Transcriptomic Changes Related to Cellular Processes with Particular Emphasis on Cell Activation in Lysosomal Storage Diseases from the Group of Mucopolysaccharidoses


 , ,
, ,  and
and 
Abstract
:1. Introduction
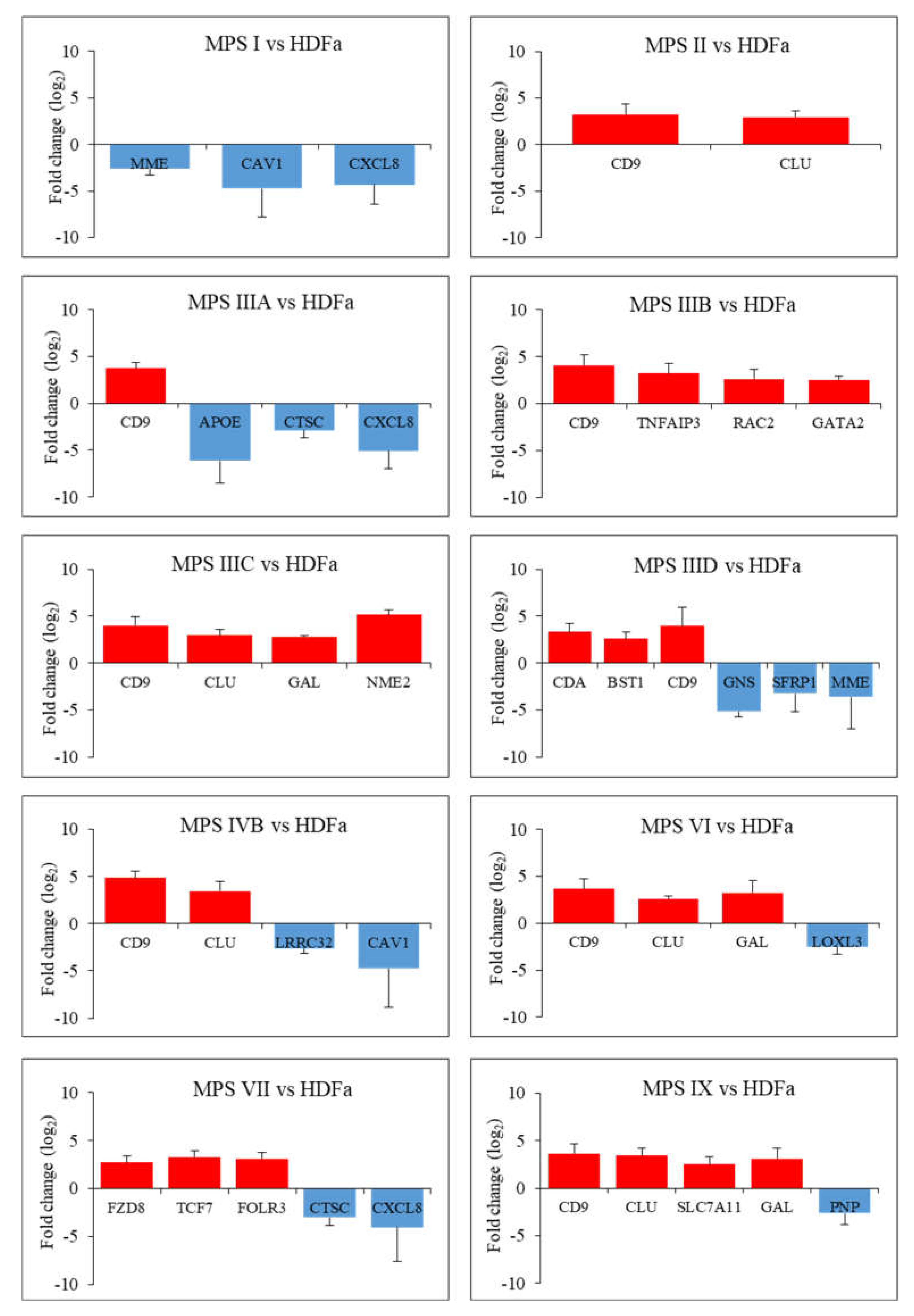
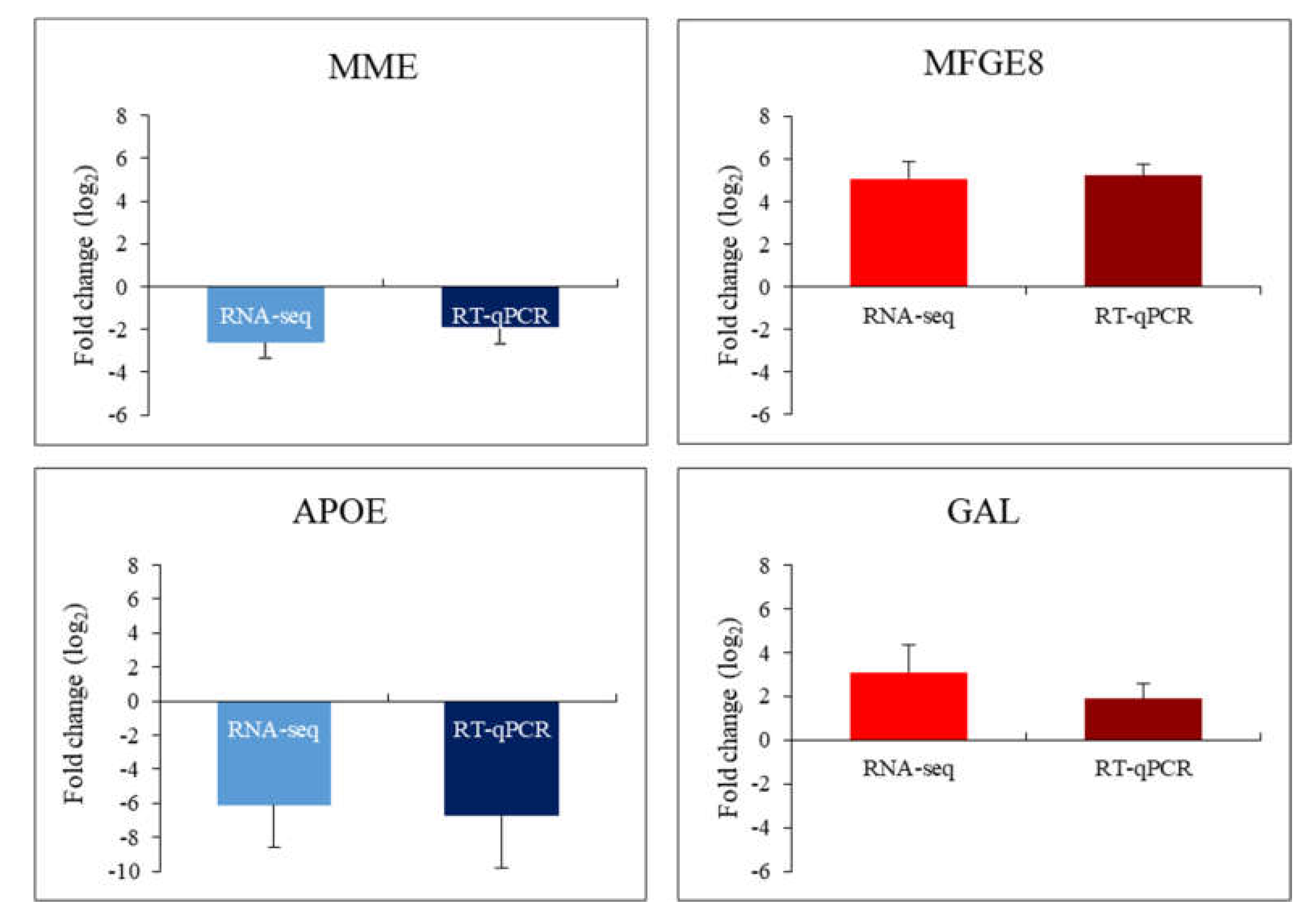
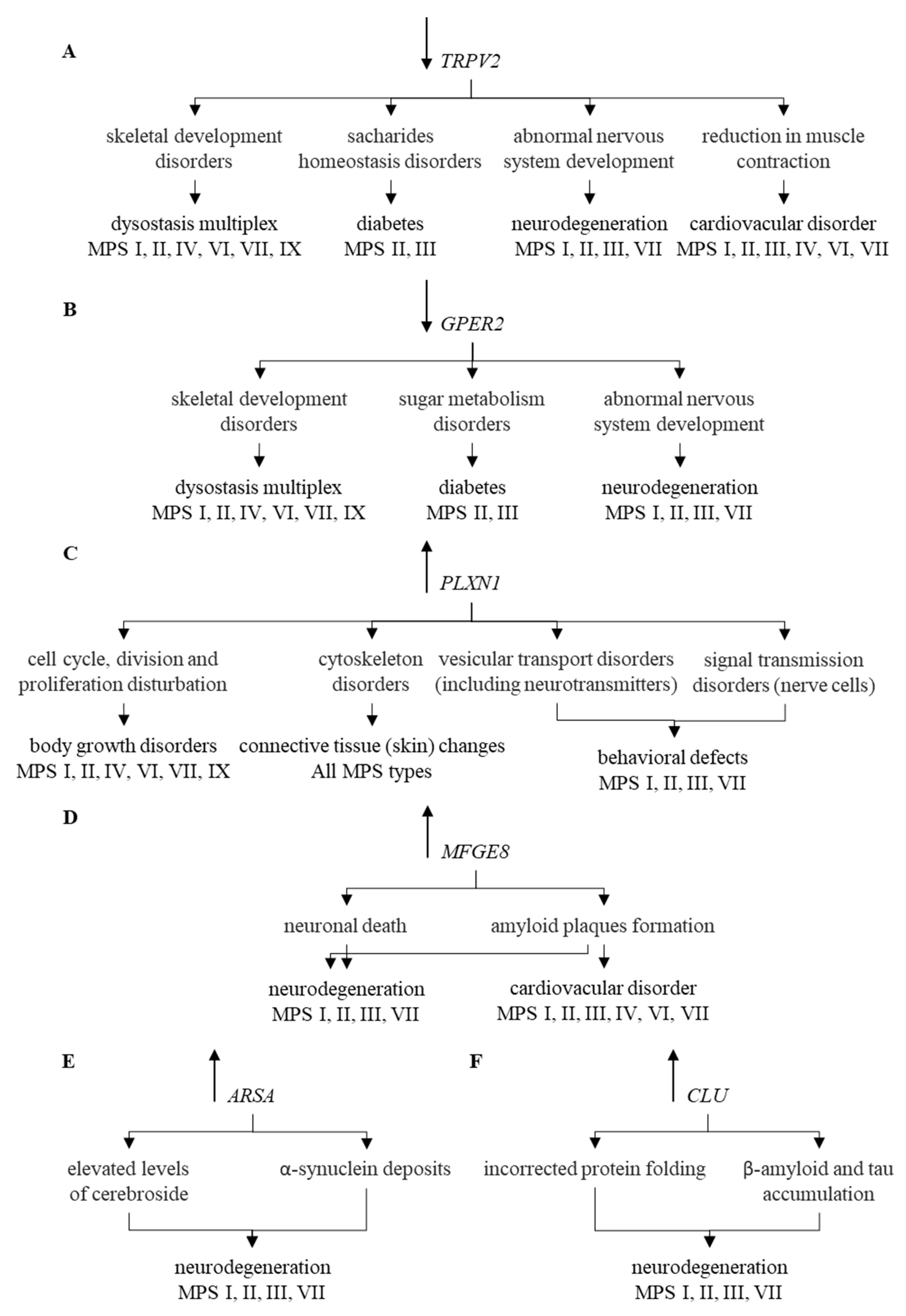
2. Results
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Cultures
4.2. RNA Isolation and Purification
4.3. RNA-Seq Analysis
4.4. Reverse Trascription qPCR
4.5. Western Blotting
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ERT | enzyme replacement therapy |
| ECM | extracellular matrix |
| FDR | false discovery rate |
| FC | fold change |
| GAG(s) | glycosaminoglycan(s) |
| LSD | lysosomal storage disease |
| MPS | mucopolysaccharidosis |
References
- Tomatsu, S.; Giugliani, R.; Harmatz, P.; Scarpa, M.; Węgrzyn, G.; Orii, T. Mucopolysaccharidoses Update (2 Volume Set); Nova Science Publishers: Hauppauge, NY, USA, 2018. [Google Scholar]
- Piotrowska, E.; Jakóbkiewicz-Banecka, J.; Tylki-Szymańska, A.; Czartoryska, B.; Wegrzyn, A.; Wegrzyn, G. Correlation between severity of mucopolysaccharidoses and combination of the residual enzyme activity and efficiency of glycosaminoglycan synthesis. Acta Paediatr. 2009, 98, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Knottnerus, S.J.G.; Nijmeijer, S.C.M.; IJlst, L.; Te Brinke, H.; van Vlies, N.; Wijburg, F.A. Prediction of phenotypic severity in mucopolysaccharidosis type IIIA. Ann. Neurol. 2017, 82, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Guarany, N.R.; Schwartz, I.V.D.; Guarany, F.C.; Giugliani, R. Functional capacity evaluation of patients with mucopolysaccharidosis. J. Pediatric Rehabil. Med. 2012, 5, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.A.; Barboza, I.C.F.; Ferraz, M.V.A.R.; Hembroff, G. Michigan Hand Outcomes Questionnaire for the Evaluation of Patients with Mucopolysaccharidosis. Bull. NYU Hosp. Jt. Dis. 2018, 76, 112–115. [Google Scholar]
- Wraith, J.E. Mucopolysaccharidoses and mucolipidoses. Handb. Clin. Neurol. 2013, 113, 1723–1729. [Google Scholar]
- Fecarotta, S.; Gasperini, S.; Parenti, G. New treatments for the mucopolysaccharidoses: from pathophysiology to therapy. Ital. J. Pediatric 2018, 44, 124. [Google Scholar] [CrossRef]
- Gabig-Cimińska, M.; Jakóbkiewicz-Banecka, J.; Malinowska, M.; Kloska, A.; Piotrowska, E.; Chmielarz, I.; Moskot, M.; Węgrzyn, A.; Węgrzyn, G. Combined Therapies for Lysosomal Storage Diseases. Curr. Mol. Med. 2015, 15, 746–771. [Google Scholar] [CrossRef]
- Parente, M.K.; Rozen, R.; Seeholzer, S.H.; Wolfe, J.H. Integrated analysis of proteome and transcriptome changes in the mucopolysaccharidosis type VII mouse hippocampus. Mol. Genet. Metab. 2016, 118, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Pshezhetsky, A.V. Crosstalk between 2 organelles: Lysosomal storage of heparan sulfate causes mitochondrial defects and neuronal death in mucopolysaccharidosis III type C. Rare Dis. 2015, 3, e1049793. [Google Scholar] [CrossRef] [Green Version]
