New Insights into Molecular Links Between Microbiota and Gastrointestinal Cancers: A Literature Review



Abstract
:1. Introduction
2. Microbiota and Host Immunity
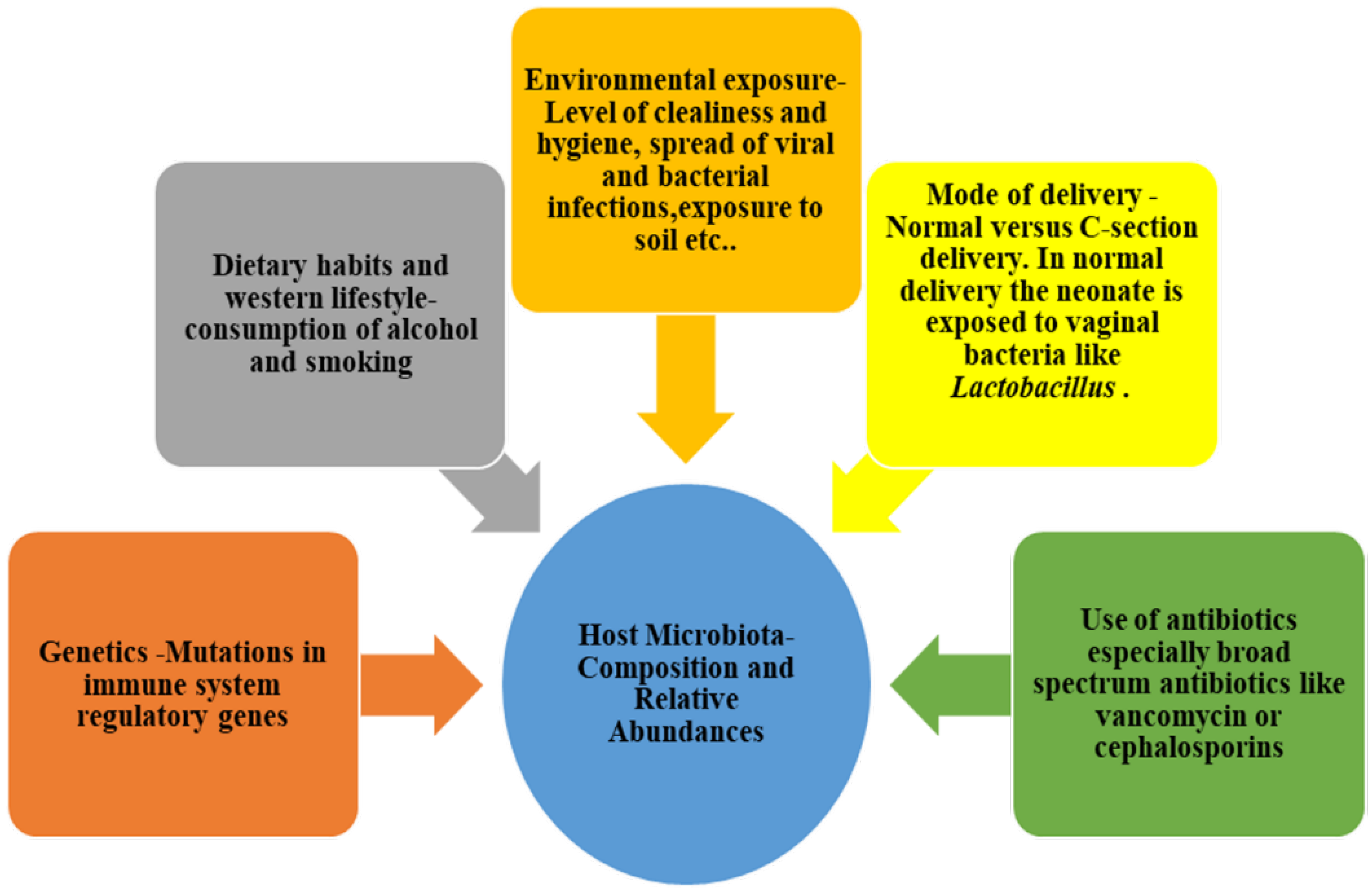
3. Microbiota Eubiosis: Characteristics and Implications
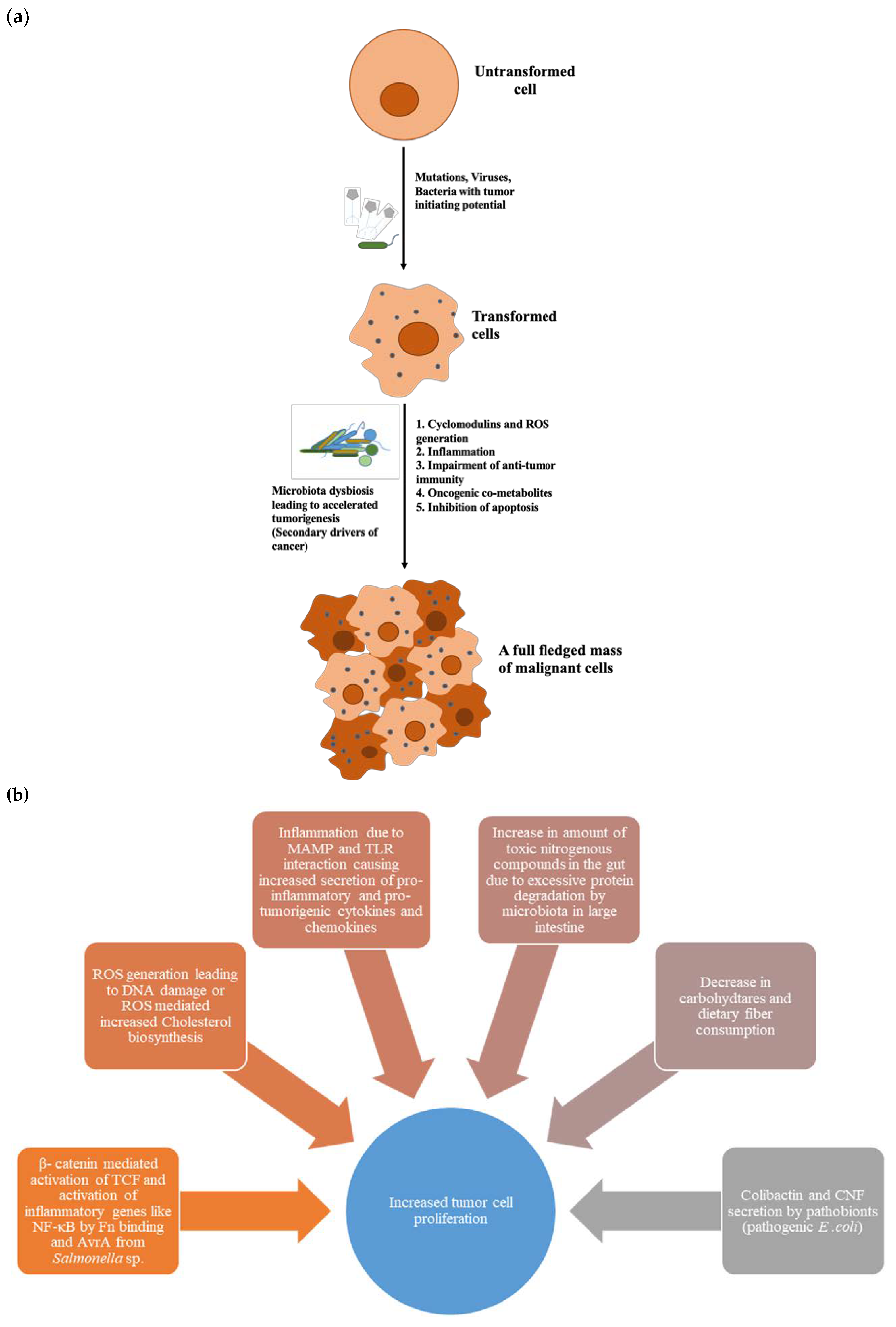
4. Dysbiosed Microbiota as Tumour Promoter
4.1. Cyclomodulins
4.2. Microbial-Associated Molecular Patterns (MAMPs) and Inflammation
4.3. Oncogenic Microbial Metabolites
5. Gut Microbiota and Gastrointestinal Cancers: Potential Molecular Mechanisms
5.1. Cancer Initiation and Progression in Stomach
5.2. Cancer Initiation and Progression in Intestine
5.2.1. Microbiota-Driven Suppression of Antitumour Immunity
5.2.2. Microbiota Role in CRC Metastasis and Recurrence
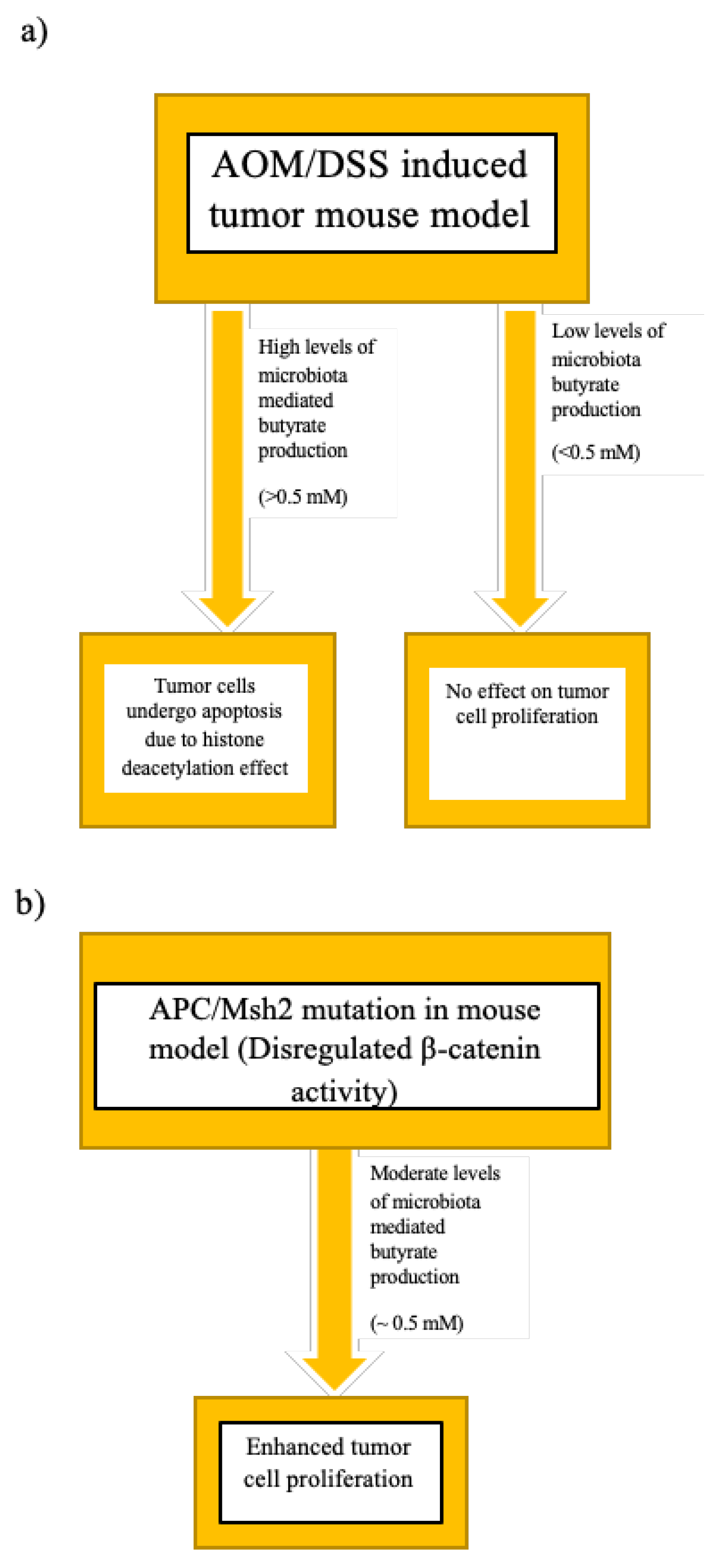
5.2.3. Dietary Habits, Host–Microbiome Cometabolism, and the Butyrate Paradox
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limon, J.J.; Skalski, J.H.; Underhill, D.M. Commensal Fungi in Health and Disease. Cell Host Microbe 2017, 22, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F. Host–Bacterial Mutualism in the Human Intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, S.R.; Pop, M.; DeBoy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic Analysis of the Human Distal Gut Microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [Green Version]
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. The Lancet Global Health 2016, 4, e609–e616. [Google Scholar] [CrossRef] [Green Version]
- Bennet, R.; Nord, C.E. Development of the Faecal Anaerobic Microflora After Caesarean Section and Treatment with Antibiotics in Newborn Infants. Infection 1987, 15, 332–336. [Google Scholar] [CrossRef]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. PNAS 2010, 107, 11971–11975. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Bello, M.G.; De Jesus-Laboy, K.M.; Shen, N.; Cox, L.M.; Amir, A.; Gonzalez, A.; Bokulich, N.A.; Song, S.J.; Hoashi, M.; Rivera-Vinas, J.I.; et al. Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat. Med. 2016, 22, 250–253. [Google Scholar] [CrossRef]
- Palmer, C.; Bik, E.M.; Digiulio, D.B.; Relman, D.A.; Brown, P.O. Development of the Human Infant Intestinal Microbiota. PLoS Biol. 2007, 5, e177. [Google Scholar] [CrossRef] [Green Version]
- Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-datchary, P. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life Resource. Cell Host Microbe 2015, 690–703. [Google Scholar]
