Short Exposure to Ethanol Diminishes Caspase-1 and ASC Activation in Human HepG2 Cells In Vitro

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Inflammasome Activation upon Ethanol Administration in Hepato-Derived Cells
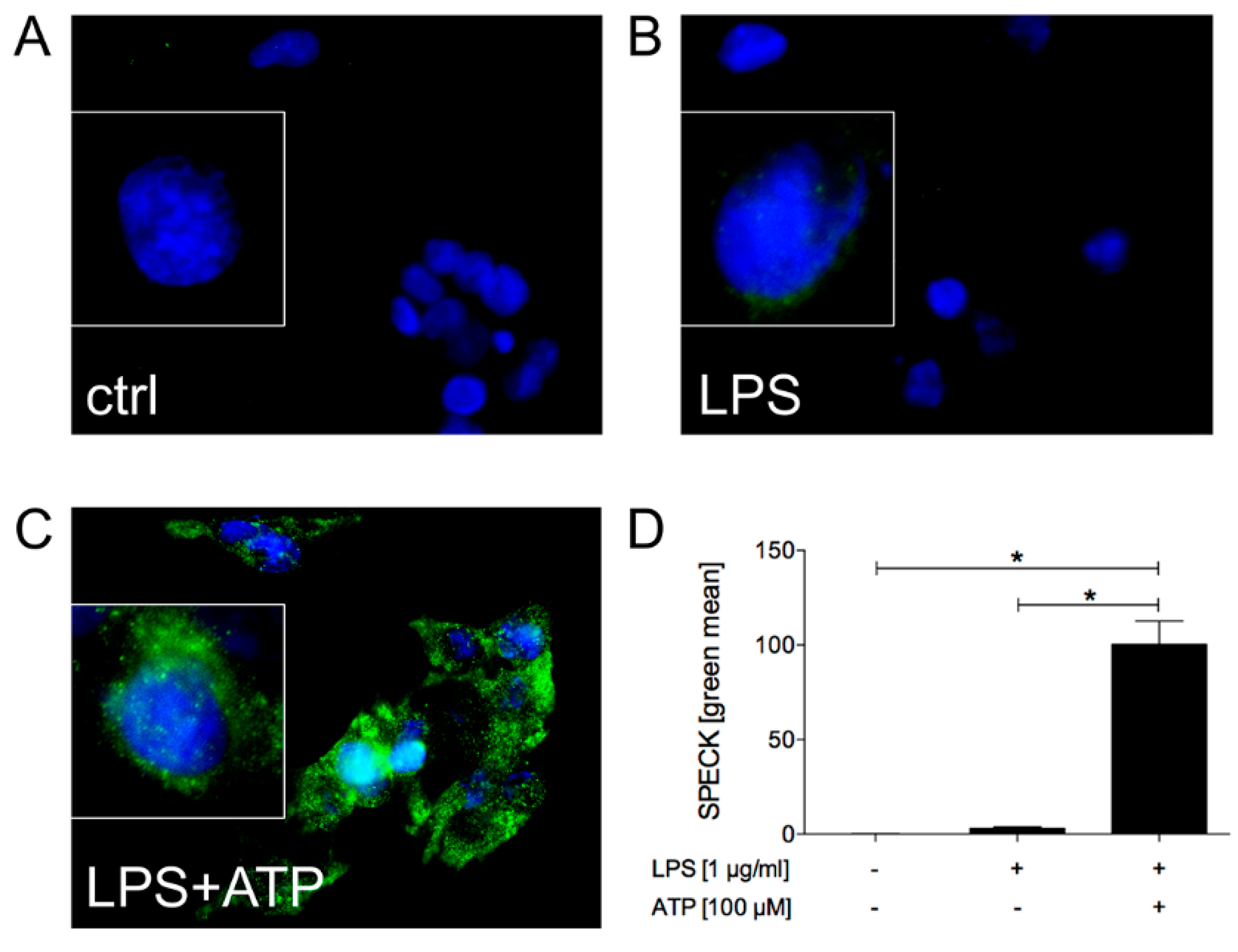
2.1.1. LPS and ATP Induce ASC Speck Formation and Activate Caspase-1 in HepG2 Cells
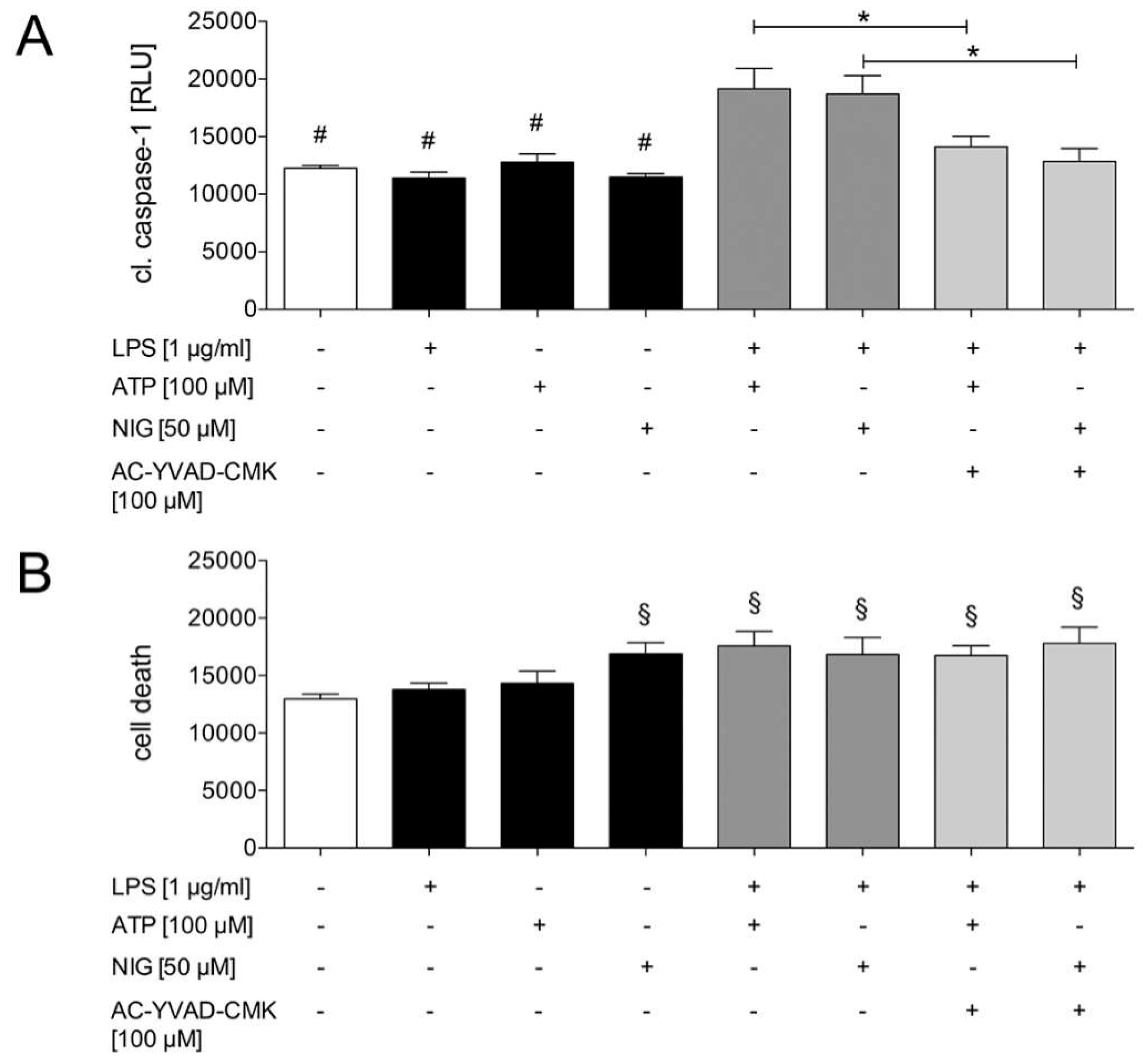
2.1.2. Caspase-1 Activation in HepG2 Cells is Mediated by the Purinergic P2X7 Receptor
2.1.3. Ethanol Diminishes Caspase-1 Activity and ASC Speck Formation
2.1.4. LPS + ATP-Induced ROS Generation Was Decreased by Ethanol Administration
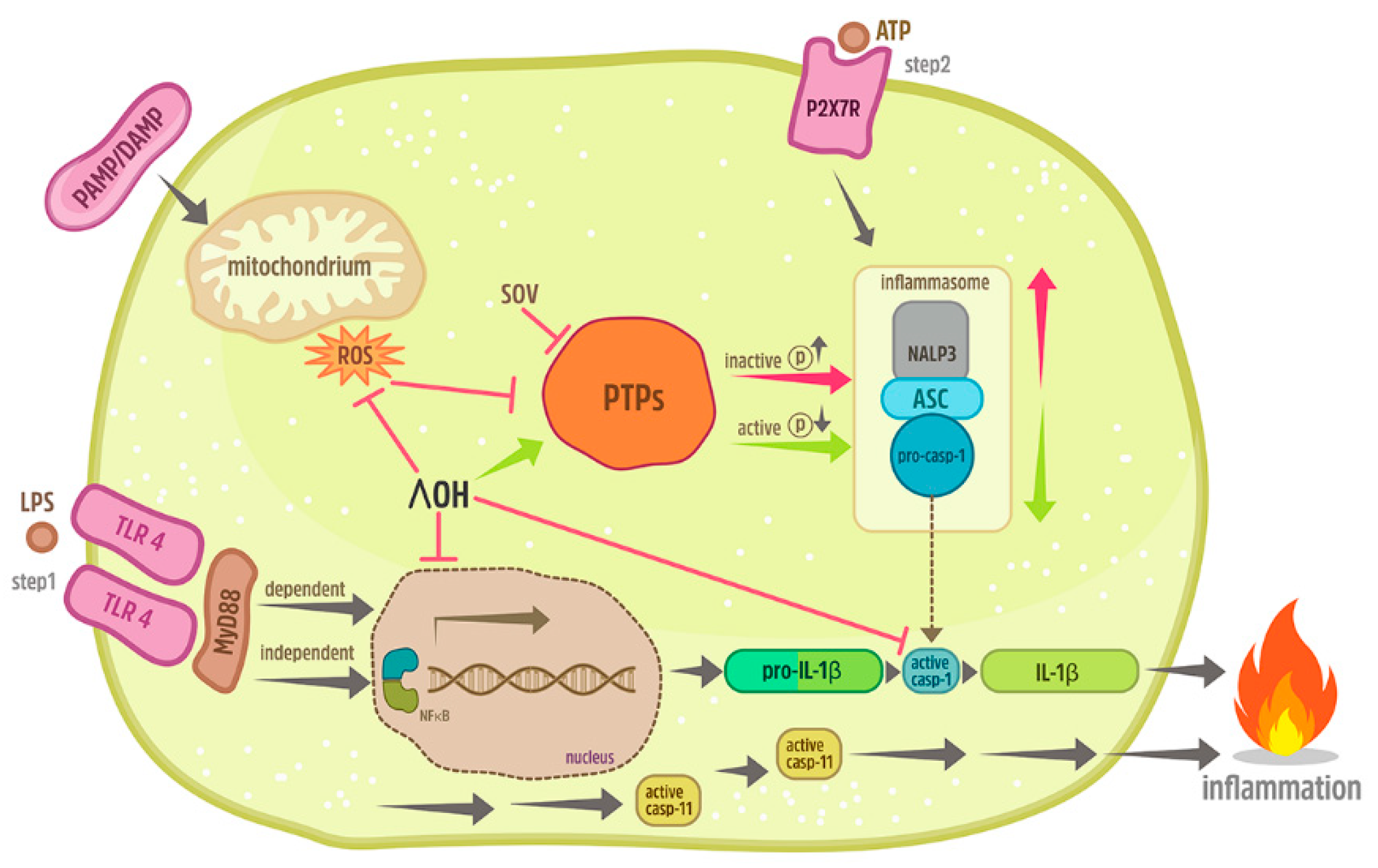
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Stimulation Protocol
4.4. Immune Cytological Staining of SPECKs
4.5. Knock Down of Inflammasome Components Via Small Interfering (si)RNA
4.6. Caspase-1 Activity Assay
4.7. Cell Viability
4.8. Determination of the NF-κB Activity
4.9. Measurement of the Intracellular ROS
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Awad, F.; Assrawi, E.; Louvrier, C.; Jumeau, C.; Georgin-Lavialle, S.; Grateau, G.; Amselem, S.; Giurgea, I.; Karabina, S.A. Inflammasome biology, molecular pathology and therapeutic implications. Pharmacol. Ther. 2018, 187, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, H.M.; Mueller, J.L.; Broide, D.H.; Wanderer, A.A.; Kolodner, R.D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 2001, 29, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2012, 493, 674–678. [Google Scholar] [CrossRef]
- Place, D.E.; Kanneganti, T.D. Recent advances in inflammasome biology. Curr. Opin. Immunol. 2017, 50, 32–38. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.L.; Stutz, A.; Alnemri, E.S.; Macdonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J.P.V. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Petrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2010, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.M.; Rouge, L.; Wiesmann, C.; Scheer, J.M. Crystal structure of procaspase-1 zymogen domain reveals insight into inflammatory caspase autoactivation. J. Biol. Chem. 