- Pshezhetsky, A.V. Lysosomal storage of heparan sulfate causes mitochondrial defects, altered autophagy, and neuronal death in the mouse model of mucopolysaccharidosis III type C. Autophagy 2016, 12, 1059–1060. [Google Scholar] [CrossRef] [Green Version]
- Ou, L.; Przybilla, M.J.; Whitley, C.B. Proteomic analysis of mucopolysaccharidosis I mouse brain with two-dimensional polyacrylamide gel electrophoresis. Mol. Genet. Metab. 2017, 120, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaffke, L.; Pierzynowska, K.; Podlacha, M.; Hoinkis, D.; Rintz, E.; Brokowska, J.; Cyske, Z.; Wegrzyn, G. Underestimated aspect of mucopolysaccharidosis pathogenesis: global changes in cellular processes revealed by transcriptomic studies. Int. J. Mol. Sci. 2020, 21, 1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierzynowska, K.; Gaffke, L.; Podlacha, M.; Węgrzyn, G. Genetic base of behavioral disorders in mucopolysaccharidoses: transcriptomic studies. Int. J. Mol. Sci. 2020, 21, 1156. [Google Scholar] [CrossRef] [Green Version]
- Brokowska, J.; Pierzynowska, K.; Gaffke, L.; Rintz, E.; Węgrzyn, G. Expression of genes involved in apoptosis is dysregulated in mucopolysaccharidoses as revealed by pilot transcriptomic analyses. Cell Biol. Int. 2020. [Google Scholar] [CrossRef] [PubMed]
- López-Maury, L.; Marguerat, S.; Bähler, J. Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nat. Rev. Genet. 2008, 9, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef]
- Murphy, K.; Travers, P.; Walport, M.; Janeway, C. Janeway’s Immunobiology; Garland Science: New York, NY, USA, 2012; ISBN 978-0-8153-4243-4. [Google Scholar]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: novel roles and mediators. Front Pharm. 2014, 5, 123. [Google Scholar] [CrossRef] [Green Version]
- Coelho, C.M.; Leevers, S.J. Do growth and cell division rates determine cell size in multicellular organisms? J. Cell. Sci. 2000, 113 Pt 17, 2927–2934. [Google Scholar]
- Krafts, K.P. Tissue repair: The hidden drama. Organogenesis 2010, 6, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Tyson, J.J.; Novak, B. Control of cell growth, division and death: information processing in living cells. Interface Focus 2014, 4, 20130070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, P.; Tyers, M. How cells coordinate growth and division. Curr. Biol. 2004, 14, R1014–R1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Deng, J.; Nan, M.-L.; Zhang, J.; Okekunle, A.; Li, J.-Y.; Yu, X.-Q.; Wang, P.-H. The Interplay Between Pattern Recognition Receptors and Autophagy in Inflammation. Adv. Exp. Med. Biol. 2019, 1209, 79–108. [Google Scholar] [PubMed]
- Canzio, D.; Maniatis, T. The generation of a protocadherin cell-surface recognition code for neural circuit assembly. Curr. Opin. Neurobiol. 2019, 59, 213–220. [Google Scholar] [CrossRef]
- Piletsky, S.; Canfarotta, F.; Poma, A.; Bossi, A.M.; Piletsky, S. Molecularly imprinted polymers for cell recognition. Trends Biotechnol. 2020, 38, 368–387. [Google Scholar] [CrossRef]