- Francis, S.S.; Selvin, S.; Metayer, C.; Wallace, A.D.; Crouse, V.; Moore, T.B.; Wiemels, J.L.; Buffler, P.A. Mode of delivery and risk of childhood leukemia. Cancer Epidemiol Biomarkers Prev. 2014, 23, 876–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How colonization by microbiota in early life shapes the immune system. Science 2016, 352, 539–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamburini, S.; Shen, N.; Wu, H.C.; Clemente, J.C. The microbiome in early life: Implications for health outcomes. Nature Publishing Group 2016, 22, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Di, Y.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef]
- Shi, C.; Jia, T.; Mendez-Ferrer, S.; Hohl, T.M.; Serbina, N.V.; Lipuma, L.; Leiner, I.; Li, M.O.; Frenette, P.S.; Pamer, E.G. Bone Marrow Mesenchymal Stem and Progenitor Cells Induce Monocyte Emigration in Response to Circulating Toll-like Receptor Ligands. Immunity 2011, 34, 590–601. [Google Scholar] [CrossRef] [Green Version]
- Tada, T.; Yamamura, S.; Kuwano, Y.; Abo, T. Level of Myelopoiesis in the Bone Marrow Is Influenced by Intestinal Flora. Cell Immunol. 1996, 173, 155–161. [Google Scholar] [CrossRef]
- Gomez, A.; Espinoza, J.L.; Harkins, D.M.; Leong, P.; Saffery, R.; Bockmann, M.; Torralba, M.; Kuelbs, C.; Kodukula, R.; Inman, J.; et al. Host Genetic Control of the Oral Microbiome in Health and Disease. Cell Host Microbe 2017, 22, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- David, L.A.; Materna, A.C.; Friedman, J.; Campos-Baptista, M.I.; Blackburn, M.C.; Perrotta, A.; Erdman, S.E.; Alm, E.J. Host lifestyle affects human microbiota on daily timescales. Genome Biol. 2014, 15, R89. [Google Scholar] [CrossRef] [Green Version]
- Graf, D.; Di Cagno, R.; Fåk, F.; Flint, H.J.; Nyman, M.; Saarela, M.; Watzl, B. Contribution of diet to the composition of the human gut microbiota. Microb Ecol. 2015, 26, 26164. [Google Scholar] [CrossRef]
- Sommer, F.; Anderson, J.M.; Bharti, R.; Raes, J.; Rosenstiel, P. The resilience of the intestinal microbiota influences health and disease. Nat. Rev. Microbiol 2017, 15, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wu, K.; Mehta, R.; Drew, D.A.; Song, M.; Lochhead, P.; Nguyen, L.H.; Izard, J.; Fuchs, C.S.; Garrett, W.S.; et al. Long-term use of antibiotics and risk of colorectal adenoma. Gut 2017, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Freifeld, A.G.; Bow, E.J.; Sepkowitz, K.A.; Boeckh, M.J.; Ito, J.I.; Mullen, C.A.; Raad, I.I.; Rolston, K.V.; Young, J.-A.H.; Wingard, J.R. Clinical Practice Guideline for the Use of Antimicrobial Agents in Neutropenic Patients with Cancer: 2010 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2011, 52, e56–e93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaac, S.; Scher, J.U.; Djukovic, A.; Jiménez, N.; Littman, D.R.; Abramson, S.B.; Pamer, E.G.; Ubeda, C. Short- and long-term effects of oral vancomycin on the human intestinal microbiota. J. Antimicrob. Chemother. 2017, 72, 128–136. [Google Scholar] [CrossRef]