2008, 284, 6546–6553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornberry, N.A.; Bull, H.G.; Calaycay, J.R.; Chapman, K.T.; Howard, A.D.; Kostura, M.J.; Miller, U.K.; Molineaux, S.M.; Weidner, J.R.; Aunins, J.; et al. A novel heterodimeric cysteine protease is required for interleukin-1βprocessing in monocytes. Nature 1992, 356, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.A.; Cookson, B.T. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol. Microbiol. 2000, 38, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Parker, R.; Kim, S.J.; Gao, B. Alcohol, adipose tissue and liver disease: Mechanistic links and clinical considerations. Nat. Rev. Gastroenterol. Hepatol. 2017, 15, 50–59. [Google Scholar] [CrossRef]
- Relja, B.; Höhn, C.; Bormann, F.; Seyboth, K.; Henrich, D.; Marzi, I.; Lehnert, M. Acute alcohol intoxication reduces mortality, inflammatory responses and hepatic injury after haemorrhage and resuscitation in vivo. Br. J. Pharmacol. 2012, 165, 1188–1199. [Google Scholar] [CrossRef] [Green Version]
- Relja, B.; Menke, J.; Wagner, N.; Auner, B.; Voth, M.; Nau, C.; Marzi, I. Effects of positive blood alcohol concentration on outcome and systemic interleukin-6 in major trauma patients. Injury 2016, 47, 640–645. [Google Scholar] [CrossRef]
- Jerrells, T.R.; Pavlik, J.A.; DeVasure, J.; Vidlak, D.; Costello, A.; Strachota, J.M.; Wyatt, T.A. Association of chronic alcohol consumption and increased susceptibility to and pathogenic effects of pulmonary infection with respiratory syncytial virus in mice. Alcohol 2007, 41, 357–369. [Google Scholar] [CrossRef] [Green Version]
- Lippai, R.; Bala, S.; Petrasek, J.; Csak, T.; Levin, I.; Kurt-Jones, E.A.; Szabo, G. Alcohol-induced IL-1β in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. J. Leukoc. Biol. 2013, 94, 171–182. [Google Scholar] [CrossRef]