- Żeromski, J.; Kaczmarek, M.; Boruczkowski, M.; Kierepa, A.; Kowala-Piaskowska, A.; Mozer-Lisewska, I. Significance and Role of Pattern Recognition Receptors in Malignancy. Arch. Immunol. Ther. Exp. 2019, 67, 133–141. [Google Scholar] [CrossRef] [Green Version]
- García-Revilla, J.; Alonso-Bellido, I.M.; Burguillos, M.A.; Herrera, A.J.; Espinosa-Oliva, A.M.; Ruiz, R.; Cruz-Hernández, L.; García-Domínguez, I.; Roca-Ceballos, M.A.; Santiago, M.; et al. Reformulating pro-oxidant microglia in neurodegeneration. J. Clin. Med. 2019, 8, 1719. [Google Scholar] [CrossRef] [Green Version]
- Barrow, A.D.; Martin, C.J.; Colonna, M. The Natural Cytotoxicity Receptors in Health and Disease. Front Immunol. 2019, 10, 909. [Google Scholar] [CrossRef] [Green Version]
- Kakkis, E.D.; Muenzer, J.; Tiller, G.E.; Waber, L.; Belmont, J.; Passage, M.; Izykowski, B.; Phillips, J.; Doroshow, R.; Walot, I.; et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N. Engl. J. Med. 2001, 344, 182–188. [Google Scholar] [CrossRef]
- Muenzer, J.; Wraith, J.E.; Beck, M.; Giugliani, R.; Harmatz, P.; Eng, C.M.; Vellodi, A.; Martin, R.; Ramaswami, U.; Gucsavas-Calikoglu, M.; et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet. Med. 2006, 8, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Gaffke, L.; Pierzynowska, K.; Piotrowska, E.; Węgrzyn, G. How close are we to therapies for Sanfilippo disease? Metab. Brain Dis. 2018, 33, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomatsu, S.; Sawamoto, K.; Shimada, T.; Bober, M.B.; Kubaski, F.; Yasuda, E.; Mason, R.W.; Khan, S.; Alméciga-Díaz, C.J.; Barrera, L.A.; et al. Enzyme replacement therapy for treating mucopolysaccharidosis type IVA (Morquio A syndrome): effect and limitations. Expert Opin. Orphan Drugs 2015, 3, 1279–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molderings, G.J.; Brettner, S.; Homann, J.; Afrin, L.B. Mast cell activation disease: a concise practical guide for diagnostic workup and therapeutic options. J. Hematol. Oncol. 2011, 4, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpert, S.D.; Koide, J.; Takada, S.; Engleman, E.G. T cell regulatory disturbances in the rheumatic diseases. Rheum. Dis. Clin. N. Am. 1987, 13, 431–445. [Google Scholar]
- Azizi, G.; Rezaei, N.; Kiaee, F.; Tavakolinia, N.; Yazdani, R.; Mirshafiey, A.; Aghamohammadi, A. T-Cell Abnormalities in Common Variable Immunodeficiency. J. Investig. Allergol. Clin. Immunol. 2016, 26, 233–243. [Google Scholar] [CrossRef] [Green Version]
- Crampton, S.P.; Voynova, E.; Bolland, S. Innate pathways to B-cell activation and tolerance. Ann. N. Y. Acad. Sci. 2010, 1183, 58–68. [Google Scholar] [CrossRef]
- Yeo, S.-Y.; Lee, K.-W.; Shin, D.; An, S.; Cho, K.-H.; Kim, S.-H. A positive feedback loop bi-stably activates fibroblasts. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Goss, R.J. Hypertrophy versus hyperplasia. Science 1966, 153, 1615–1620. [Google Scholar] [CrossRef]
- Wagner, K.R.; Cohen, J.S. Myostatin-Related Muscle Hypertrophy. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1498/ (accessed on 24 November 2019).