- Capurso, G.; Lahner, E. The interaction between smoking, alcohol and the gut microbiome. Best Pr. Res. Clin. Gastroenterol. 2017, 31, 579–588. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Zhou, M.; Kang, C.; Lang, H.; Chen, M.; Hui, S.; Wang, B.; Mi, M. Crosstalk between gut microbiota and Sirtuin-3 in colonic inflammation and tumorigenesis. Exp. Mol. Med. 2018, 50, 21. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Zhang, C.; Xing, Y.; Xue, G.; Zhang, Q.; Pan, F.; Wu, G.; Hu, Y.; Guo, Q.; Lu, A.; et al. Remodelling of the gut microbiota by hyperactive NLRP3 induces regulatory T cells to maintain homeostasis. Nat. Commun. 2017, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Kissoon-singh, V.; Coria, A.L.; Moreau, F.; Chadee, K. Probiotic mixture VSL # 3 reduces colonic inflammation and improves intestinal barrier function in Muc2 mucin-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2017, 312, 34–45. [Google Scholar]
- Velcich, A. Colorectal Cancer in Mice Genetically Deficient in the Mucin Muc2. Science 2002, 295, 1726–1729. [Google Scholar] [CrossRef]
- Wu, M.; Wu, Y.; Li, J.; Bao, Y.; Guo, Y.; Yang, W. The Dynamic Changes of Gut Microbiota in Muc2 Deficient Mice. Int. J. Mol. 2018, 19, 2809. [Google Scholar] [CrossRef] [Green Version]
- Moschen, A.R.; Gerner, R.R.; Wang, J.; Klepsch, V.; Adolph, T.E.; Reider, S.J.; Hackl, H.; Pfister, A.; Schilling, J.; Moser, P.L.; et al. Lipocalin 2 Protects from Inflammation and Tumorigenesis Associated with Gut Microbiota Alterations. Cell Host Microbe 2016, 19, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Coker, O.O.; Nakatsu, G.; Wu, W.K.K.; Zhao, L.; Chen, Z.; Chan, F.K.L.; Kristiansen, K.; Sung, J.J.Y.; Wong, S.H.; et al. Multi-cohort analysis of colorectal cancer metagenome identified altered bacteria across populations and universal bacterial markers. Microbiome 2018, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- Ellermann, M.; Arthur, J.C. Siderophore-mediated iron acquisition and modulation of host–bacterial interactions. Free Radic. Biol. Med. 2017, 105, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma–carcinoma sequence. Nat. Commun 2015, 6, 6528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flemer, B.; Warren, R.D.; Barrett, M.P.; Cisek, K.; Das, A.; Jeffery, I.B.; Hurley, E.; Riordain, M.O.; Shanahan, F.; Toole, P.W.O. The oral microbiota in colorectal cancer is distinctive and predictive. Gut 2017, 67, 1454–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flemer, B.; Lynch, D.B.; Brown, J.M.R.; Jeffery, I.B.; Ryan, F.J.; Claesson, M.J.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 2017, 66, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Jacqueline, C.; Brazier, L.; Faugère, D.; Renaud, F.; Roche, B. Can intestinal microbiota be associated with non-intestinal cancers? Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Goedert, J.J.; Hua, X.; Bielecka, A.; Okayasu, I.; Milne, G.L.; Jones, G.S.; Fujiwara, M.; Sinha, R.; Wan, Y.; Xu, X.; et al. Postmenopausal breast cancer and oestrogen associations with the IgA-coated and IgA-noncoated faecal microbiota. Br. J. Cancer 2018, 118, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Fernández, M.F.; Reina-Pérez, I.; Astorga, J.M.; Rodríguez-Carrillo, A.; Plaza-Díaz, J.; Fontana, L. Breast Cancer and Its Relationship with the Microbiota. Int. J. Environ. Res. Public Health 2018, 15, 1747. [Google Scholar]
- Hsieh, Y.Y.; Tung, S.Y.; Pan, H.Y.; Yen, C.W.; Xu, H.W.; Lin, Y.J.; Deng, Y.F.; Hsu, W.T.; Wu, C.S.; Li, C. Increased Abundance of Clostridium and Fusobacterium in Gastric Microbiota of Patients with Gastric Cancer in Taiwan. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, J.; Chen, T.; Li, Q.; Peng, W.; Li, H.; Tang, X.; Fu, X. Fusobacterium nucleatum Potentiates Intestinal Tumorigenesis in Mice via a Toll-Like Receptor 4/p21-Activated Kinase 1 Cascade. Dig. Dis Sci 2018, 63, 1210–1218. [Google Scholar] [CrossRef] [PubMed]
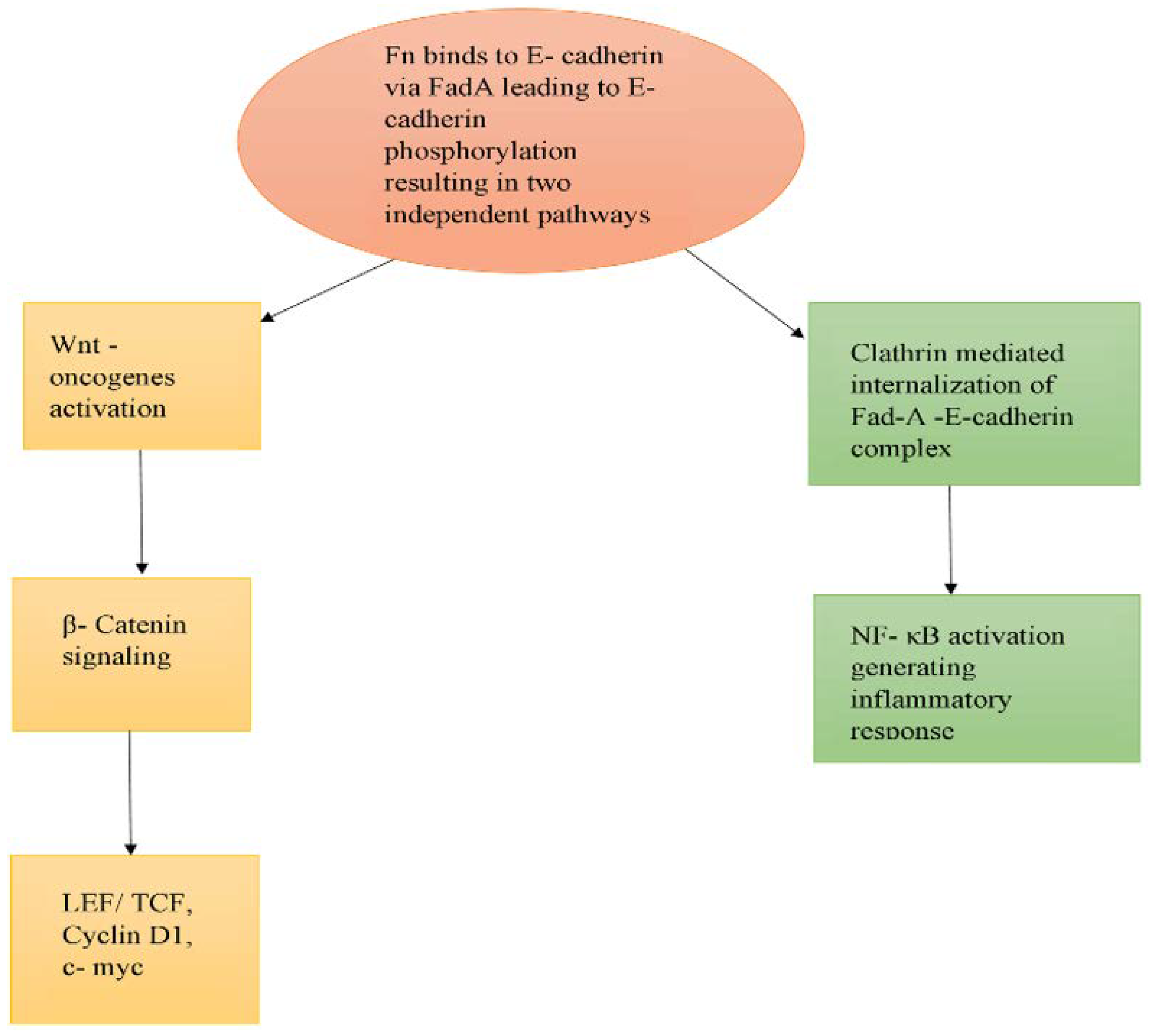
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abed, J.; Emgård, J.E.M.; Zamir, G.; Faroja, M.; Almogy, G.; Grenov, A.; Sol, A.; Naor, R.; Pikarsky, E.; Atlan, K.A.; et al. Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by Binding to Tumor-Expressed Gal-GalNAc. Cell Host Microbe 2016, 20, 215–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, A.C.; Shields, C.E.D.; Wu, S.; Huso, D.L.; Wu, X.; Murray-Stewart, T.R.; Hacker-Prietz, A.; Rabizadeh, S.; Woster, P.M.; Sears, C.L.; et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. PNAS 2011, 108, 15354–15359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sears, C.L.; Geis, A.L.; Housseau, F. Bacteroides fragilis subverts mucosal biology: From symbiont to colon carcinogenesis. J. Clin. Invest. 2014, 124, 4166–4172. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef]
- Bonnet, M.; Buc, E.; Sauvanet, P.; Darcha, C.; Dubois, D.; Pereira, B.; Dechelotte, P.; Bonnet, R.; Pezet, D.; Darfeuille-Michaud, A. Colonization of the Human Gut by E. coli and Colorectal Cancer Risk. Clin. Cancer Res. 2014, 20, 859–867. [Google Scholar] [CrossRef] [Green Version]
- Cuevas-Ramos, G.; Petit, C.R.; Marcq, I.; Boury, M.; Oswald, E.; Nougayrede, J.-P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. PNAS 2010, 107, 11537–11542. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Wu, S.; Zhang, Y.; Xia, Y.; Liu, X.; Zheng, Y.; Chen, H.; Schaefer, K.L.; Zhou, Z.; Bissonnette, M.; et al. Enteric bacterial protein AvrA promotes colonic tumorigenesis and activates colonic beta-catenin signaling pathway. Oncogenesis 2014, 3, e105. [Google Scholar] [CrossRef] [Green Version]
