Advances in Enzymatic Synthesis of D-Amino Acids




Abstract
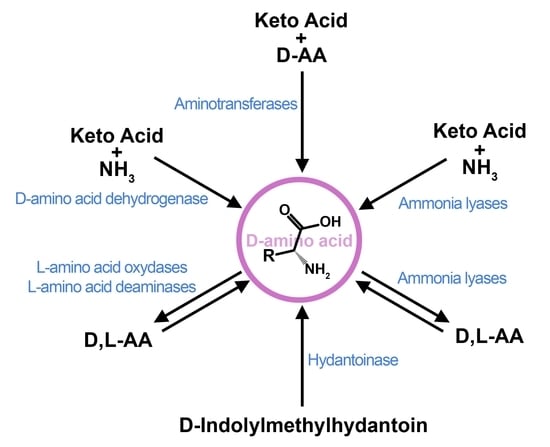
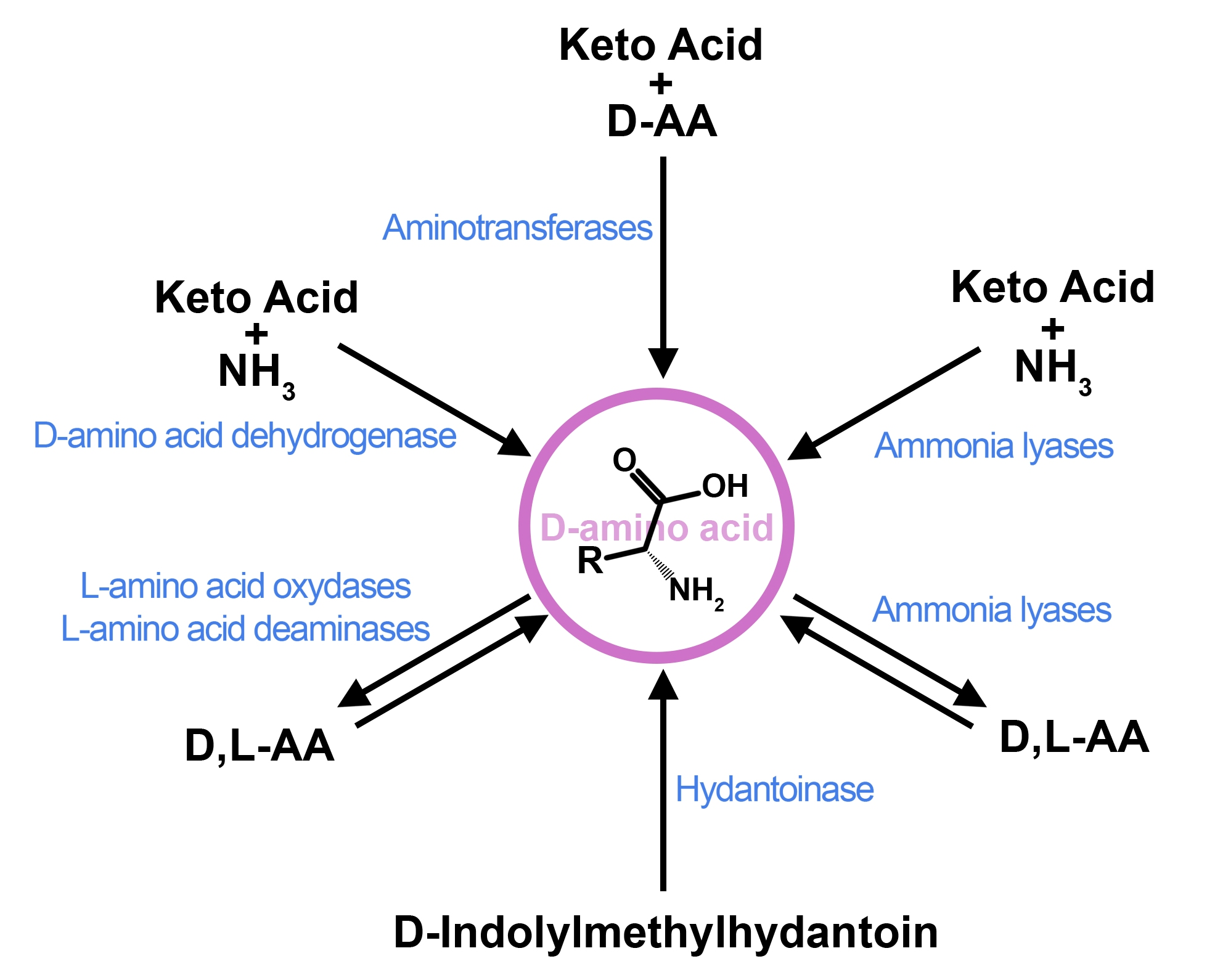
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Aminotransferases
2.1. D-Amino Acids Synthesis
2.2. Resolution of Racemic Mixtures
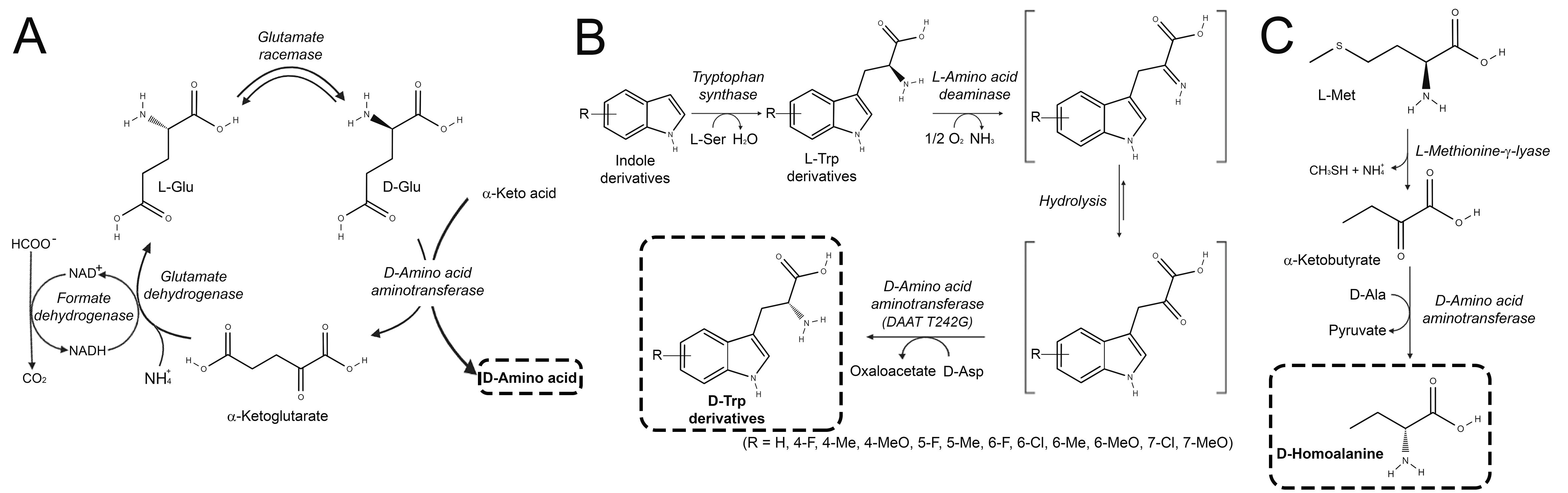
3. D-Amino Acid Dehydrogenases
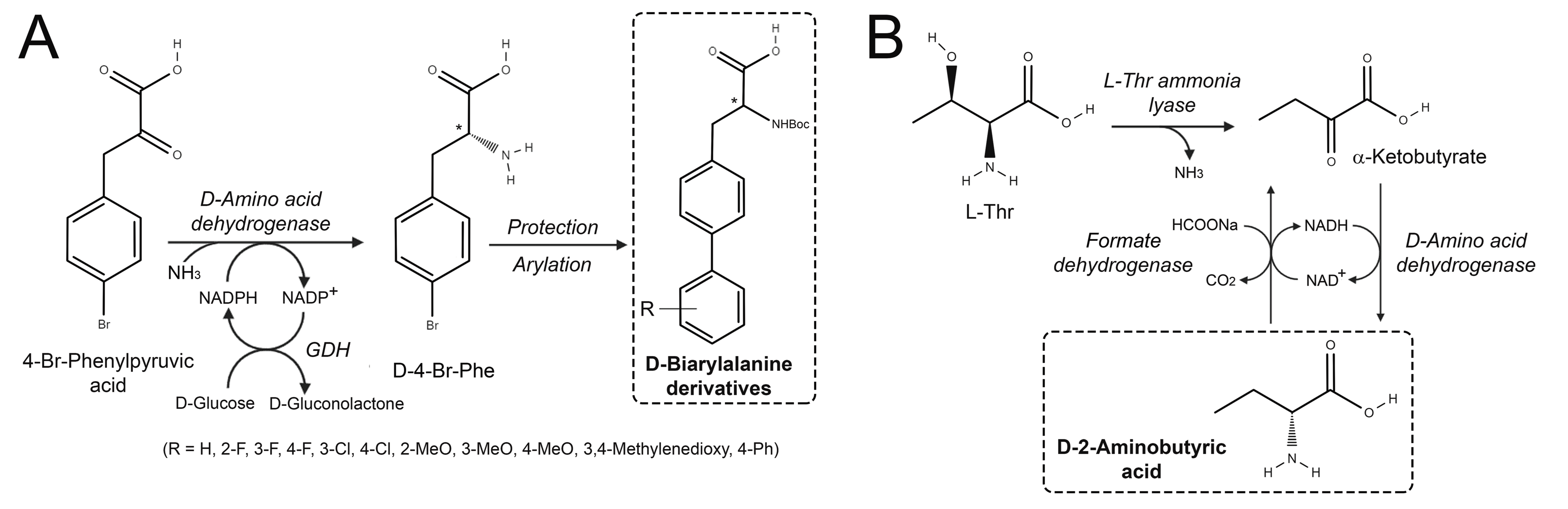
3.1. Synthesis of D-Amino Acids