- Aruna, R.; Geetha, A.; Suguna, P. Rutin modulates ASC expression in NLRP3 inflammasome: A study in alcohol and cerulein-induced rat model of pancreatitis. Mol. Cell. Biochem. 2014, 396, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Diehl, A.M. Cytokines in Alcoholic and Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2000, 343, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Valles, S.L.; Blanco, A.; Azorín, I.; Guasch, R.; Pascual, M.; Gomez-Lechon, M.J.; Renau-Piqueras, J.; Guerri, C. Chronic Ethanol Consumption Enhances Interleukin-1-Mediated Signal Transduction in Rat Liver and in Cultured Hepatocytes. Alcohol. Clin. Exp. Res. 2003, 27, 1979–1986. [Google Scholar] [CrossRef]
- Petrášek, J.; Bala, S.; Csak, T.; Lippai, R.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef] [Green Version]
- Nurmi, K.; Virkanen, J.; Rajamäki, K.; Niemi, K.; Kovanen, P.T.; Eklund, K.K. Ethanol Inhibits Activation of NLRP3 and AIM2 Inflammasomes in Human Macrophages–A Novel Anti-Inflammatory Action of Alcohol. PLoS ONE 2013, 8, e78537. [Google Scholar] [CrossRef] [Green Version]
- Hoyt, L.; Ather, J.; Randall, M.; Depuccio, D.; Landry, C.C.; Wewers, M.D.; Gavrilin, M.; Poynter, M. Ethanol and Other Short-Chain Alcohols Inhibit NLRP3 Inflammasome Activation through Protein Tyrosine Phosphatase Stimulation. J. Immunol. 2016, 197, 1322–1334. [Google Scholar] [CrossRef] [Green Version]
- Mörs, K.; Hörauf, J.A.; Kany, S.; Wagner, N.; Sturm, R.; Woschek, M.; Perl, M.; Marzi, I.; Relja, B. Ethanol Decreases Inflammatory Response in Human Lung Epithelial Cells by Inhibiting the Canonical NF-kB-Pathway. Cell. Physiol. Biochem. 2017, 43, 17–30. [Google Scholar]
- Relja, B.; Omid, N.; Kontradowitz, K.; Jurida, K.; Oppermann, E.; Störmann, P.; Werner, I.; Juengel, E.; Seebach, C.; Marzi, I. Decreased Inflammatory Responses of Human Lung Epithelial Cells after Ethanol Exposure Are Mimicked by Ethyl Pyruvate. Mediat. Inflamm. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- Relja, B.; Omid, N.; Wagner, N.; Mörs, K.; Werner, I.; Juengel, E.; Perl, M.; Marzi, I. Ethanol, ethyl and sodium pyruvate decrease the inflammatory responses of human lung epithelial cells via Akt and NF-κB in vitro but have a low impact on hepatocellular cells. Int. J. Mol. Med. 2015, 37, 517–525. [Google Scholar] [CrossRef]
