. 1995, 27, 119–137. [Google Scholar] [CrossRef]
- Jendroska, K.; Poewe, W.; Daniel, S.E.; Pluess, J.; Iwerssen-Schmidt, H.; Paulsen, J.; Barthel, S.; Schelosky, L.; Cervos-Navarr, J.; DeArmond, S.J. Ischemic stress induces deposition of amyloid beta immunoreactivity in human brain. Acta Neuropathol. 1995, 90, 461–466. [Google Scholar] [CrossRef]
- Wisniewski, H.M.; Maslinska, D. Beta-protein immunoreactivity in the human brain after cardiac arrest. Folia Neuropathol. 1996, 34, 65–71. [Google Scholar]
- Jendroska, K.; Hoffmann, O.M.; Patt, S. Amyloid β peptide and precursor protein (APP) in mild and severe brain ischemia. Ann. N. Y. Acad. Sci. 1997, 826, 401–405. [Google Scholar] [CrossRef]
- Qi, J.; Wu, H.; Yang, Y.; Wand, D.; Chen, Y.; Gu, Y.; Liu, T. Cerebral ischemia and Alzheimer’s disease: The expression of amyloid-β and apolipoprotein E in human hippocampus. J. Alzheimers Dis. 2007, 12, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Maślińska, D.; Laure-Kamionowska, M.; Taraszewska, A.; Deręgowski, K.; Maśliński, S. Immunodistribution of amyloid beta protein (Aβ) and advanced glycation end-product receptors (RAGE) in choroid plexus and ependyma of resuscitated patients. Folia Neuropathol. 2011, 49, 295–300. [Google Scholar] [PubMed]
- Pluta, R.; Bogucka-Kocka, A.; Ułamek-Kozioł, M.; Bogucki, J.; Czuczwar, S.J. Ischemic tau protein gene induction as an additional key factor driving development of Alzheimer’s phenotype changes in CA1 area of hippocampus in an ischemic model of Alzheimer’s disease. Pharmacol. Rep. 2018, 70, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Dewar, D.; Graham, D.I.; Teasdale, G.M.; McCulloch, J. Alz-50 and ubiquitin immunoreactivity is induced by permanent focal cerebral ischaemia in the cat. Acta Neuropathol. 1993, 86, 623–629. [Google Scholar] [CrossRef]
- Dewar, D.; Graham, D.I.; Teasdale, G.M.; McCulloch, J. Cerebral ischemia induces alterations in tau and ubiquitin proteins. Dementia 1994, 5, 168–173. [Google Scholar] [CrossRef]
- Geddes, J.W.; Schwab, C.; Craddock, S.; Wilson, J.L.; Pettigrew, L.C. Alterations in tau immunostaining in the rat hippocampus following transient cerebral ischemia. J. Cereb. Blood Flow Metab. 1994, 14, 554–564. [Google Scholar] [CrossRef] [Green Version]
- Dewar, D.; Dawson, D. Tau protein is altered by focal cerebral ischaemia in the rat: An immunohistochemical and immunoblotting study. Brain Res. 1995, 684, 70–78. [Google Scholar] [CrossRef]
- Uchihara, T.; Tsuchiya, K.; Kondo, H.; Hayama, T.; Ikeda, K. Widespread appearance of Alz-50 immunoreactive neurons in the human brain with cerebral infarction. Stroke 1995, 26, 2145–2148. [Google Scholar] [CrossRef]
- Irving, E.A.; Nicoll, J.; Graham, D.I.; Dewar, D. Increased tau immunoreactivity in oligodendrocytes following human stroke and head injury. Neurosci. Lett. 1996, 213, 189–192. [Google Scholar] [CrossRef]
- Irving, E.A.; Yatsushiro, K.; McCulloch, J.; Dewar, D. Rapid alteration of tau in oligodendrocytes after focal ischemic injury in the rat: Involvement of free radicals. J. Cereb. Blood Flow Metab. 1997, 17, 612–622. [Google Scholar] [CrossRef]
- Uchihara, T.; Nakamura, A.; Arai, T.; Ikeda, K.; Tsuchiya, K. Microglial tau undergoes phosphorylation-independent modification after ischemia. Glia 2004, 45, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Bitsch, A.; Horn, C.; Kemmling, Y.; Seipelt, M.; Hellenbrand, U.; Stiefel, M.; Ciesielczyk, B.; Cepek, L.; Bahn, E.; Ratzka, P.; et al. Serum tau protein level as a marker of axonal damage in acute ischemic stroke. Eur. Neurol. 2002, 47, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, M.T.; Lins, H.; Skalej, M.; Wallesch, C.W.; Goertler, M. Neuron-specific enolase and tau protein as neurobiochemical markers of neuronal damage are related to early clinical course and long-term outcome in acute ischemic stroke. Clin. Neurol. Neurosurg. 2006, 108, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Kurzepa, J.; Bielewicz, J.; Grabarska, A.; Stelmasiak, Z.; Stryjecka-Zimmer, M.; Bartosik-Psujek, H. Matrix metalloproteinase-9 contributes to the increase of tau protein in serum during acute ischemic stroke. J. Clin. Neurosci. 2010, 17, 997–999. [Google Scholar] [CrossRef]