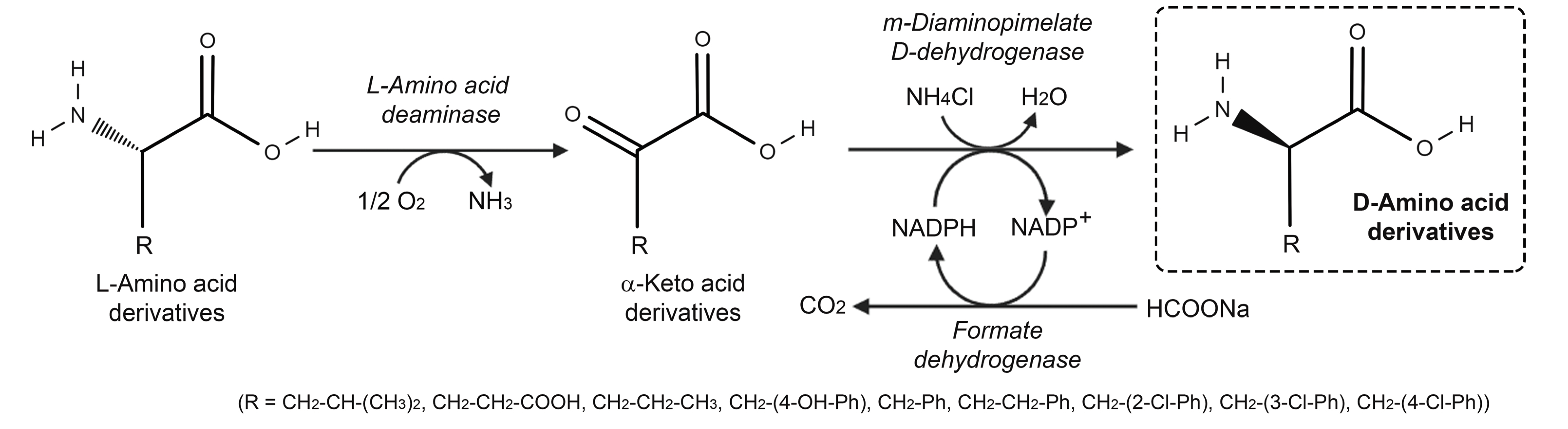
3.2. Stereoinversion of L-Amino Acids
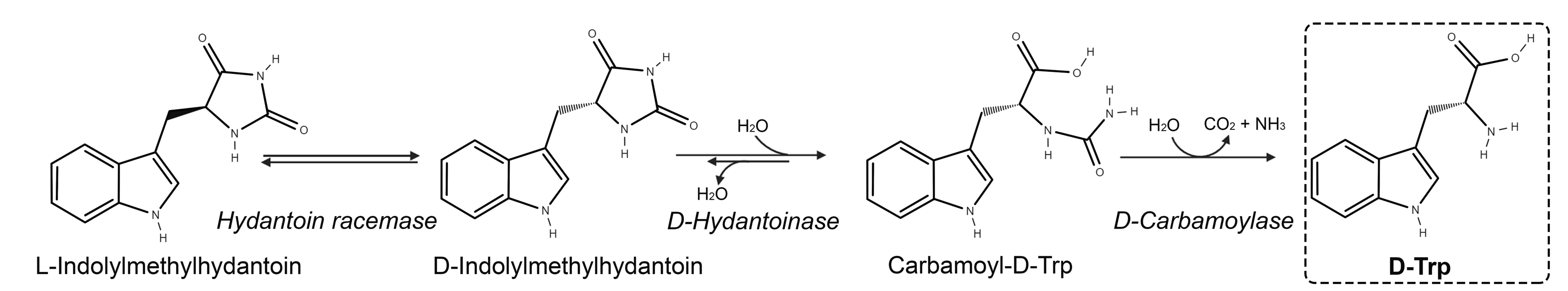
4. Hydantoinase Process
5. Ammonia Lyases
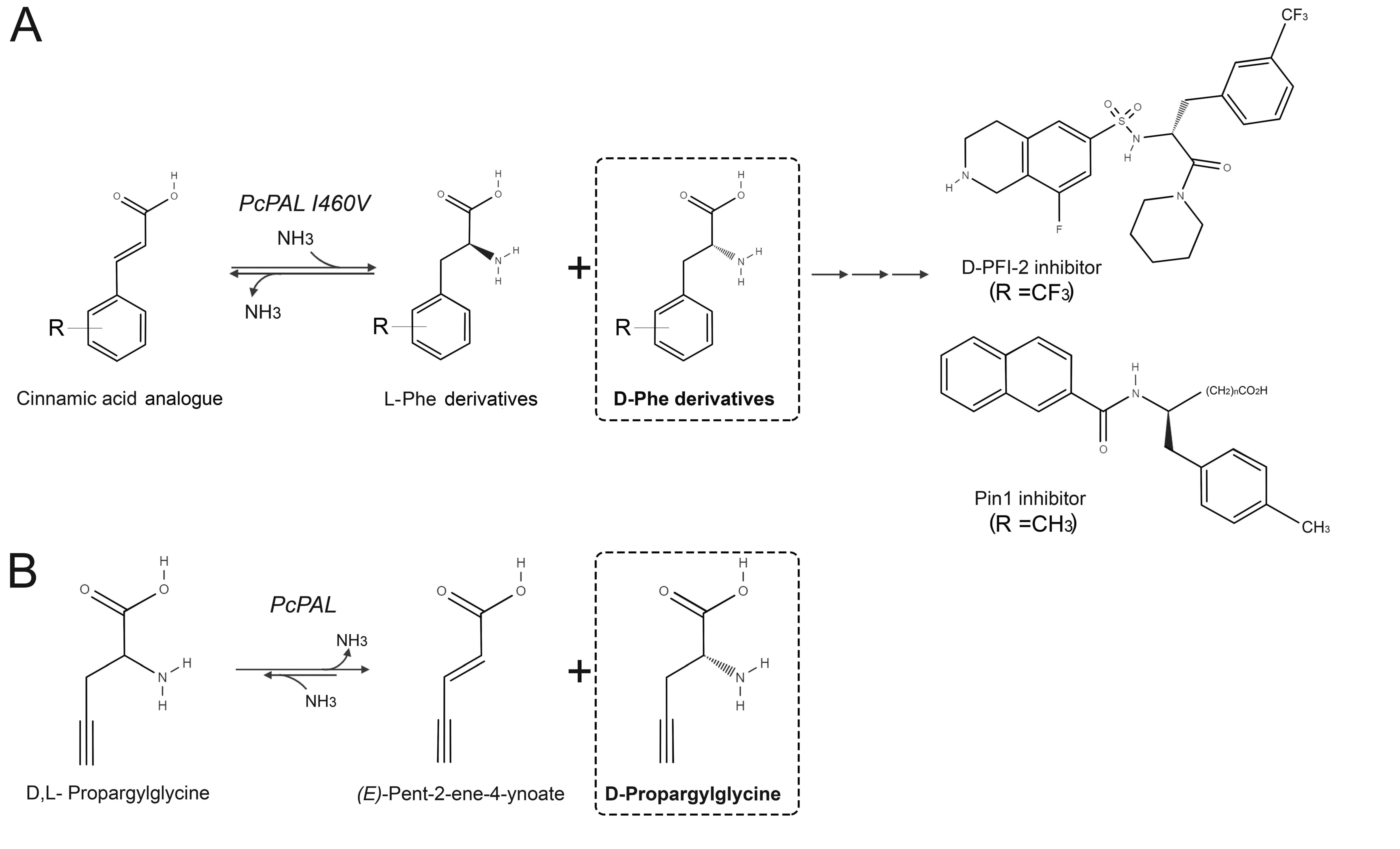
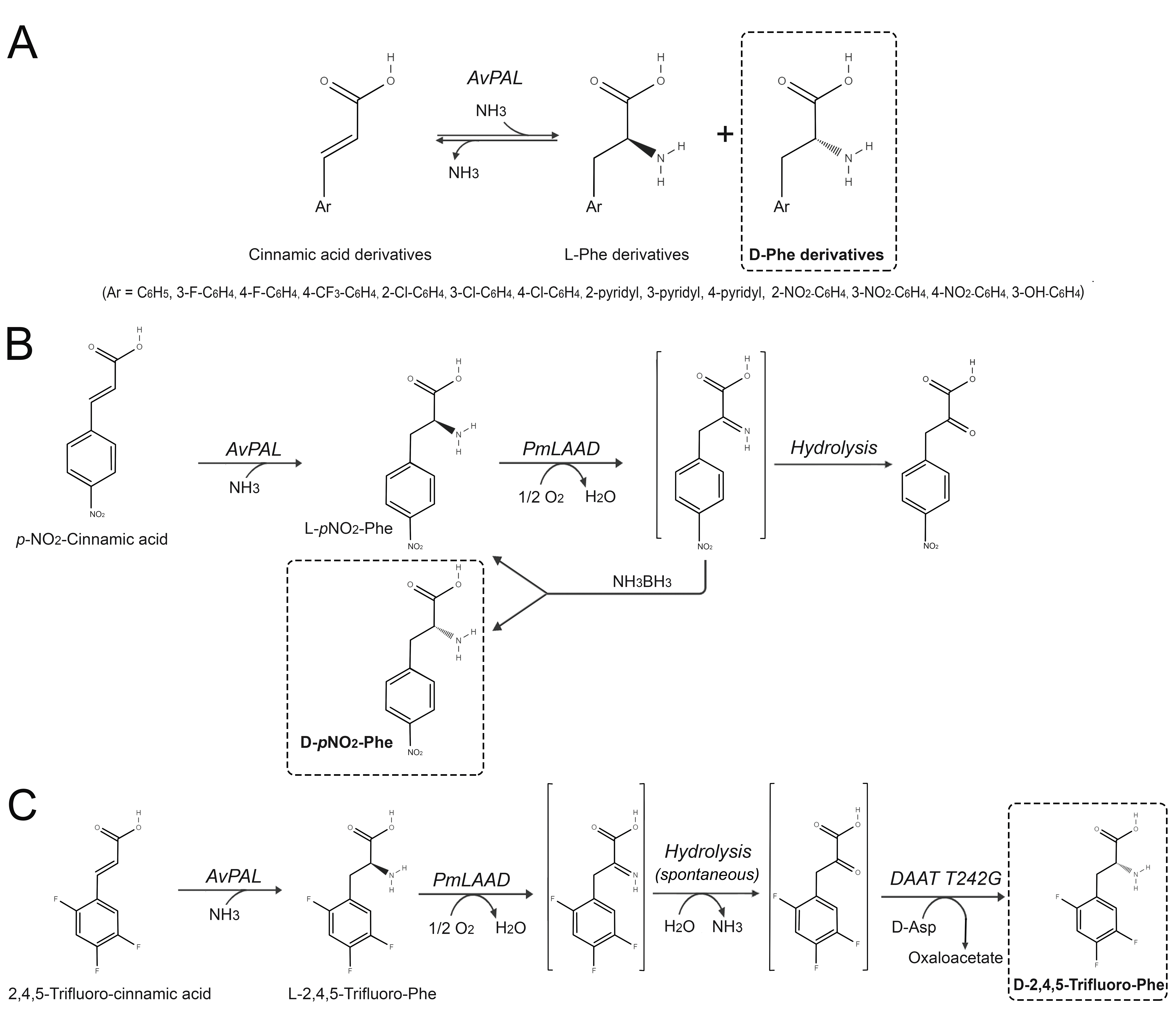
5.1. Production of D-Amino Acids by Enantioselective Deamination
5.2. Production of D-Amino Acids by Enantioselective Hydroamination