- Di Domenico, E.G.; Cavallo, I.; Pontone, M.; Toma, L.; Ensoli, F. Biofilm Producing Salmonella Typhi: Chronic Colonization and Development of Gallbladder Cancer. Int. J. Mol. Sci. 2017, 18, 1887. [Google Scholar] [CrossRef]
- Koshiol, J.; Wozniak, A.; Cook, P.; Adaniel, C.; Acevedo, J.; Azócar, L.; Hsing, A.W.; Roa, J.C.; Pasetti, M.F.; Miquel, J.F.; et al. Salmonella enterica serovar Typhi and gallbladder cancer: A case-control study and meta-analysis. Cancer Med. 2016, 5, 3235–3310. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, H.; Chu, E.S.H.; Zhang, X.; Sheng, J.; Nakatsu, G.; Ng, S.C.; Chan, A.W.H.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Peptostreptococcus anaerobius Induces Intracellular Cholesterol Biosynthesis in Colon Cells to Induce Proliferation and Causes Dysplasia in Mice. Gastroenterology 2017, 152, 1419–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, J.V.; Kosaka, T.; Sheppard, B.J.; Fox, J.G.; Schauer, D.B. Bacterial Infection Promotes Colon Tumorigenesis in Apc Min/+ Mice. J. Infect. Dis. 2001, 184, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Mao, Q.; Jiang, F.; Yin, R.; Wang, J.; Xia, W.; Dong, G.; Ma, W.; Yang, Y.; Xu, L.; Hu, J. Interplay between the lung microbiome and lung cancer. Cancer Lett. 2018, 415, 40–48. [Google Scholar] [CrossRef]
- Pilaniya, V.; Gera, K.; Kunal, S.; Shah, A. Pulmonary tuberculosis masquerading as metastatic lung disease. Eur. Respir. Rev. 2016, 25, 97–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.-Y.; Li, X.-L.; Yu, X.-S.; Guan, P.; Yin, Z.-H.; He, Q.-C.; Zhou, B.-S. Facts and fiction of the relationship between preexisting tuberculosis and lung cancer risk: A systematic review. Int. J. Cancer 2009, 125, 2936–2944. [Google Scholar] [CrossRef]
- Fan, X.; Alekseyenko, A.V.; Wu, J.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Abnet, C.C.; Stolzenberg-Solomon, R.; Miller, G.; et al. Human oral microbiome and prospective risk for pancreatic cancer: A population-based nested case-control study. Gut 2018, 67, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Curtis, M.A. Periodontal Microbiology—The Lid’s off the Box Again. J. Dent. Res. 2014, 93, 840–842. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Zhi, A.; Lai, P.F.H.; Wang, G.; Xia, Y.; Xiong, Z.; Zhang, H.; Che, N.; Ai, L. The oral microbiota – a mechanistic role for systemic diseases. Br. Dent. J. 2018, 224, 447–455. [Google Scholar] [CrossRef]