- Bielewicz, J.; Kurzepa, J.; Czekajska-Chehab, E.; Stelmasiak, Z.; Bartosik-Psujek, H. Does serum tau protein predict the outcome of patients with ischemic stroke? J. Mol. Neurosci. 2011, 43, 241–245. [Google Scholar] [CrossRef]
- Lasek-Bal, A.; Jedrzejowska-Szypulka, H.; Rozycka, J.; Bal, W.; Kowalczyk, A.; Holecki, M.; Dulawa, J.; Lewin-Kowalik, J. The presence of tau protein in blood as a potential prognostic factor in stroke patients. J. Physiol. Pharmacol. 2016, 67, 691–696. [Google Scholar]
- De Vos, A.; Bjerke, M.; Brouns, R.; De Roeck, N.; Jacobs, D.; Van den Abbeele, L.; Guldolf, K.; Zetterberg, H.; Blennow, K.; Engelborghs, S.; et al. Neurogranin and tau in cerebrospinal fluid and plasma of patients with acute ischemic stroke. BMC Neurol. 2017, 17, 170. [Google Scholar] [CrossRef] [Green Version]
- Hesse, C.; Rosengren, L.; Andreasen, N.; Davidsson, P.; Vanderstichele, H.; Vanmechelen, E.; Blennow, K. Transient increase in total tau but not phospho-tau in human cerebrospinal fluid after acute stroke. Neurosci. Lett. 2001, 297, 187–190. [Google Scholar] [CrossRef]
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic brain injury and Alzheimer’s disease: The cerebrovascular link. EBioMedicine 2018, 28, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Stetler, R.A.; Leak, R.K.; Shi, Y.; Li, Y.; Yu, W.; Bennett, M.V.L.; Chen, J. Oxidative stress and DNA damage after cerebral ischemia: Potential therapeutic targets to repair the genome and improve stroke recovery. Neuropharmacology 2018, 134, 208–217. [Google Scholar] [CrossRef]
- Kato, T.; Hirano, A.; Katagiri, T.; Sasaki, H.; Yamada, S. Neurofibrillary tangle formation in the nucleus basalis of Meynert ipsilateral to a massive cerebral infarct. Ann. Neurol. 1988, 23, 620–623. [Google Scholar]
- Wen, Y.; Yang, S.; Liu, R.; Brun-Zinkernagel, A.M.; Koulen, P.; Simpkins, J.W. Transient cerebral ischemia induces aberrant neuronal cell cycle re-entry and Alzheimer’s disease-like tauopathy in female rats. J. Biol. Chem. 2004, 279, 22684–22692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.; Yang, S.H.; Liu, R.; Perez, E.J.; Brun-Ziukemagel, A.M.; Koulen, P.; Simpkins, J.W. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim. Biophys. Acta 2007, 1772, 473–483. [Google Scholar] [CrossRef]
- Wen, Y.; Yang, S.; Liu, R.; Simpkins, J.W. Transient cerebral ischemia induces site-specific hyperphosphorylation of tau protein. Brain Res. 2004, 1022, 30–38. [Google Scholar] [CrossRef]
- Michalicova, A.; Banks, W.A.; Legath, J.; Kovac, A. Tauopathies—Focus on changes at the neurovascular unit. Curr. Alzheimer Res. 2017, 14, 790–801. [Google Scholar] [CrossRef]
- Banks, W.A.; Kovac, A.; Majerova, P.; Bullock, K.M.; Shi, M.; Zhang, J. Tau proteins cross the blood-brain barrier. J. Alzheimers Dis. 2017, 55, 411–419. [Google Scholar] [CrossRef]
- Fujii, H.; Takahashi, T.; Mukai, T.; Tanaka, S.; Hosomi, N.; Maruyama, H.; Sakai, N.; Matsumoto, M. Modifications of tau protein after cerebral ischemia and reperfusion in rats are similar to those occurring in Alzheimer’s disease - Hyperphosphorylation and cleavage of 4- and 3-repeat tau. J. Cereb. Blood Flow Metab. 2017, 37, 2441–2457. [Google Scholar] [CrossRef] [Green Version]