- Chandrasekar, A.; Heuvel, F.O.; Palmer, A.; Linkus, B.; Ludolph, A.C.; Boeckers, T.M.; Relja, B.; Huber-Lang, M.; Roselli, F. Acute ethanol administration results in a protective cytokine and neuroinflammatory profile in traumatic brain injury. Int. Immunopharmacol. 2017, 51, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Baroja-Mazo, A.; Martín-Sánchez, F.; Gomez, A.I.; Martínez, C.M.; Amores-Iniesta, J.; Compan, V.; Barberà-Cremades, M.; Yagüe, J.; Ruiz-Ortiz, E.; Antón, J.; et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 2014, 15, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Xaus, J.; Comalada, M.; Valledor, A.F.; Lloberas, J.; López-Soriano, F.; Argilés, J.M.; Bogdan, C.; Celada, A. LPS induces apoptosis in macrophages mostly through the autocrine production of TNF-alpha. Blood 2000, 95, 3823–3831. [Google Scholar] [CrossRef] [PubMed]
- Raza, H.; John, A.; Shafarin, J. Potentiation of LPS-Induced Apoptotic Cell Death in Human Hepatoma HepG2 Cells by Aspirin via ROS and Mitochondrial Dysfunction: Protection by N-Acetyl Cysteine. PLoS ONE 2016, 11, e0159750. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Takeuchi, O.; Fujita, T.; Inoue, J.I.; Mühlradt, P.F.; Sato, S.; Hoshino, K.; Akira, S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 2001, 167, 5887–5894. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of Adaptor TRIF in the MyD88-Independent Toll-Like Receptor Signaling Pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Vanaja, S.K.; Waggoner, L.; Sokolovska, A.; Becker, C.; Stuart, L.M.; Leong, J.M.; Fitzgerald, K.A. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 2012, 150, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Goral, J.; Choudhry, M.A.; Kovacs, E.J. Acute ethanol exposure inhibits macrophage IL-6 production: Role of p38 and ERK1/2 MAPK. J. Leukoc. Biol. 2003, 75, 553–559. [Google Scholar] [CrossRef]
- Hara, H.; Tsuchiya, K.; Kawamura, I.; Fang, R.; Hernandez-Cuellar, E.; Shen, Y.; Mizuguchi, J.; Schweighoffer, E.; Tybulewicz, V.L.; Mitsuyama, M. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 2013, 14, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Song, N.; Liu, Z.S.; Xue, W.; Bai, Z.F.; Wang, Q.Y.; Dai, J.; Liu, X.; Huang, Y.J.; Cai, H.; Zhan, X.Y.; et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol. Cell 2017, 68, 185–197.e6. [Google Scholar] [CrossRef] [Green Version]