5.3. Production of D-Amino Acids by One-Pot Deracemization Reaction
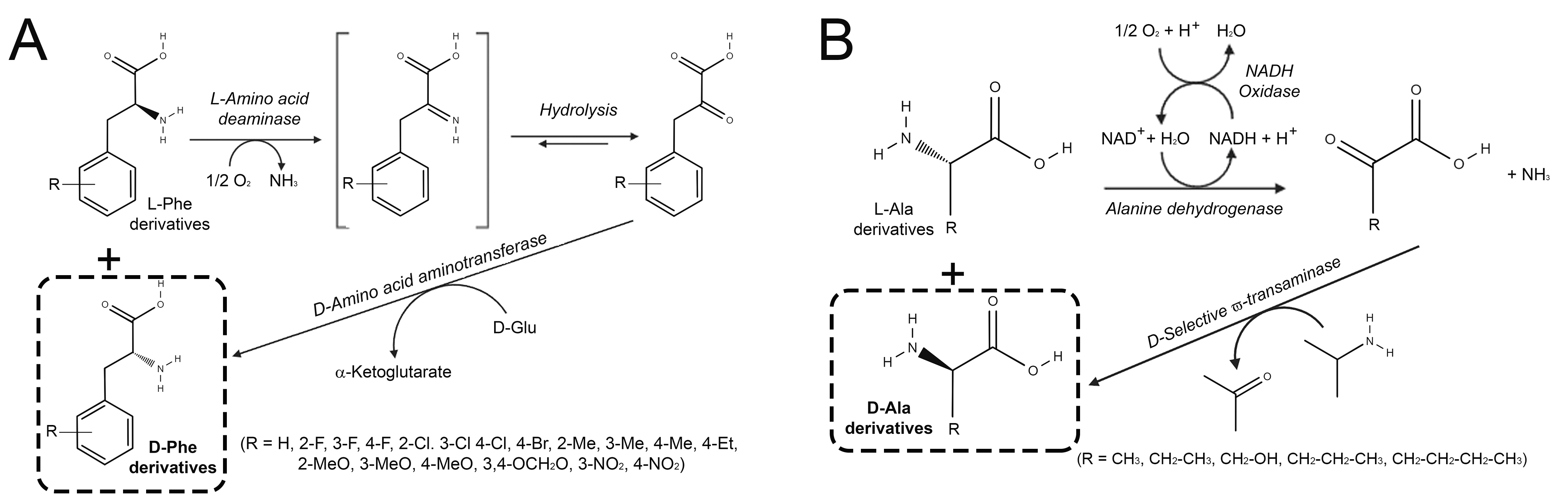
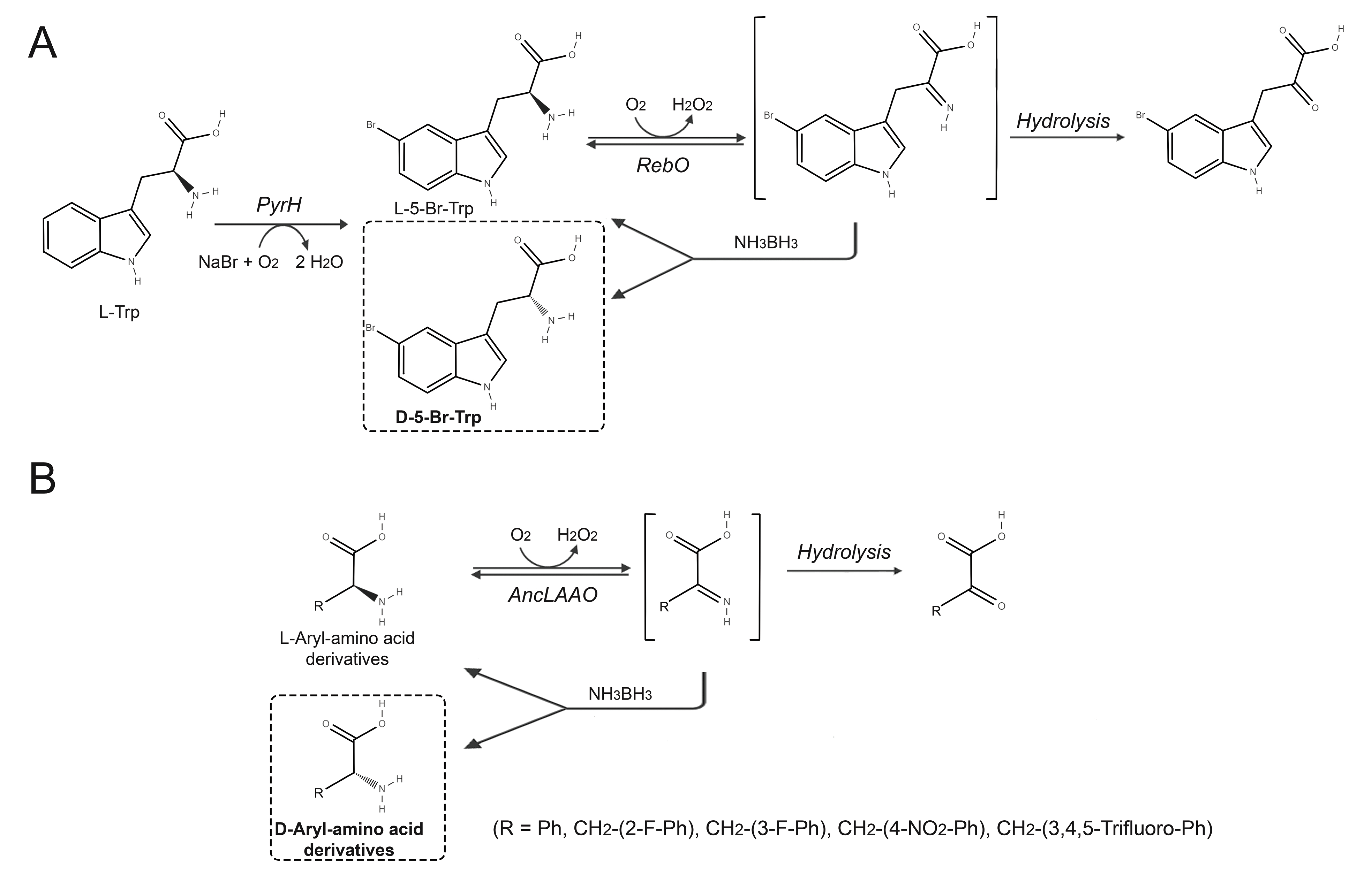
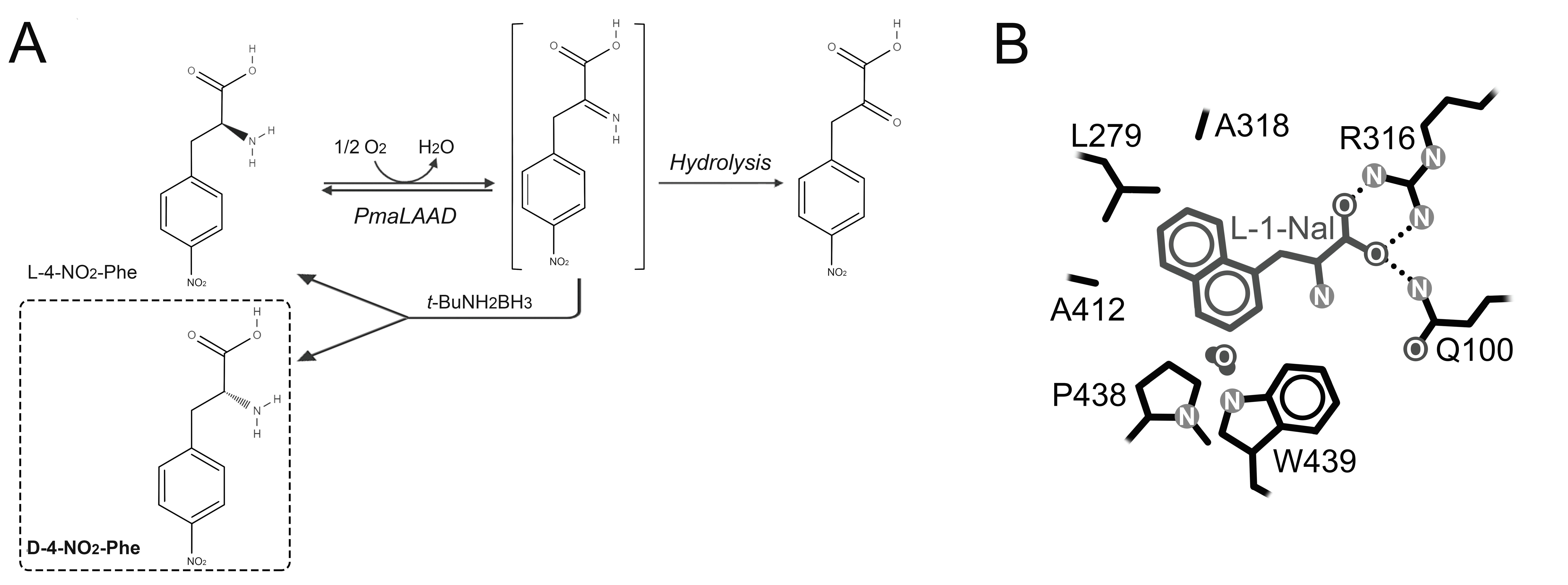
6. L-Amino Acid Oxidases and L-Amino Acid Deaminases
6.1. Production of D-Amino Acids by Enantioselective Deamination/Deracemization Using L-Amino Acid Oxidase
6.2. Production of D-Amino Acids by Enantioselective Deamination/Deracemization Using L-Amino Acid Deaminase
7. Conclusions
- -
- the full conversion of enantiomeric solutions of amino acids into the D-enantiomer;
- -
- the production of a variety of unnatural D-AA derivatives by means of novel enzymes, both isolated from natural sources (e.g., LAADs) and generated by protein engineering (DAADHs and ARTA);
- -
- the optimization of the hydantoinase process by the identification of well-suited enzymes that rendered it one of the most promising strategies to achieve economic and enantioselective synthesis of D-AAs;
- -
- -
- the establishment of an attractive synthetic method for D-2-aminobutyric acid by the reductive amination of 2-oxobutyric acid by DAADH [25];
- -
- the enantioselective deamination of L-AAs by enzymes immobilized on magnetic nanoparticles in a microfluidic reactor [43].