- Basurto-Islas, G.; Gu, J.H.; Tung, Y.C.; Liu, F.; Iqbal, K. Mechanism of tau hyperphosphorylation involving lysosomal enzyme asparagine endopeptidase in a mouse model of brain ischemia. J. Alzheimers Dis. 2018, 63, 821–833. [Google Scholar] [CrossRef]
- Kovalska, M.; Tothova, B.; Kovalska, L.; Tatarkova, Z.; Kalenska, D.; Tomascova, A.; Adamkov, M.; Lehotsky, J. Association of induced hyperhomocysteinemia with Alzheimer’s disease-like neurodegeneration in rat cortical neurons after global ischemia-reperfusion injury. Neurochem. Res. 2018, 43, 1766–1778. [Google Scholar] [CrossRef]
- Shackelford, D.A.; Yeh, R.Y. Dephosphorylation of tau during transient forebrain ischemia in the rat. Mol. Chem. Neuropathol. 1998, 34, 103–120. [Google Scholar] [CrossRef] [PubMed]
- Mailliot, C.; Podevin-Dimster, V.; Rosenthal, R.E.; Sergeant, N.; Delacourte, A.; Fiskum, G.; Buee, L. Rapid tau protein dephosphorylation and differential rephosphorylation during cardiac arrest-induced cerebral ischemia and reperfusion. J. Cereb. Blood Flow Metab. 2000, 20, 543–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morioka, M.; Kawano, T.; Yano, S.; Kai, Y.; Tsuiki, H.; Yoshinaga, Y.; Matsumoto, J.; Maeda, T.; Hamada, J.; Yamamoto, H.; et al. Hyperphosphorylation at serine 199/202 of tau factor in the gerbil hippocampus after transient forebrain ischemia. Biochem. Biophys. Res. Commun. 2006, 347, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Majd, S.; Power, J.H.; Koblar, S.A.; Grantham, H.J. Early glycogen synthase kinase-3 and protein phosphatase 2A independent tau dephosphorylation during global brain ischaemia and reperfusion following cardiac arrest and the role of the adenosine monophosphate kinase pathway. Eur. J. Neurosci. 2016, 44, 1987–1997. [Google Scholar] [PubMed] [Green Version]
- Khan, S.; Yuldasheva, N.Y.; Batten, T.F.C.; Pickles, A.R.; Kellett, K.A.B.; Saha, S. Tau pathology and neurochemical changes associated with memory dysfunction in an optimized murine model of global cerebral ischaemia – A potential model for vascular dementia? Neurochem. Int. 2018, 118, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuo, Q.Z.; Lei, P.; Jackman, K.A.; Li, X.L.; Xiong, H.; Li, X.L.; Liuyang, Z.Y.; Roisman, L.; Zhang, S.T.; Ayton, S.; et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 2017, 22, 1520–1530. [Google Scholar] [CrossRef]
- Bi, M.; Gladbach, A.; van Eersel, J.; Ittner, A.; Przybyla, M.; van Hummel, A.; Chua, S.W.; van der Hoven, J.; Lee, W.S.; Müller, J.; et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat. Commun. 2017, 8, 473. [Google Scholar] [CrossRef] [Green Version]
- Magnoni, S.; Esparza, T.J.; Conte, V.; Carbonara, M.; Carrabba, G.; Holtzman, D.M.; Zipfel, G.J.; Stocchetti, N.; Brody, D.L. Tau elevations in the brain extracellular space correlate with reduced amyloid-β levels and predict adverse clinical outcomes after severe traumatic brain injury. Brain 2012, 135, 1268–1280. [Google Scholar] [CrossRef] [Green Version]
- Ułamek-Kozioł, M.; Kocki, J.; Bogucka-Kocka, A.; Petniak, A.; Gil-Kulik, P.; Januszewski, S.; Bogucki, J.; Jabłoński, M.; Furmaga-Jabłońska, W.; Brzozowska, J.; et al. Dysregulation of autophagy, mitophagy and apoptotic genes in the medial temporal lobe cortex in an ischemic model of Alzheimer’s disease. J. Alzheimers Dis. 2016, 54, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Krajewski, S.; Mai, J.K.; Krajewska, M.; Sikorska, M.; Mossakowski, M.J.; Reed, J.C. Upregulation of bax protein levels in neurons following cerebral ischemia. J. Neurosci. 1995, 15, 6364–6376. [Google Scholar] [CrossRef]