- Fulton, R.M.; Hutchinson, E.C.; Jones, A.M. Ventricular weight in cardiac hypertrophy. Br. Heart J. 1952, 14, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Bhide, M.R.; Mucha, R.; Mikula, I.; Kisova, L.; Skrabana, R.; Novak, M.; Mikula, I. Novel mutations in TLR genes cause hyporesponsiveness to Mycobacterium avium subsp. paratuberculosis infection. BMC Genet. 2009, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Murakami, S.; Okada, H. Lymphocyte-fibroblast interactions. Crit. Rev. Oral. Biol. Med. 1997, 8, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiattarella, G.G.; Cerulo, G.; De Pasquale, V.; Cocchiaro, P.; Paciello, O.; Avallone, L.; Belfiore, M.P.; Iacobellis, F.; Di Napoli, D.; Magliulo, F.; et al. The murine model of mucopolysaccharidosis IIIB develops cardiopathies over time leading to heart failure. PLoS ONE 2015, 10, e0131662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parfrey, N.A.; Hutchins, G.M. Hepatic fibrosis in the mucopolysaccharidoses. Am. J. Med. 1986, 81, 825–829. [Google Scholar] [CrossRef]
- Caciotti, A.; Garman, S.C.; Rivera-Colón, Y.; Procopio, E.; Catarzi, S.; Ferri, L.; Guido, C.; Martelli, P.; Parini, R.; Antuzzi, D.; et al. GM1 gangliosidosis and Morquio B disease: an update on genetic alterations and clinical findings. Biochim. Biophys. Acta 2011, 1812, 782–790. [Google Scholar] [CrossRef]
- Congedi, S.; Orzalesi, M.; Di Pede, C.; Benini, F. Pain in Mucopolysaccharidoses: Analysis of the Problem and Possible Treatments. Int. J. Mol. Sci. 2018, 19, 3063. [Google Scholar] [CrossRef] [Green Version]
- Parker, H.; Bigger, B.W. The role of innate immunity in mucopolysaccharide diseases. J. Neurochem. 2019, 148, 639–651. [Google Scholar] [CrossRef] [Green Version]
- Oussoren, E.; Brands, M.M.M.G.; Ruijter, G.J.G.; van der Ploeg, A.T.; Reuser, A.J.J. Bone, joint and tooth development in mucopolysaccharidoses: relevance to therapeutic options. Biochim. Biophys. Acta 2011, 1812, 1542–1556. [Google Scholar] [CrossRef] [Green Version]
- Stepien, K.M.; Stewart, F.J.; Hendriksz, C.J. The factors affecting lipid profile in adult patients with Mucopolysaccharidosis. Mol. Genet. Metab. Rep. 2017, 12, 35–40. [Google Scholar] [CrossRef]
- Bohan, E.M. Diabetes mellitus and the Hurler syndrome. A case diabetic coma with deliberate production of Insulin shock to produce recovery. Med. Times 1963, 91, 502–509. [Google Scholar]
- Kubaski, F.; Kecskemethy, H.H.; Harcke, H.T.; Tomatsu, S. Bone mineral density in mucopolysaccharidosis IVB. Mol. Genet. Metab. Rep. 2016, 8, 80–84. [Google Scholar] [CrossRef]
- Aguettaz, E.; Bois, P.; Cognard, C.; Sebille, S. Stretch-activated TRPV2 channels: Role in mediating cardiopathies. Prog. Biophys. Mol. Biol. 2017, 130, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, C.; Abu-Tair, T.; Gökce, S.; Lampe, C.; Reinke, J.; Mengel, E.; Hennermann, J.B.; Wiethoff, C.M. Heart and Cardiovascular Involvement in Patients with Mucopolysaccharidosis Type IVA (Morquio-A Syndrome). PLoS ONE 2016, 11, e0162612. [Google Scholar] [CrossRef] [PubMed]
- Braunlin, E.A.; Harmatz, P.R.; Scarpa, M.; Furlanetto, B.; Kampmann, C.; Loehr, J.P.; Ponder, K.P.; Roberts, W.C.; Rosenfeld, H.M.; Giugliani, R. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J. Inherit. Metab. Dis. 2011, 34, 1183–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilius, B. TRP channels in disease. Biochim. Biophys. Acta 2007, 1772, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Kajta, M.; Beyer, C. Cellular strategies of estrogen-mediated neuroprotection during brain development. Endocrine 2003, 21, 3–9. [Google Scholar] [CrossRef]
- Bigger, B.W.; Begley, D.J.; Virgintino, D.; Pshezhetsky, A.V. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol. Genet. Metab. 2018, 125, 322–331. [Google Scholar] [CrossRef]
- Sharma, G.; Prossnitz, E.R. GPER/GPR30 Knockout mice: effects of GPER on metabolism. Methods Mol. Biol. 2016, 1366, 489–502. [Google Scholar]
- Sharma, G.; Hu, C.; Brigman, J.L.; Zhu, G.; Hathaway, H.J.; Prossnitz, E.R. GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology 2013, 154, 4136–4145. [Google Scholar] [CrossRef] [Green Version]
- Mårtensson, U.E.A.; Salehi, S.A.; Windahl, S.; Gomez, M.F.; Swärd, K.; Daszkiewicz-Nilsson, J.; Wendt, A.; Andersson, N.; Hellstrand, P.; Grände, P.-O.; et al. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology 2009, 150, 687–698. [Google Scholar] [CrossRef]
- Filardo, E.J.; Thomas, P. Minireview: G protein-coupled estrogen receptor-1, GPER-1: its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology 2012, 153, 2953–2962. [Google Scholar] [CrossRef] [Green Version]
- Ford, J.; Hajibeigi, A.; Long, M.; Hahner, L.; Gore, C.; Hsieh, J.-T.; Clegg, D.; Zerwekh, J.; Oz, O.K. GPR30 deficiency causes increased bone mass, mineralization, and growth plate proliferative activity in male mice. J. Bone Miner. Res. 2011, 26, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomatsu, S.; Alméciga-Díaz, C.J.; Montaño, A.M.; Yabe, H.; Tanaka, A.; Dung, V.C.; Giugliani, R.; Kubaski, F.; Mason, R.W.; Yasuda, E.; et al. Therapies for the bone in mucopolysaccharidoses. Mol. Genet. Metab. 2015, 114, 94–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kecskemethy, H.H.; Kubaski, F.; Harcke, H.T.; Tomatsu, S. Bone mineral density in MPS IV A (Morquio syndrome type A). Mol. Genet. Metab. 2016, 117, 144–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deroo, B.J.; Korach, K.S. Estrogen receptors and human disease. J. Clin. Investig. 2006, 116, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Doherty, G.J.; McMahon, H.T. Mediation, modulation, and consequences of membrane-cytoskeleton interactions. Annu. Rev. Biophys. 2008, 37, 65–95. [Google Scholar] [CrossRef] [Green Version]
- Wójciak-Stothard, B.; Curtis, A.S.; Monaghan, W.; McGrath, M.; Sommer, I.; Wilkinson, C.D. Role of the cytoskeleton in the reaction of fibroblasts to multiple grooved substrata. Cell Motil. Cytoskelet. 1995, 31, 147–158. [Google Scholar] [CrossRef]
- Hota, P.K.; Buck, M. Plexin structures are coming: opportunities for multilevel investigations of semaphorin guidance receptors, their cell signaling mechanisms, and functions. Cell. Mol. Life Sci. 2012, 69, 3765–3805. [Google Scholar] [CrossRef]
- Alto, L.T.; Terman, J.R. Semaphorins and their Signaling Mechanisms. Methods Mol. Biol. 2017, 1493, 1–25. [Google Scholar]
- Akiyama, H.; Fukuda, T.; Tojima, T.; Nikolaev, V.O.; Kamiguchi, H. Cyclic nucleotide control of microtubule dynamics for axon guidance. J. Neurosci. 2016, 36, 5636–5649. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.A. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology (Oxford) 2011, 50 (Suppl. 5), v13–v18. [Google Scholar] [CrossRef] [Green Version]