- Spalinger, M.R.; Kasper, S.; Gottier, C.; Lang, S.; Atrott, K.; Vavricka, S.R.; Scharl, S.; Raselli, T.; Frey-Wagner, I.; Gutte, P.M.; et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J. Clin. Investig. 2016, 126, 4388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvestad, R.M.; Grosshans, D.R.; Coultrap, S.J.; Nakazawa, T.; Yamamoto, T.; Browning, M.D. Tyrosine Dephosphorylation and Ethanol Inhibition ofN-Methyl-d-aspartate Receptor Function. J. Biol. Chem. 2003, 278, 11020–11025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.M.; Persechini, P.; Ojcius, D.M. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2006, 282, 2871–2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, H.; Arai, T.; Endo, N.; Yamashita, K.; Fukuda, K.; Sasada, M.; Uchiyama, T. LPS-induced ROS generation and changes in glutathione level and their relation to the maturation of human monocyte-derived dendritic cells. Life Sci. 2006, 78, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, A.G.; Rivers-Auty, J.; Daniels, M.; White, C.; Schwalbe, C.H.; Schilling, T.; Hammadi, H.; Jaiyong, P.; Spencer, N.G.; England, H.; et al. Boron-Based Inhibitors of the NLRP3 Inflammasome. Cell Chem. Biol. 2017, 24, 1321–1335.e5. [Google Scholar] [CrossRef] [Green Version]
- Ding, C.; Zhao, Y.; Shi, X.; Zhang, N.; Zu, G.; Li, Z.; Zhou, J.; Gao, D.; Lv, L.; Tian, X.; et al. New insights into salvianolic acid A action: Regulation of the TXNIP/NLRP3 and TXNIP/ChREBP pathways ameliorates HFD-induced NAFLD in rats. Sci. Rep. 2016, 6, 28734. [Google Scholar] [CrossRef]
- Donato, M.T.; Tolosa, L.; Gomez-Lechon, M.J. Culture and Functional Characterization of Human Hepatoma HepG2 Cells. Adv. Struct. Saf. Stud. 2014, 1250, 77–93. [Google Scholar] [CrossRef]