- Sadowski, M.; Wisniewski, H.M.; Jakubowska-Sadowska, K.; Tarnawski, M.; Lazarewicz, J.W.; Mossakowski, M.J. Pattern of neuronal loss in the rat hippocampus following experimental cardiac arrest-induced ischemia. J. Neurol. Sci. 1999, 168, 13–20. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Reddy, P.H. Structure based design and molecular docking studies for phosphorylated tau inhibitors in Alzheimer’s disease. Cells 2019, 8, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradeepkiran, J.A.; Reddy, A.P.; Reddy, P.H. Pharmacophore-based models for therapeutic drugs against phosphorylated tau in Alzheimer’s disease. Drug Discov. Today 2019, 24, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Pradeepkiran, J.A.; Reddy, A.P.; Yin, X.; Manczak, M.; Reddy, P.H. Protective effects of BACE1 inhibitory ligand molecules against amyloid beta-induced synaptic and mitochondrial toxicities in Alzheimer’s disease. Hum. Mol. Genet. 2020, 29, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J.; Smith, M.A. Compounding artefacts with uncertainty, and an amyloid cascade hypothesis that is ‘too big to fail’. J. Pathol. 2011, 224, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Tse, K.H.; Herrup, K. Re-imagining Alzheimer’s disease—The diminishing importance of amyloid and a glimpse of what lies ahead. J. Neurochem. 2017, 143, 432–444. [Google Scholar] [CrossRef] [Green Version]
- Kametani, F.; Hasegawa, M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research, Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, H. Tau as a potential therapeutic target for ischemic stroke. Aging 2019, 11, 12827–12843. [Google Scholar] [CrossRef]
- Radenovic, L.; Andjus, P.R. Stroke and Alzheimer’s disease: Common mechanisms and therapeutic approaches. In Brain Ischemia: Alzheimer’s Disease Mechanisms; Pluta, R., Ed.; Nova, Science Publishers, Inc.: New York, NY, USA, 2019; pp. 251–264. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Survival | 2 Days | 7 Days | 30 Days | |
|---|---|---|---|---|
| Genes | ||||
| APP | ↓ | ↑ | ↑ | |
| BACE1 | ↑ | ↑ | ↓ | |
| PSEN1 | ↑ | ↑ | ↓ | |
| PSEN2 | ↑ | ↑ | ↓ | |
| MAPT | ↑ | ↓ | ↓ | |
| Survival | 2 Days | 7 Days | 30 Days | |
|---|---|---|---|---|
| Genes | ||||
| APP | ↑ | ↑ | ↑ | |
| ADAM10 | ↓ | ↓ | ↓ | |
| BACE1 | ↓ | ↓ | ↑ | |
| PSEN1 | ↑ | ↑ | ↓ | |
| PSEN2 | ↓ | ↓ | ↑ | |
| MAPT | ↓ | ↑ | ↑ | |
| Survival | 2 Days | 7 Days | 30 Days | |
|---|---|---|---|---|
| Genes | ||||
| APP | ↓ | ↑ | ↑ | |
| BACE1 | ↑ | ↓ | ↓ | |
| PSEN1 | ↓ | ↓ | ↑ | |
| PSEN2 | ↑ | ↑ | ↓ | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Shared Genomic and Proteomic Contribution of Amyloid and Tau Protein Characteristic of Alzheimer’s Disease to Brain Ischemia. Int. J. Mol. Sci. 2020, 21, 3186. https://doi.org/10.3390/ijms21093186
Pluta R, Ułamek-Kozioł M, Januszewski S, Czuczwar SJ. Shared Genomic and Proteomic Contribution of Amyloid and Tau Protein Characteristic of Alzheimer’s Disease to Brain Ischemia. International Journal of Molecular Sciences. 2020; 21(9):3186. https://doi.org/10.3390/ijms21093186
Chicago/Turabian StylePluta, Ryszard, Marzena Ułamek-Kozioł, Sławomir Januszewski, and Stanisław J. Czuczwar. 2020. "Shared Genomic and Proteomic Contribution of Amyloid and Tau Protein Characteristic of Alzheimer’s Disease to Brain Ischemia" International Journal of Molecular Sciences 21, no. 9: 3186. https://doi.org/10.3390/ijms21093186
APA StylePluta, R., Ułamek-Kozioł, M., Januszewski, S., & Czuczwar, S. J. (2020). Shared Genomic and Proteomic Contribution of Amyloid and Tau Protein Characteristic of Alzheimer’s Disease to Brain Ischemia. International Journal of Molecular Sciences, 21(9), 3186. https://doi.org/10.3390/ijms21093186