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AncLAAO | Ancestral variant of L-amino acid oxidase from Pseudoalteromonas piscicida |
| ARTA | ω-transaminase from Arthrobacter sp. |
| AvPAL | Phenylalanine ammonia lyase from Anabena variabilis |
| DAADH | D-amino acid dehydrogenase |
| D-AAs | D-amino acids |
| DAAT | D-amino acid aminotransferase |
| LAAD | L-amino acid deaminase |
| LAAO | L-amino acid oxidase |
| L-AAs | L-amino acids |
| m-DAPDH | meso-diaminopimelate D-dehydrogenase |
| MIO | 4-methylideneimidazol-5-one |
| PAL | Phenylalanine ammonia lyase |
| PcPAL | Phenylalanine ammonia lyase from Petroselinum crispum |
| PLP | Pyridoxal 5′-phosphate cofactor |
| PmaLAAD | L-amino acid deaminase from Proteus myxofaciens |
| PmLAAD | L-amino acid deaminase from Proteus mirabilis |
| RebO | L-amino acid oxidase from the actinomycete Lechevalieria aerocolonigenes |
| α-MBA | (S)-α-methylbenzylamine |
References
- Pollegioni, P.; Servi, S. Unnatural Amino Acids: Methods and Protocol; Humana Press: New York, NY, USA, 2012. [Google Scholar] [CrossRef]
- DAAIR Center—D-Amino Acids International Research Center. Available online: https://www.d-aminoacids.com/ (accessed on 1 March 2020).
- Fujii, N. D-amino acid in elderly tissues. Biol. Pharm. Bull. 2005, 28, 1585–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, M. Origin, microbiology, nutrition, and pharmacology of D-amino acids. Chem. Biodivers. 2010, 7, 1491–1530. [Google Scholar] [CrossRef] [PubMed]
- Pollegioni, L.; Sacchi, S. Metabolism of the neuromodulator D-serine. Cell. Mol. Life Sci. 2010, 67, 2387–2404. [Google Scholar] [CrossRef] [PubMed]
- Marcone, G.L.; Rosini, E.; Crespi, E.; Pollegioni, L. D-amino acids in foods. Appl. Microbiol. Biotechnol. 2020, 104, 555–574. [Google Scholar] [CrossRef]
- Schmid, A.; Hollmann, F.; Park, J.B.; Bühler, B. The use of enzymes in the chemical industry in Europe. Curr. Opin. Biotechnol. 2002, 13, 359–366. [Google Scholar] [CrossRef]
- Gao, X.; Ma, Q.; Zhu, H. Distribution, industrial applications, and enzymatic synthesis of D-amino acids. Appl. Microbiol. Biotechnol. 2015, 99, 3341–3349. [Google Scholar] [CrossRef]
- Xue, Y.P.; Cao, C.H.; Zheng, Y.G. Enzymatic asymmetric synthesis of chiral amino acids. Chem. Soc. Rev. 2018, 47, 1516–1561. [Google Scholar] [CrossRef]
- Mehta, P.K.; Christen, P. The molecular evolution of pyridoxal-5’-phosphate-dependent enzymes. Adv. Enzymol. Relat. Areas Mol. Biol. 2000, 74, 129–184. [Google Scholar] [CrossRef]
- Nakajima, N.; Tanizawa, K.; Tanaka, H.; Soda, K. Enantioselective synthesis of various D-amino acids by a multi-enzyme system. J. Biotechnol. 1988, 8, 243–248. [Google Scholar] [CrossRef]
- Parmeggiani, F.; Casamajo, R.A.; Walton, C.J.W.; Galman, J.L.; Turner, N.J.; Chica, R.A. One-pot biocatalytic synthesis of substituted D-tryptophans from indoles enabled by an engineered aminotransferase. ACS Catal. 2019, 9, 3482–3486. [Google Scholar] [CrossRef]
- Silva, M.V.M.; Costa, I.C.R.; de Souza, R.O.M.A.; Bornscheuer, U.T. Biocatalytic cascade reaction for the asymmetric synthesis of L- and D-homoalanine. ChemCatChem 2019, 11, 407–411. [Google Scholar] [CrossRef]
- Bea, H.; Lee, S.; Yun, H. Asymmetric synthesis of (R)-3-fluoroalanine from 3-fluoropyruvate using omega-transaminase. Biotechnol. Bioproc. 2011, 16, 291–296. [Google Scholar] [CrossRef]
- Park, E.S.; Dong, J.Y.; Shin, J.K. Biocatalytic asymmetric synthesis of unnatural amino acids through the cascade transfer of amino groups from primary amines onto keto acids. ChemCatChem 2013, 5, 3538–3542. [Google Scholar] [CrossRef]
- Park, E.S.; Dong, J.Y.; Shin, J.K. Active site model of (R)-selective ω-transaminase and its application to the production of D-amino acids. Appl. Microbiol. Biotechnol. 2014, 98, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.X.; Liu, S.P.; Cheng, S.; Zhang, L.; Ding, Z.Y.; Gu, Z.H.; Shi, G.Y. Screening, characterization and utilization of D-amino acid aminotransferase to obtain D-phenylalanine. Appl. Biochem. Microbiol. 2015, 51, 695–703. [Google Scholar] [CrossRef]
- Walton, C.J.W.; Parmeggiani, F.; Barber, J.E.B.; McCann, J.L.; Turner, N.J.; Chica, R.A. Engineered aminotransferase for the production of D-phenylalanine derivatives using biocatalytic cascades. ChemCatChem 2018, 10, 470–474. [Google Scholar] [CrossRef]
- Han, S.W.; Shin, J.S. One-pot preparation of D-amino acids through biocatalytic deracemization using alanine dehydrogenase and ω-transaminase. Catal. Lett. 2018, 148, 3678–3684. [Google Scholar] [CrossRef]
- Vedha-Peters, K.; Gunawardana, M.; Rozzell, J.D.; Novick, S.J. Creation of a broad-range and highly stereoselective D-amino acid dehydrogenase for the one-step synthesis of D-amino acids. J. Am. Chem. Soc. 2006, 128, 10923–10929. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Parmeggiani, F.; Weise, N.; Flitsch, S.; Turner, N. Chemoenzymatic synthesis of optically pure L- and D-biarylalanines through biocatalytic asymmetric amination and palladium-catalyzed arylation. ACS Catal. 2015, 5, 5410–5413. [Google Scholar] [CrossRef]