- Relja, B.; Omid, N.; Schaible, A.; Perl, M.; Meier, S.; Oppermann, E.; Lehnert, M.; Marzi, I. Pre- or post-treatment with ethanol and ethyl pyruvate results in distinct anti-inflammatory responses of human lung epithelial cells triggered by interleukin-6. Mol. Med. Rep. 2015, 12, 2991–2998. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, A.S.M.; Palmblad, J. Effects of Ethanol on NF-κB Activation, Production of Myeloid Growth Factors, and Adhesive Events in Human Endothelial Cells. J. Infect. Dis. 2001, 184, 761–769. [Google Scholar] [CrossRef] [Green Version]
- Mörs, K.; Kany, S.; Hörauf, J.A.; Wagner, N.; Neunaber, C.; Perl, M.; Relja, B. Suppression of the interleukin-1ß-induced inflammatory response of human Chang liver cells by acute and subacute exposure to alcohol: An in vitro study. Croat. Med. J. 2018, 59, 46–55. [Google Scholar] [CrossRef]
- Karmakar, M.; Katsnelson, M.A.; Dubyak, G.R.; Pearlman, E. Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1β secretion in response to ATP. Nat. Commun. 2016, 7, 10555. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hörauf, J.-A.; Kany, S.; Janicova, A.; Xu, B.; Vrdoljak, T.; Sturm, R.; Dunay, I.R.; Martin, L.; Relja, B. Short Exposure to Ethanol Diminishes Caspase-1 and ASC Activation in Human HepG2 Cells In Vitro. Int. J. Mol. Sci. 2020, 21, 3196. https://doi.org/10.3390/ijms21093196
Hörauf J-A, Kany S, Janicova A, Xu B, Vrdoljak T, Sturm R, Dunay IR, Martin L, Relja B. Short Exposure to Ethanol Diminishes Caspase-1 and ASC Activation in Human HepG2 Cells In Vitro. International Journal of Molecular Sciences. 2020; 21(9):3196. https://doi.org/10.3390/ijms21093196
Chicago/Turabian StyleHörauf, Jason-Alexander, Shinwan Kany, Andrea Janicova, Baolin Xu, Teodora Vrdoljak, Ramona Sturm, Ildiko Rita Dunay, Lukas Martin, and Borna Relja. 2020. "Short Exposure to Ethanol Diminishes Caspase-1 and ASC Activation in Human HepG2 Cells In Vitro" International Journal of Molecular Sciences 21, no. 9: 3196. https://doi.org/10.3390/ijms21093196
APA StyleHörauf, J. -A., Kany, S., Janicova, A., Xu, B., Vrdoljak, T., Sturm, R., Dunay, I. R., Martin, L., & Relja, B. (2020). Short Exposure to Ethanol Diminishes Caspase-1 and ASC Activation in Human HepG2 Cells In Vitro. International Journal of Molecular Sciences, 21(9), 3196. https://doi.org/10.3390/ijms21093196