- Tran, M.C.; Lam, J.M. Cutaneous Manifestations of Mucopolysaccharidoses. Pediatric Dermatol. 2016, 33, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Salvalaio, M.; D’Avanzo, F.; Rigon, L.; Zanetti, A.; D’Angelo, M.; Valle, G.; Scarpa, M.; Tomanin, R. Brain RNA-Seq Profiling of the Mucopolysaccharidosis Type II Mouse Model. Int. J. Mol. Sci. 2017, 18, 1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintremil, S.; Ferrer, F.M.; Puente, J.; Pando, M.E.; Valenzuela, M.A. Roles of semaphorins in neurodegenerative diseases. In Neurons-Dendrites and Axons; Arnada-Abreu, G.E., Hernandez-Aguilar, M.E., Eds.; Intech Open: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Pasterkamp, R.J.; Giger, R.J. Semaphorin function in neural plasticity and disease. Curr. Opin. Neurobiol. 2009, 19, 263–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, L.A.T.; Fritz, J.; Pierdant-Mancera, M.; Bagnard, D. Current drug design to target the Semaphorin/Neuropilin/Plexin complexes. Cell Adh. Migr. 2016, 10, 700–708. [Google Scholar] [CrossRef] [Green Version]
- Atabai, K.; Jame, S.; Azhar, N.; Kuo, A.; Lam, M.; McKleroy, W.; Dehart, G.; Rahman, S.; Xia, D.D.; Melton, A.C.; et al. Mfge8 diminishes the severity of tissue fibrosis in mice by binding and targeting collagen for uptake by macrophages. J. Clin. Investig. 2009, 119, 3713–3722. [Google Scholar] [CrossRef] [Green Version]
- Li, E.; Noda, M.; Doi, Y.; Parajuli, B.; Kawanokuchi, J.; Sonobe, Y.; Takeuchi, H.; Mizuno, T.; Suzumura, A. The neuroprotective effects of milk fat globule-EGF factor 8 against oligomeric amyloid β toxicity. J Neuroinflammation 2012, 9, 148. [Google Scholar] [CrossRef] [Green Version]
- Neniskyte, U.; Brown, G.C. Lactadherin/MFG-E8 is essential for microglia-mediated neuronal loss and phagoptosis induced by amyloid β. J. Neurochem. 2013, 126, 312–317. [Google Scholar] [CrossRef]
- Lyons, J.A.; Dickson, P.I.; Wall, J.S.; Passage, M.B.; Ellinwood, N.M.; Kakkis, E.D.; McEntee, M.F. Arterial pathology in canine mucopolysaccharidosis-I and response to therapy. Lab. Investig. 2011, 91, 665–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beard, H.; Hassiotis, S.; Gai, W.-P.; Parkinson-Lawrence, E.; Hopwood, J.J.; Hemsley, K.M. Axonal dystrophy in the brain of mice with Sanfilippo syndrome. Exp. Neurol. 2017, 295, 243–255. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Galvin, J.E.; Lee, V.M.; Rorke, L.B.; Dickson, D.W.; Wolfe, J.H.; Jones, M.Z.; Trojanowski, J.Q. Accumulation of intracellular amyloid-beta peptide (A beta 1-40) in mucopolysaccharidosis brains. J. Neuropathol. Exp. Neurol. 1999, 58, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Martins, C.; Hůlková, H.; Dridi, L.; Dormoy-Raclet, V.; Grigoryeva, L.; Choi, Y.; Langford-Smith, A.; Wilkinson, F.L.; Ohmi, K.; DiCristo, G.; et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 2015, 138, 336–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantopoulos, G.; Iqbal, K.; Dekaban, A.S. Mucopolysaccharidosis types IH, IS, II, and IIIA: glycosaminoglycans and lipids of isolated brain cells and other fractions from autopsied tissues. J. Neurochem. 1980, 34, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Karnati, R.; Talla, V.; Peterson, K.; Laurie, G.W. Lacritin and other autophagy associated proteins in ocular surface health. Exp. Eye Res. 2016, 144, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, M.T.; Fluharty, A.L.; Harris, S.E.; Kihara, H. The accumulation of cerebroside sulfates by fibroblasts in culture from patients with late infantile metachromatic leukodystrophy. Arch. Biochem. Biophys. 1970, 138, 646–652. [Google Scholar] [CrossRef]
- Shah, S.N.; Johnson, R.C.; Stone, R.K.; Mahon-Haft, H. Prevalence of partial cerebroside sulfate sulfatase (arylsulfatase A) defect in adult psychiatric patients. Biol. Psychiatry 1985, 20, 50–57. [Google Scholar] [CrossRef]
- Lee, J.S.; Kanai, K.; Suzuki, M.; Kim, W.S.; Yoo, H.S.; Fu, Y.; Kim, D.-K.; Jung, B.C.; Choi, M.; Oh, K.W.; et al. Arylsulfatase A, a genetic modifier of Parkinson’s disease, is an α-synuclein chaperone. Brain 2019, 142, 2845–2859. [Google Scholar] [CrossRef]
- Winder-Rhodes, S.E.; Garcia-Reitböck, P.; Ban, M.; Evans, J.R.; Jacques, T.S.; Kemppinen, A.; Foltynie, T.; Williams-Gray, C.H.; Chinnery, P.F.; Hudson, G.; et al. Genetic and pathological links between Parkinson’s disease and the lysosomal disorder Sanfilippo syndrome. Mov. Disord. 2012, 27, 312–315. [Google Scholar] [CrossRef]
- Lehri-Boufala, S.; Ouidja, M.-O.; Barbier-Chassefière, V.; Hénault, E.; Raisman-Vozari, R.; Garrigue-Antar, L.; Papy-Garcia, D.; Morin, C. New roles of glycosaminoglycans in α-synuclein aggregation in a cellular model of Parkinson sisease. PLoS ONE 2015, 10, e0116641. [Google Scholar] [CrossRef]
- Gregory, J.M.; Whiten, D.R.; Brown, R.A.; Barros, T.P.; Kumita, J.R.; Yerbury, J.J.; Satapathy, S.; McDade, K.; Smith, C.; Luheshi, L.M.; et al. Clusterin protects neurons against intracellular proteotoxicity. Acta Neuropathol. Commun. 2017, 5, 81. [Google Scholar] [CrossRef]
- Brown, R. The effects of clusterin on the aggregation and pathogenicity of TDP-43, a protein implicated in amyotrophic lateral sclerosis. In University of Wollongong Thesis Collection 1954–2016; The University of Wollongogn Press: Wollongong, Australia, 2015. [Google Scholar]
- Beeg, M.; Stravalaci, M.; Romeo, M.; Carrá, A.D.; Cagnotto, A.; Rossi, A.; Diomede, L.; Salmona, M.; Gobbi, M. Clusterin Binds to Aβ1–42 Oligomers with High Affinity and Interferes with Peptide Aggregation by Inhibiting Primary and Secondary Nucleation. J. Biol. Chem. 2016, 291, 6958–6966. [Google Scholar] [CrossRef] [Green Version]
- Oda, T.; Wals, P.; Osterburg, H.H.; Johnson, S.A.; Pasinetti, G.M.; Morgan, T.E.; Rozovsky, I.; Stine, W.B.; Snyder, S.W.; Holzman, T.F. Clusterin (apoJ) alters the aggregation of amyloid beta-peptide (A beta 1-42) and forms slowly sedimenting A beta complexes that cause oxidative stress. Exp. Neurol. 1995, 136, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Lidström, A.M.; Bogdanovic, N.; Hesse, C.; Volkman, I.; Davidsson, P.; Blennow, K. Clusterin (apolipoprotein J) protein levels are increased in hippocampus and in frontal cortex in Alzheimer’s disease. Exp. Neurol. 1998, 154, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Thambisetty, M.; Simmons, A.; Velayudhan, L.; Hye, A.; Campbell, J.; Zhang, Y.; Wahlund, L.-O.; Westman, E.; Kinsey, A.; Güntert, A.; et al. Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch. Gen. Psychiatry 2010, 67, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Debure, L.; Vayssiere, J.-L.; Rincheval, V.; Loison, F.; Le Drean, Y.; Michel, D. Intracellular clusterin causes juxtanuclear aggregate formation and mitochondrial alteration. J. Cell. Sci. 2003, 116, 3109–3121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansanwal, P.; Li, L.; Sarwal, M.M. Inhibition of intracellular clusterin attenuates cell death in nephropathic cystinosis. J. Am. Soc. Nephrol. 2015, 26, 612–625. [Google Scholar] [CrossRef]
- Khalid, O.; Vera, M.U.; Gordts, P.L.; Ellinwood, N.M.; Schwartz, P.H.; Dickson, P.I.; Esko, J.D.; Wang, R.Y. Immune-mediated inflammation may contribute to the pathogenesis of cardiovascular disease in mucopolysaccharidosis type I. PLoS ONE 2016, 11, e0150850. [Google Scholar] [CrossRef] [Green Version]
- Jackson Laboratories. Available online: https://www.jax.org/strain/005642 (accessed on 25 November 2019).