- Gao, X.; Chen, X.; Liu, W.; Feng, J.; Wu, Q.; Hua, L.C.; Zhu, D. A novel meso-diaminopimelate dehydrogenase from Symbiobacterium thermophilum: Overexpression, characterization, and potential for D-amino acid synthesis. Appl. Environ. Microbiol. 2012, 78, 8595–8600. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Huang, F.; Feng, J.; Chen, X.; Zhang, H.; Wang, Z.; Wu, Q.; Zhu, D. Engineering the meso-diaminopimelate dehydrogenase from Symbiobacterium thermophilum by site saturation mutagenesis for D-phenylalanine synthesis. Appl. Environ. Microbiol. 2013, 79, 5078–5081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Chen, X.; Feng, J.; Wu, Q.; Zhu, D. Structure-guided engineering of meso-diaminopimelate dehydrogenase for enantioselective reductive amination of sterically bulky 2-keto acids. Catal. Sci. Technol. 2018, 8, 4994–5002. [Google Scholar] [CrossRef]
- Chen, X.; Cui, Y.; Cheng, X.; Feng, J.; Wu, Q.; Zhu, D. Highly atom economic synthesis of D-2-aminobutyric acid through an in vitro tri-enzymatic catalytic system. ChemistryOpen 2017, 6, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Akita, H.; Suzuki, H.; Doi, K.; Ohshima, T. Efficient synthesis of D-branched-chain amino acids and their labeled compounds with stable isotopes using D-amino acid dehydrogenase. Appl. Microbiol. Biotechnol. 2014, 98, 1135–1143. [Google Scholar] [CrossRef]
- Hayashi, J.; Seto, T.; Akita, H.; Watanabe, M.; Hoshino, T.; Yoneda, K.; Ohshima, T.; Sakuraba, H. Structure-based engineering of an artificially generated NADP+-dependent D-amino acid dehydrogenase. Appl. Environ. Microbiol. 2017, 83, e00491-17. [Google Scholar] [CrossRef] [Green Version]
- Akita, H.; Hayashi, J.; Sakuraba, H.; Ohshima, T. Artificial thermostable D-amino acid dehydrogenase: Creation and application. Front. Microbiol. 2018, 9, 1760. [Google Scholar] [CrossRef]
- Hanson, R.L.; Johnston, R.M.; Goldberg, S.L.; Parker, W.L.; Goswami, A. Enzymatic preparation of an R-amino acid intermediate for a γ-secretase inhibitor. Org. Process Res. Dev. 2013, 17, 693–700. [Google Scholar] [CrossRef]
- Zhang, D.; Xiaoran, J.; Zhang, W.; Yao, N.; Yan, X. Highly selective synthesis of D-amino acids from readily available L-amino acids by a one-pot biocatalytic stereoinversion cascade. RSC Adv. 2019, 9, 29927–29935. [Google Scholar] [CrossRef] [Green Version]
- Parmeggiani, F.; Ahmed, S.T.; Thompson, M.P.; Weise, N.J.; Galman, J.L.; Gahloth, D.; Dunstan, M.S.; Leys, D.; Turner, N.J. Single-biocatalyst synthesis of enantiopure D-arylalanines exploiting an engineered D-amino acid dehydrogenase. Adv. Synth. Catal. 2016, 358, 3298–3306. [Google Scholar] [CrossRef]
- Martínez-Rodríguez, S.; Martínez-Gómez, A.I.; Rodríguez-Vico, F.; Clemente-Jiménez, J.M.; Las Heras-Vázquez, F.J. Natural occurrence and industrial applications of D-amino acids: An overview. Chem. Biodivers. 2010, 7, 1531–1548. [Google Scholar] [CrossRef]
- Chen, M.; Shi, C.; Zhao, J.; Gao, Z.; Zhang, C. Application and microbial preparation of D-valine. World J. Microbiol. Biotechnol. 2016, 32, 171. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, G.; Han, R.; Dong, J.; Ni, Y. Identification of D-carbamoylase for biocatalytic cascade synthesis of D-tryptophan featuring high enantioselectivity. Bioresour. Technol. 2018, 249, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Barros, J.; Dixon, R.A. Plant phenylalanine/tyrosine ammonia-lyases. Trends Plant Sci. 2020, 25, 66–79. [Google Scholar] [CrossRef]
- Parmeggiani, F.; Weise, N.J.; Ahmed, S.T.; Turner, N.J. Synthetic and therapeutic applications of ammonia-lyases and aminomutases. Chem. Rev. 2018, 118, 73–118. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.T.; Weise, N.J.; Parmeggiani, F.; Turner, N.J. Applications of phenylalanine ammonia lyases in synthesis access to enantiopure arylalanines from laboratory scale to industrial production. Chim. OGGI Chem. Today 2017, 35, 30–33. [Google Scholar]
- Nakatsu, Y.; Matsunaga, Y.; Ueda, K.; Yamamotoya, T.; Inoue, Y.; Inoue, M.K.; Mizuno, Y.; Kushiyama, A.; Ono, H.; Fujishiro, M.; et al. Development of Pin1 inhibitors and their potential as therapeutic agents. Curr. Med. Chem. 2020, 27. [Google Scholar] [CrossRef]
- Tork, S.D.; Nagy, E.Z.A.; Cserepes, L.; Bordea, D.M.; Nagy, B.; Toşa, M.I.; Paizs, C.; Bencze, L.C. The production of L- and D-phenylalanines using engineered phenylalanine ammonia lyases from Petroselinum crispum. Sci. Rep. 2019, 9, 20123. [Google Scholar] [CrossRef]
- Zhu, L.; Zhou, L.; Huang, N.; Cui, W.; Liu, Z.; Xiao, K.; Zhou, Z. Efficient preparation of enantiopure D-phenylalanine through asymmetric resolution using immobilized phenylalanine ammonia-lyase from Rhodotorula glutinis JN-1 in a recirculating packed-bed reactor. PLoS ONE 2014, 9, e108586. [Google Scholar] [CrossRef] [Green Version]
- Bartha-Vári, J.H.; Toşa, M.I.; Irimie, F.D.; Weiser, D.; Boros, Z.; Vértessy, B.G.; Paizs, C.; Poppe, L. Immobilization of phenylalanine ammonia-lyase on single-walled carbon nanotubes for stereoselective biotransformations in batch and continuous-flow modes. ChemCatChem 2015, 7, 1122–1128. [Google Scholar] [CrossRef] [Green Version]
- Ender, F.; Weiser, D.; Nagy, B.; Bencze, C.L.; Paizs, C.; Pálovics, P.; Poppe, L. Microfluidic multiple cell chip reactor filled with enzyme-coated magnetic nanoparticles—An efficient and flexible novel tool for enzyme catalyzed biotransformations. J. Flow Chem. 2016, 6, 43–52. [Google Scholar] [CrossRef]
- Weiser, D.; Bencze, L.C.; Bánóczi, G.; Ender, F.; Kiss, R.; Kókai, E.; Szilágyi, A.; Vértessy, B.G.; Farkas, Ö.; Paizs, C.; et al. Phenylalanine ammonia-lyase-catalyzed deamination of an acyclic amino acid: Enzyme mechanistic studies aided by a novel microreactor filled with magnetic nanoparticles. ChemBioChem 2015, 16, 2283–2288. [Google Scholar] [CrossRef] [PubMed]