- Dicksved, J.; Lindberg, M.; Rosenquist, M.; Enroth, H.; Jansson, J.K.; Engstrand, L. Molecular characterization of the stomach microbiota in patients with gastric cancer and in controls. J. Med. Microbiol. 2009, 58, 509–516. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, J.; Xin, Y.; Geng, C.; Tian, Z.; Yu, X.; Dong, Q. Bacterial overgrowth and diversi fi cation of microbiota in gastric cancer. Eur. J. Gastroenterol. Hepatol. 2016, 28, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; Crabtree, J.; Mattick, J.S.A.; Anderson, K.E.; Hotopp, J.C.D. Distinguishing potential bacteria-tumor associations from contamination in a secondary data analysis of public cancer genome sequence data. Microbiome 2017, 5, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef]
- El-Aouar Filho, R.A.; Nicolas, A.; De Paula Castro, T.L.; Deplanche, M.; De Carvalho Azevedo, V.A.; Goossens, P.L.; Taieb, F.; Lina, G.; Le Loir, Y.; Berkova, N. Heterogeneous Family of Cyclomodulins: Smart Weapons That Allow Bacteria to Hijack the Eukaryotic Cell Cycle and Promote Infections. Front. Cell. Infect. Microbiol. 2017, 7, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, S.M.; Zhu, Q.; Zhu, L.; Liu, Z.; Karki, R.; Malik, A.; Sharma, D.; Li, L.; Malireddi, R.K.S.; Gurung, P.; et al. Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell 2015, 162, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Morin, P.J.; Maouyo, D.; Sears, C.L. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology 2003, 124, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Harmey, D.; Stenbeck, G.; Nobes, C.D.; Lax, A.J.; Grigoriadis, A.E. Regulation of Osteoblast Differentiation by Pasteurella Multocida Toxin (PMT): A Role for Rho GTPase in Bone Formation. J. Bone Miner. Res. 2004, 19, 661–670. [Google Scholar] [CrossRef]
- Buc, E.; Dubois, D.; Sauvanet, P.; Raisch, J.; Delmas, J.; Darfeuille-Michaud, A.; Pezet, D.; Bonnet, R. High Prevalence of Mucosa-Associated, E. coli Producing Cyclomodulin and Genotoxin in Colon Cancer. PLoS ONE 2013, 8, e56964. [Google Scholar] [CrossRef] [Green Version]
- Nougayrède, J.-P.; Taieb, F.; Rycke, J.D.; Oswald, E. Cyclomodulins: Bacterial effectors that modulate the eukaryotic cell cycle. TIM 2005, 13, 103–110. [Google Scholar]
- Horiguchi, Y. Escherichia coli cytotoxic necrotizing factors and Bordetella dermonecrotic toxin: The dermonecrosis-inducing toxins activating Rho small GTPases. Toxicon 2001, 39, 1619–1627. [Google Scholar] [CrossRef]
- Lax, A. The Pasteurella multocida Toxin: A New Paradigm for the Link Between Bacterial Infection and Cancer. In Pasteurella multocida; Aktories, K., Orth, J.H.C., Adler, B., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 361, pp. 131–144. ISBN 978-3-642-31016-4. [Google Scholar]