- Faye, C.; Moreau, C.; Chautard, E.; Jetne, R.; Fukai, N.; Ruggiero, F.; Humphries, M.J.; Olsen, B.R.; Ricard-Blum, S. Molecular interplay between endostatin, integrins, and heparan sulfate. J. Biol. Chem. 2009, 284, 22029–22040. [Google Scholar] [CrossRef] [Green Version]
- Parente, M.K.; Rozen, R.; Cearley, C.N.; Wolfe, J.H. Dysregulation of gene expression in a lysosomal storage disease varies between brain regions implicating unexpected mechanisms of neuropathology. PLoS ONE 2012, 7, e32419. [Google Scholar] [CrossRef]
- Provenzale, J.M.; Nestrasil, I.; Chen, S.; Kan, S.-H.; Le, S.Q.; Jens, J.K.; Snella, E.M.; Vondrak, K.N.; Yee, J.K.; Vite, C.H.; et al. Diffusion tensor imaging and myelin composition analysis reveal abnormal myelination in corpus callosum of canine mucopolysaccharidosis I. Exp. Neurol. 2015, 273, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shipp, M.A.; Vijayaraghavan, J.; Schmidt, E.V.; Masteller, E.L.; D’Adamio, L.; Hersh, L.B.; Reinherz, E.L. Common acute lymphoblastic leukemia antigen (CALLA) is active neutral endopeptidase 24.11 (“enkephalinase”): direct evidence by cDNA transfection analysis. Proc. Natl. Acad. Sci. USA 1989, 86, 297–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sargın, Z.G.; Erin, N.; Tazegul, G.; Elpek, G.Ö.; Yıldırım, B. Profound loss of neprilysin accompanied by decreased levels of neuropeptides and increased CRP in ulcerative colitis. PLoS ONE 2017, 12, e0189526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Elia, E.; Iacovoni, A.; Vaduganathan, M.; Lorini, F.L.; Perlini, S.; Senni, M. Neprilysin inhibition in heart failure: mechanisms and substrates beyond modulating natriuretic peptides. Eur. J. Heart Fail. 2017, 19, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, Y.; Hashiguchi, A.; Yuan, J.; Yoshimura, A.; Mitsui, J.; Ishiura, H.; Tanaka, M.; Ishihara, S.; Tanabe, H.; Nozuma, S.; et al. Mutations in MME cause an autosomal-recessive Charcot-Marie-Tooth disease type 2. Ann. Neurol. 2016, 79, 659–672. [Google Scholar] [CrossRef] [Green Version]
- Lupo, V.; Frasquet, M.; Sánchez-Monteagudo, A.; Pelayo-Negro, A.L.; García-Sobrino, T.; Sedano, M.J.; Pardo, J.; Misiego, M.; García-García, J.; Sobrido, M.J.; et al. Characterising the phenotype and mode of inheritance of patients with inherited peripheral neuropathies carrying MME mutations. J. Med. Genet. 2018, 55, 814–823. [Google Scholar] [CrossRef]
- Wewer Albrechtsen, N.J.; Kuhre, R.E.; Pedersen, J.; Knop, F.K.; Holst, J.J. The biology of glucagon and the consequences of hyperglucagonemia. Biomark. Med. 2016, 10, 1141–1151. [Google Scholar] [CrossRef] [Green Version]
- Michaels, L.A.; Ohene-Frempong, K.; Zhao, H.; Douglas, S.D. Serum levels of substance P are elevated in patients with sickle cell disease and increase further during vaso-occlusive crisis. Blood 1998, 92, 3148–3151. [Google Scholar] [CrossRef]
- Miners, J.S.; Van Helmond, Z.; Chalmers, K.; Wilcock, G.; Love, S.; Kehoe, P.G. Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 2006, 65, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Miners, S.; van Helmond, Z.; Barker, R.; Passmore, P.A.; Johnston, J.A.; Todd, S.; McGuinness, B.M.; Panza, F.; Seripa, D.; Solfrizzi, V.; et al. Genetic variation in MME in relation to neprilysin protein and enzyme activity, Aβ levels, and Alzheimer’s disease risk. Int. J. Mol. Epidemiol. Genet. 2012, 3, 30–38. [Google Scholar]
- Madani, R.; Poirier, R.; Wolfer, D.P.; Welzl, H.; Groscurth, P.; Lipp, H.-P.; Lu, B.; El Mouedden, M.; Mercken, M.; Nitsch, R.M.; et al. Lack of neprilysin suffices to generate murine amyloid-like deposits in the brain and behavioral deficit in vivo. J. Neurosci. Res. 2006, 84, 1871–1878. [Google Scholar] [CrossRef]
- Hersh, L.B.; Rodgers, D.W. Neprilysin and amyloid beta peptide degradation. Curr. Alzheimer Res. 2008, 5, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Farris, W.; Schütz, S.G.; Cirrito, J.R.; Shankar, G.M.; Sun, X.; George, A.; Leissring, M.A.; Walsh, D.M.; Qiu, W.Q.; Holtzman, D.M.; et al. Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am. J. Pathol. 2007, 171, 241–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wang, J.; Zhang, S.; Liu, Z. Neprilysin gene transfer: A promising therapeutic approach for Alzheimer’s disease. J. Neurosci. Res. 2015, 93, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Nalivaeva, N.N.; Belyaev, N.D.; Zhuravin, I.A.; Turner, A.J. The Alzheimer’s amyloid-degrading peptidase, neprilysin: can we control it? Int. J. Alzheimers Dis. 2012, 2012, 383796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marr, R.A.; Rockenstein, E.; Mukherjee, A.; Kindy, M.S.; Hersh, L.B.; Gage, F.H.; Verma, I.M.; Masliah, E. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J. Neurosci. 2003, 23, 1992–1996. [Google Scholar] [CrossRef] [Green Version]
- Spencer, B.; Marr, R.A.; Gindi, R.; Potkar, R.; Michael, S.; Adame, A.; Rockenstein, E.; Verma, I.M.; Masliah, E. Peripheral delivery of a CNS targeted, metalo-protease reduces aβ toxicity in a mouse model of Alzheimer’s disease. PLoS ONE 2011, 6, e16575. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, J.; Liu, J.; Liu, F. A novel system for in vivo neprilysin gene delivery using a syringe electrode. J. Neurosci. Methods 2010, 193, 226–231. [Google Scholar] [CrossRef]
- Xu, X.-J.; Hökfelt, T.; Wiesenfeld-Hallin, Z. Galanin and spinal pain mechanisms: where do we stand in 2008? Cell. Mol. Life Sci. 2008, 65, 1813–1819. [Google Scholar] [CrossRef]
- Xu, X.J.; Hökfelt, T.; Bartfai, T.; Wiesenfeld-Hallin, Z. Galanin and spinal nociceptive mechanisms: recent advances and therapeutic implications. Neuropeptides 2000, 34, 137–147. [Google Scholar] [CrossRef]
- Lang, R.; Gundlach, A.L.; Kofler, B. The galanin peptide family: receptor pharmacology, pleiotropic biological actions, and implications in health and disease. Pharmacol. Ther. 2007, 115, 177–207. [Google Scholar] [CrossRef]
- Counts, S.E.; Perez, S.E.; Mufson, E.J. Galanin in Alzheimer’s disease: neuroinhibitory or neuroprotective? Cell. Mol. Life Sci. 2008, 65, 1842–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; MacTavish, D.; Kar, S.; Jhamandas, J.H. Galanin attenuates beta-amyloid (Abeta) toxicity in rat cholinergic basal forebrain neurons. Neurobiol. Dis. 2006, 21, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; La Vecchia, C.; de Groh, M.; Negri, E.; Morrison, H.; Mery, L. Canadian Cancer Registries Epidemiology Research Group Dietary cholesterol intake and cancer. Ann. Oncol. 2012, 23, 491–500. [Google Scholar] [CrossRef]
- Cruz, P.M.R.; Mo, H.; McConathy, W.J.; Sabnis, N.; Lacko, A.G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: a review of scientific findings, relevant to future cancer therapeutics. Front Pharmacol. 2013, 4, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvente-Poirot, S.; Poirot, M. Cholesterol metabolism and cancer: the good, the bad and the ugly. Curr. Opin. Pharmacol. 2012, 12, 673–676. [Google Scholar] [CrossRef]
- Plump, A.S.; Smith, J.D.; Hayek, T.; Aalto-Setälä, K.; Walsh, A.; Verstuyft, J.G.; Rubin, E.M.; Breslow, J.L. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 1992, 71, 343–353. [Google Scholar] [CrossRef]
- Gordon, I.; Grauer, E.; Genis, I.; Sehayek, E.; Michaelson, D.M. Memory deficits and cholinergic impairments in apolipoprotein E-deficient mice. Neurosci. Lett. 1995, 199, 1–4. [Google Scholar] [CrossRef]
- Rashidi, O.M.; Nazar, F.A.; Alama, M.N.; Awan, Z.A. Interpreting the Mechanism of APOE (p. Leu167del) Mutation in the incidence of familial hypercholesterolemia; an in-silico approach. Open Cardiovasc Med. J. 2017, 11, 84–93. [Google Scholar] [CrossRef]
- Huang, R.; Zong, X.; Nadesan, P.; Huebner, J.L.; Kraus, V.B.; White, J.P.; White, P.J.; Baht, G.S. Lowering circulating apolipoprotein E levels improves aged bone fracture healing. JCI Insight 2019, 4, 129144. [Google Scholar] [CrossRef]
- Orgeig, S.; Paget, T.; Duplock, S.; Snel, M.; Hemsley, K.; Parkinson-Lawrence, E. Changes in lipid metabolism in mucopolysaccharidosis (MPS) IIIA mouse lung tissue and pulmonary surfactant. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef]
- McGlynn, R.; Dobrenis, K.; Walkley, S.U. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J. Comp. Neurol. 2004, 480, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Fountoulakis, M.; Cairns, N.J.; Lubec, G. Human brain nucleoside diphosphate kinase activity is decreased in Alzheimer’s disease and Down syndrome. Biochem. Biophys. Res. Commun. 2002, 296, 970–975. [Google Scholar] [PubMed]
- Qiu, Y.; Feng, Y.; Hammes, H.-P.; Skolnik, E.; Wieland, T.; Zhao, D. Deficiency in Nucleoside Diphosphate Kinase B Aggravates the Development of Diabetic Retinopathy through Upregulation of Angiopoietin-2 via FOXO1-Virtual Meeting | EASD. Available online: https://www.easd.org/virtualmeeting/home.html#!resources/deficiency-in-nucleoside-diphosphate-kinase-b-aggravates-the-development-of-diabetic-retinopathy-through-upregulation-of-angiopoietin-2-via-foxo1--2 (accessed on 24 November 2019).