- Lovelock, S.L.; Lloyd, R.C.; Turner, N.J. Phenylalanine ammonia lyase catalysed synthesis of amino acids by an MIO-cofactor independent pathway. Angew. Chem. Int. Ed. Engl. 2014, 53, 4652–4656. [Google Scholar] [CrossRef] [PubMed]
- Parmeggiani, F.; Lovelock, S.L.; Weise, N.J.; Ahmed, S.T.; Turner, N.J. Synthesis of D- and L-phenylalanine derivatives by phenylalanine ammonia lyases: A multienzymatic cascade process. Angew. Chem. Int. Ed. Engl. 2015, 54, 4608–4611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Feng, G.; Ge, F.; Song, P.; Wang, T.; Liu, Y.; Tao, Y.; Zhou, Z. One-pot enzymatic synthesis of D-arylalanines using phenylalanine ammonia lyase and L-amino acid deaminase. Appl. Biochem. Biotechnol. 2019, 187, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Parmeggiani, F.; Casamajo, R.A.; Colombo, D.; Ghezzi, M.C.; Galman, J.L.; Chica, R.A.; Brenna, E.; Turner, N.J. Biocatalytic retrosynthesis approaches to D-(2,4,5-trifluorophenyl)alanine, key precursor of the antidiabetic sitagliptin. Green Chem. 2019, 21, 4368–4379. [Google Scholar] [CrossRef]
- Molla, G.; Melis, R.; Pollegioni, L. Breaking the mirror: L-amino acid deaminase, a novel stereoselective biocatalyst. Biotechnol. Adv. 2017, 35, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Motta, P.; Molla, G.; Pollegioni, L.; Nardini, M. Structure-function relationships in L-amino acid deaminase, a flavoprotein belonging to a novel class of biotechnologically relevant enzymes. J. Biol. Chem. 2016, 291, 10457–10475. [Google Scholar] [CrossRef] [Green Version]
- Motta, P.; Pollegioni, L.; Molla, G. Properties of L-amino acid deaminase: En route to optimize bioconversion reactions. Biochimie 2019, 158, 199–207. [Google Scholar] [CrossRef]
- Pollegioni, L.; Molla, G. New biotech applications from evolved D-amino acid oxidases. Trends Biotechnol. 2011, 29, 276–283. [Google Scholar] [CrossRef]
- Pollegioni, L.; Motta, P.; Molla, G. L-amino acid oxidase as biocatalyst: A dream too far? Appl. Microbiol. Biotechnol. 2013, 97, 9323–9341. [Google Scholar] [CrossRef]
- Schnepel, C.; Kemker, I.; Sewald, N. One-pot synthesis of D-halotryptophans by dynamic stereoinversion using a specific L-amino acid oxidase. ACS Catal. 2019, 9, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Siddiq, M.A.; Hochberg, G.K.; Thornton, J.W. Evolution of protein specificity: Insights from ancestral protein reconstruction. Curr. Opin. Struct. Biol. 2017, 47, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.; Minamino, Y.; Hasebe, F.; Ito, S. Deracemization and stereoinversion to aromatic D-amino acid derivatives with ancestral L-amino acid oxidase. ACS Catal. 2019, 9, 10152–10158. [Google Scholar] [CrossRef]
- Alexandre, F.R.; Pantaleone, D.P.; Taylor, P.P.; Fotheringham, I.G.; Ager, D.J.; Turner, N.J. Amine-boranes: Effective reducing agents for the deracemisation of D,L-amino acids using L-amino acid oxidase from Proteus myxofaciens. Tetrahedron Lett. 2002, 43, 707–710. [Google Scholar] [CrossRef]
- Hanson, R.L.; Davis, B.L.; Goldberg, S.L.; Johnston, R.M.; Parker, W.L.; Tully, T.P.; Montana, M.A.; Patel, R.N. Enzymatic preparation of a D-amino acid from a racemic amino acid or keto acid. Org. Process Res. Dev. 2008, 12, 1119–1129. [Google Scholar] [CrossRef]
- Liu, L.; Zhu, Y.; Chen, Y.; Chen, H.; Fan, C.; Mo, Q.; Yuan, J. One-pot cascade biotransformation for efficient synthesis of benzyl alcohol and its analogs. Chem. Asian J. 2020. [Google Scholar] [CrossRef]
- Wu, L.; Guo, X.; Wu, G.; Liu, P.; Liu, Z. Efficient enzymatic synthesis of α-keto acids by redesigned substrate-binding pocket of the L-amino acid deaminase (PmiLAAD). Enzyme Microb. Technol. 2020, 132, 109393. [Google Scholar] [CrossRef]
- Wu, L.; Guo, X.; Wu, G.; Liu, P.; Liu, Z. Efficient production of α-keto acids by immobilized E. coli-pETduet-1-PmiLAAO in a jacketed packed-bed reactor. R. Soc. Open Sci. 2019, 6, 182035. [Google Scholar] [CrossRef] [Green Version]
- Rosini, E.; Melis, R.; Molla, G.; Tessaro, D.; Pollegioni, L. Deracemization and stereoinversion of α-amino acids by L-amino acid deaminase. Adv. Synth. Catal. 2017, 359, 3773–3781. [Google Scholar] [CrossRef]
- Melis, R.; Rosini, E.; Pirillo, V.; Pollegioni, L.; Molla, G. In Vitro evolution of an L-amino acid deaminase active on L-1-naphthylalanine. Catal. Sci. Technol. 2018, 8, 5359–5367. [Google Scholar] [CrossRef]
- Tessaro, D.; Pollegioni, L.; Piubelli, L.; D’Arrigo, P.; Servi, S. Systems biocatalysis: An artificial metabolism for interconversion of functional groups. ACS Catal. 2015, 5, 1604–1608. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pollegioni, L.; Rosini, E.; Molla, G. Advances in Enzymatic Synthesis of D-Amino Acids. Int. J. Mol. Sci. 2020, 21, 3206. https://doi.org/10.3390/ijms21093206
Pollegioni L, Rosini E, Molla G. Advances in Enzymatic Synthesis of D-Amino Acids. International Journal of Molecular Sciences. 2020; 21(9):3206. https://doi.org/10.3390/ijms21093206
Chicago/Turabian StylePollegioni, Loredano, Elena Rosini, and Gianluca Molla. 2020. "Advances in Enzymatic Synthesis of D-Amino Acids" International Journal of Molecular Sciences 21, no. 9: 3206. https://doi.org/10.3390/ijms21093206
APA StylePollegioni, L., Rosini, E., & Molla, G. (2020). Advances in Enzymatic Synthesis of D-Amino Acids. International Journal of Molecular Sciences, 21(9), 3206. https://doi.org/10.3390/ijms21093206