- De Luca, A.; Iaquinto, G. Helicobacter pylori and gastric diseases: A dangerous association. Cancer Letters 2004, 213, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Elinav, E.; Huber, S.; Strowig, T.; Hao, L.; Hafemann, A.; Jin, C.; Wunderlich, C.; Wunderlich, T.; Eisenbarth, S.C.; et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. PNAS 2013, 110, 9862–9867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuffrè, M.; Campigotto, M.; Campisciano, G.; Comar, M.; Crocè, L.S. A Story of Liver and Gut Microbes: How Does the Intestinal Flora Affect Liver Disease? AM. J. Physiol.-Gastrointest. Liver Physiol. 2020. [Google Scholar] [CrossRef]
- Yu, L.-X.; Schwabe, R.F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol Hepatol. 2017, 14, 527–539. [Google Scholar] [CrossRef]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome – mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef] [Green Version]
- Drewes, J.L.; White, J.R.; Dejea, C.M.; Fathi, P.; Iyadorai, T.; Vadivelu, J.; Roslani, A.C.; Wick, E.C.; Mongodin, E.F.; Loke, M.F.; et al. High-resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. npj Biofilms Microbiomes 2017, 3, 34. [Google Scholar] [CrossRef] [Green Version]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A microbial signature for Crohn’s disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.A.; Yang, J.; Dou, R.; Masugi, Y.; Song, M.; et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 2016, 65, 1973–1980. [Google Scholar] [CrossRef] [Green Version]
- Tahara, T.; Yamamoto, E.; Suzuki, H.; Maruyama, R.; Chung, W.; Garriga, J.; Jelinek, J.; Yamano, H.-o.; Sugai, T.; An, B.; et al. Fusobacterium in Colonic Flora and Molecular Features of Colorectal Carcinoma. Cancer Res. 2014, 74, 1311–1318. [Google Scholar] [CrossRef] [Green Version]
- Ianiro, G.; Molina-Infante, J.; Gasbarrini, A. Gastric Microbiota. Helicobacter 2015, 20, 68–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tegtmeyer, N.; Wessler, S.; Backert, S. Role of the cag -pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS J. 2011, 278, 1190–1202. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeyer, N.; Backert, S. Role of Abl and Src family kinases in actin-cytoskeletal rearrangements induced by the Helicobacter pylori CagA protein. Eur. J. Cell Biol. 2011, 90, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xu, S.; Zhu, Y. Helicobacter pylori CagA: A Critical Destroyer of the Gastric Epithelial Barrier. Dig. Dis. Sci. 2013, 58, 1830–1837. [Google Scholar] [CrossRef] [PubMed]
- Tohidpour, A. CagA-mediated Pathogenesis of Helicobacter pylori. Microb. Pathog. 2016, 93, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Aoyama, T.; Kashiwabara, K.; Oba, K.; Honda, M.; Sadahiro, S.; Hamada, C.; Maeda, H.; Mayanagi, S.; Kanda, M.; Sakamoto, J.; et al. Clinical impact of tumor location on the colon cancer survival and recurrence: Analyses of pooled data from three large phase III randomised clinical trials. Cancer Med. 2017, 6, 2523–2530. [Google Scholar] [CrossRef]