- Igawa, M.; Rukstalis, D.B.; Tanabe, T.; Chodak, G.W. High levels of nm23 expression are related to cell proliferation in human prostate cancer. Cancer Res. 1994, 54, 1313–1318. [Google Scholar]
- Lacombe, M.L.; Sastre-Garau, X.; Lascu, I.; Vonica, A.; Wallet, V.; Thiery, J.P.; Véron, M. Overexpression of nucleoside diphosphate kinase (Nm23) in solid tumours. Eur. J. Cancer 1991, 27, 1302–1307. [Google Scholar] [CrossRef]
- Allard, L.; Burkhard, P.R.; Lescuyer, P.; Burgess, J.A.; Walter, N.; Hochstrasser, D.F.; Sanchez, J.-C. PARK7 and nucleoside diphosphate kinase A as plasma markers for the early diagnosis of stroke. Clin. Chem. 2005, 51, 2043–2051. [Google Scholar] [CrossRef] [PubMed]
- Lutz, S.; Mura, R.; Baltus, D.; Movsesian, M.; Kübler, W.; Niroomand, F. Increased activity of membrane-associated nucleoside diphosphate kinase and inhibition of cAMP synthesis in failing human myocardium. Cardiovasc. Res. 2001, 49, 48–55. [Google Scholar] [CrossRef] [Green Version]
- King, J.D.; Lee, J.; Riemen, C.E.; Neumann, D.; Xiong, S.; Foskett, J.K.; Mehta, A.; Muimo, R.; Hallows, K.R. Role of binding and nucleoside diphosphate kinase A in the regulation of the cystic fibrosis transmembrane conductance regulator by AMP-activated protein kinase. J. Biol. Chem. 2012, 287, 33389–33400. [Google Scholar] [CrossRef] [Green Version]
- Borthwick, L.A.; Kerbiriou, M.; Taylor, C.J.; Cozza, G.; Lascu, I.; Postel, E.H.; Cassidy, D.; Trouvé, P.; Mehta, A.; Robson, L.; et al. Role of Interaction and Nucleoside Diphosphate Kinase B in Regulation of the Cystic Fibrosis Transmembrane Conductance Regulator Function by cAMP-Dependent Protein Kinase A. PLoS ONE 2016, 11, e0149097. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Macwan, I.; Singh, S.; Silwal, S.; Patra, P. A Computational Approach for Understanding the Interactions between Graphene Oxide and Nucleoside Diphosphate Kinase with Implications for Heart Failure. Nanomaterials (Basel) 2018, 8, 57. [Google Scholar] [CrossRef] [Green Version]
- Glass, D.; Viñuela, A.; Davies, M.N.; Ramasamy, A.; Parts, L.; Knowles, D.; Brown, A.A.; Hedman, A.K.; Small, K.S.; Buil, A.; et al. Gene expression changes with age in skin, adipose tissue, blood and brain. Genome Biol. 2013, 14, R75. [Google Scholar] [CrossRef] [Green Version]
- Marthandan, S.; Priebe, S.; Baumgart, M.; Groth, M.; Cellerino, A.; Guthke, R.; Hemmerich, P.; Diekmann, S. Similarities in gene expression profiles during in vitro aging of primary human embryonic lung and foreskin fibroblasts. Biomed. Res. Int. 2015, 2015, 731938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleischer, J.G.; Schulte, R.; Tsai, H.H.; Tyagi, S.; Ibarra, A.; Shokhirev, M.N.; Huang, L.; Hetzer, M.W.; Navlakha, S. Predicting age from the transcriptome of human dermal fibroblasts. Genome Biol. 2018, 19, 221. [Google Scholar] [CrossRef] [PubMed]
- Pierzynowska, K.; Gaffke, L.; Cyske, Z.; Węgrzyn, G. Genistein induces degradation of mutant huntingtin in fibroblasts from Huntington’s disease patients. Metab. Brain Dis. 2019, 34, 715–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MPS Type | Stored GAG(s) | Defective Enzyme |
|---|---|---|
| MPS I | HS, DS | α-L-iduronidase |
| MPS II | HS, DS | 2-iduronate sulfatase |
| MPS IIIA | HS | N-sulfoglucosamine sulfhydrolase |
| MPS IIIB | HS | α-N-acetylglucosaminidase |
| MPS IIIC | HS | Acetyl-CoA:α-glycosaminide acetyltransferase |
| MPS IIID | HS | N-acetylglucosamine 6-sulfatase |
| MPS IVA | KS, CS | N-acetylgalactosaminide 6-sulfatase |
| MPS IVB | KS | β-galactosidase-1 |
| MPS VI | DS | N-acetylgalactosamine 4-sulfatase |
| MPS VII | HS, DS, CS | β-glucuronidase |
| MPS IX | HA | Hyaluronidase-1 |
| Cell Line | Race | Sex | Age (Years) | Locus of Mutated Gene | Mutation Type |
|---|---|---|---|---|---|
| MPS I | Caucasian | F | 1 | IDUA, 4p16.3 | Homozygote p.Trp402Ter/p.Trp402Ter |
| MPS II | Caucasian (ethnicity: Haitian) | M | 3 | IDS, Xp28 | Hemizygote p.His70ProfsTer29 |
| MPS IIIA | Caucasian | F | 3 | SGSH, 17q25.3 | Complex heterozygote p.Glu447Lys/p.Arg245His |
| MPS IIIB | Caucasian | M | 7 | NAGLU, 17q21 | Homozygote p.Arg626Ter/p.Arg626Ter |
| MPS IIIC | unknown | M | 8 | HGSNAT, 8p11.1 | ND |
| MPS IIID | Asian Indian | M | 7 | GNS, 12q14 | Homozygote p.Arg355Ter/p.Arg355Ter |
| MPS IVA | Caucasian (ethnicity: Mexican) | F | 7 | GALNS, 16q24.3 | ND |
| MPS IVB | Caucasian | F | 4 | GLB1, 3p22.3 | Complex heterozygote p.Trp273Leu/p.Trp509Cys |
| MPS VI | Black | F | 3 | ARSB, 4q14.1 | ND |
| MPS VII | African American | M | 3 | GUSB, 7q21.11 | Complex heterozygote p.Trp627Cys/p.Arg356Ter |
| MPS IX | unknown | F | 14 | HYAL1, 3p.21.3 | ND |
| Transcripts | No. of Transcripts with Changed Expression Levels in MPS vs. HDFa Line | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| I | II | IIIA | IIIB | IIIC | IIID | IVA | IVB | VI | VII | IX | |
| Cell activation (GO:0001775) | |||||||||||
| Up-regulated | 18 | 16 | 19 | 33 | 24 | 16 | 8 | 29 | 8 | 25 | 22 |
| Down-regulated | 35 | 11 | 38 | 27 | 32 | 18 | 10 | 47 | 11 | 20 | 31 |
| Total | 53 | 27 | 57 | 60 | 56 | 34 | 18 | 76 | 19 | 45 | 53 |
| Cell growth (GO:0016049) | |||||||||||
| Up-regulated | 7 | 2 | 9 | 15 | 14 | 7 | 4 | 11 | 6 | 10 | 14 |
| Down-regulated | 11 | 0 | 14 | 10 | 11 | 9 | 4 | 8 | 2 | 7 | 9 |
| Total | 18 | 2 | 23 | 25 | 25 | 16 | 8 | 19 | 8 | 17 | 23 |
| Cell division (GO:0051301) | |||||||||||
| Up-regulated | 9 | 2 | 7 | 6 | 6 | 5 | 2 | 10 | 1 | 10 | 2 |
| Down-regulated | 11 | 6 | 8 | 14 | 11 | 19 | 0 | 7 | 1 | 16 | 18 |
| Total | 18 | 8 | 15 | 20 | 17 | 24 | 2 | 17 | 2 | 26 | 20 |
| Cell recognition (GO:0008037) | |||||||||||
| Up-regulated | 3 | 2 | 5 | 3 | 4 | 4 | 2 | 5 | 0 | 3 | 3 |
| Down-regulated | 3 | 1 | 0 | 1 | 3 | 2 | 2 | 1 | 3 | 3 | 3 |
| Total | 6 | 3 | 5 | 4 | 7 | 6 | 4 | 6 | 3 | 6 | 6 |
| Gene | Value of Fold Change (FC) of Transcript Level in MPS Types | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| I | II | IIIA | IIIB | IIIC | IIID | IVA | IVB | VI | VII | IX | |
| Cell activation | |||||||||||
| DNAJC3 | 0.6 ± 0.0 | - | - | 0.7 ± 0.1 | 0.6 ± 0.0 | - | 0.7 ± 0.1 | 0.6 ± 0.0 | 0.7 ± 0.1 | 0.9 ± 0.1 | 0.7 ± 0.0 |
| GATA2 | 2.6 ± 0.5 | - | - | 5.8 ± 0.9 | 4.7 ± 0.7 | 3.4 ± 0.6 | - | 4.9 ± 0.6 | - | 3.5 ± 0.5 | 3.1 ± 0.3 |
| GPER1 | 0.4 ± 0.1 | - | 0.4 ± 0.1 | - | 0.3 ± 0.1 | 0.4 ± 0.0 | 0.5 ± 0.1 | 0.4 ± 0.1 | - | - | 0.3 ± 0.1 |
| PRKCD | 0.4 ± 0.1 | - | 0.4 ± 0.1 | - | 0.6 ± 0.1 | - | 0.6 ± 0.1 | 0.5 ± 0.1 | 0.4 ± 0.1 | - | - |
| DYNC1LI1 | 0.7 ± 0.0 | 0.7 ± 1.9 | 0.6 ± 0.0 | 0.6 ± 0.1 | 0.7 ± 0.0 | - | - | 0.7 ± 0.0 | - | 0.7 ± 0.1 | 0.6 ± 0.1 |
| RAP1A | - | 0.7 ± 0.0 | 0.6 ± 0.0 | 0.7 ± 0.1 | - | - | - | 0.7 ± 0.0 | 0.8 ± 0.0 | 0.7 ± 0.1 | 0.7 ± 0.0 |
| CD9 | - | 9.3 ± 3.3 | 13.5 ± 2.1 | 17.1 ± 4.6 | 15.9 ± 3.7 | 15.5 ± 7.7 | - | 28.9 ± 4.0 | 12.4 ± 3.8 | - | 12.8 ± 3.6 |
| TMEM30A | - | 0.7 ± 0.1 | 0.6 ± 0.0 | - | 0.6 ± 0.0 | - | 0.7 ± 0.1 | 0.6 ± 0.1 | - | 0.8 ± 0.0 | - |
| CLU | 6.4 ± 0.7 | 7.9 ± 1.6 | - | 3.4 ± 0.7 | 7.8 ± 1.6 | - | - | 11.0 ± 3.2 | 6.0 ± 0.8 | 4.2 ± 1.4 | 10.9 ± 1.4 |
| Cell growth | |||||||||||
| EXOSC9 | 0.2 ± 0.1 | - | 0.5 ± 0.2 | 0.3 ± 0.2 | 0.2 ± 0.1 | 0.3 ± 0.1 | 0.3 ± 0.3 | 0.5 ± 0.2 | - | - | 0.3 ± 0.2 |
| PLXNA1 | 1.8 ± 0.0 | - | 2.3 ± 0.3 | 1.7 ± 0.1 | 1.6 ± 0.1 | - | - | 1.9 ± 0.1 | - | - | 1.8 ± 0.1 |
| TMEM97 | 0.4 ± 0.1 | - | - | 0.2 ± 0.1 | 0.4 ± 0.1 | 0.3 ± 0.1 | - | 0.5 ± 0.1 | - | 0.3 ± 0.1 | 0.2 ± 0.1 |
| KAZALD1 | - | - | 3.9 ± 0.5 | 2.5 ± 0.4 | 2.6 ± 0.5 | - | 3.7 ± 0.5 | 2.9 ± 0.3 | 3.3 ± 0.6 | - | - |
| TRPV2 | 0.1 ± 0.1 | - | 0.2 ± 0.1 | 0.1 ± 0.0 | 0.4 ± 0.1 | - | - | 0.4 ± 0.1 | - | 0.1 ± 0.1 | 0.1 ± 0.0 |
| Cell recognition | |||||||||||
| ARSA | 2.0 ± 0.4 | 2.3 ± 0.3 | 2.2 ± 0.3 | 2.1 ± 0.3 | - | 2.8 ± 0.6 | 2.0 ± 0.3 | 1.8 ± 0.2 | - | 2.6 ± 0.4 | 2.2 ± 0.2 |
| MFGE8 | 6.2 ± 1.2 | 3.1 ± 0.9 | 14.9 ± 3.1 | 8.3 ± 1.8 | 3.2 ± 0.9 | - | - | 6.8 ± 1.0 | - | - | 4.2 ± 0.5 |
| Cell division | |||||||||||
| DYNC1LI1 | 0.7 ± 0.0 | 0.7 ± 0.1 | 0.6 ± 0.0 | 0.6 ± 0.1 | 0.7 ± 0.0 | - | - | 0.7 ± 0.0 | - | 0.7 ± 0.1 | 0.6 ± 0.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rintz, E.; Gaffke, L.; Podlacha, M.; Brokowska, J.; Cyske, Z.; Węgrzyn, G.; Pierzynowska, K. Transcriptomic Changes Related to Cellular Processes with Particular Emphasis on Cell Activation in Lysosomal Storage Diseases from the Group of Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 3194. https://doi.org/10.3390/ijms21093194
Rintz E, Gaffke L, Podlacha M, Brokowska J, Cyske Z, Węgrzyn G, Pierzynowska K. Transcriptomic Changes Related to Cellular Processes with Particular Emphasis on Cell Activation in Lysosomal Storage Diseases from the Group of Mucopolysaccharidoses. International Journal of Molecular Sciences. 2020; 21(9):3194. https://doi.org/10.3390/ijms21093194
Chicago/Turabian StyleRintz, Estera, Lidia Gaffke, Magdalena Podlacha, Joanna Brokowska, Zuzanna Cyske, Grzegorz Węgrzyn, and Karolina Pierzynowska. 2020. "Transcriptomic Changes Related to Cellular Processes with Particular Emphasis on Cell Activation in Lysosomal Storage Diseases from the Group of Mucopolysaccharidoses" International Journal of Molecular Sciences 21, no. 9: 3194. https://doi.org/10.3390/ijms21093194
APA StyleRintz, E., Gaffke, L., Podlacha, M., Brokowska, J., Cyske, Z., Węgrzyn, G., & Pierzynowska, K. (2020). Transcriptomic Changes Related to Cellular Processes with Particular Emphasis on Cell Activation in Lysosomal Storage Diseases from the Group of Mucopolysaccharidoses. International Journal of Molecular Sciences, 21(9), 3194. https://doi.org/10.3390/ijms21093194