- Mima, K.; Cao, Y.; Chan, A.T.; Qian, Z.R.; Nowak, J.A.; Masugi, Y.; Shi, Y.; Song, M.; da Silva, A.; Gu, M.; et al. Fusobacterium nucleatum in Colorectal Carcinoma Tissue According to Tumor Location. Clin. Transl. Gastroenterol. 2016, 7, e200. [Google Scholar] [CrossRef]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [Green Version]
- O’Keefe, S.J.D. Diet, microorganisms and their metabolites, and colon cancer. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 691–706. [Google Scholar]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppenhagen-Glazer, S.; Sol, A.; Abed, J.; Naor, R.; Zhang, X.; Han, Y.W.; Bachrach, G. Fap2 of Fusobacterium nucleatum Is a Galactose-Inhibitable Adhesin Involved in Coaggregation, Cell Adhesion, and Preterm Birth. Infect. Immun. 2015, 83, 1104–1113. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Weng, W.; Peng, J.; Hong, L.; Yang, L.; Toiyama, Y.; Gao, R.; Liu, M.; Yin, M.; Pan, C.; et al. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor−κB, and Up-regulating Expression of MicroRNA-21. Gastroenterology 2017, 152, 851–C866. [Google Scholar] [CrossRef] [Green Version]
- Tomkovich, S.; Yang, Y.; Winglee, K.; Gauthier, J.; Mühlbauer, M.; Sun, X.; Mohamadzadeh, M.; Liu, X.; Martin, P.; Wang, G.P.; et al. Locoregional Effects of Microbiota in a Preclinical Model of Colon Carcinogenesis. Cancer Res. 2017, 77, 2620–2632. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.C.; Gharaibeh, R.Z.; Mühlbauer, M.; Perez-Chanona, E.; Uronis, J.M.; McCafferty, J.; Fodor, A.A.; Jobin, C. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat. Commun 2014, 5, 4724. [Google Scholar] [CrossRef] [Green Version]
- Dalmasso, G.; Cougnoux, A.; Delmas, J.; Darfeuille-Michaud, A.; Bonnet, R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes 2014, 5, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 Protein of Fusobacterium nucleatum to Human Inhibitory Receptor TIGIT Protects Tumors from Immune Cell Attack. Immunity 2015, 42, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Nathenson, M.J.; Conley, A.P.; Sausville, E. Immunotherapy: A New (and Old) Approach to Treatment of Soft Tissue and Bone Sarcomas. Oncologist 2018, 23, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 1448, 1443–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivaprakasam, S.; Gurav, A.; Paschall, A.V.; Coe, G.L.; Chaudhary, K.; Cai, Y.; Kolhe, R.; Martin, P.; Browning, D.; Huang, L.; et al. An essential role of Ffar2 (Gpr43) in dietary fibre-mediated promotion of healthy composition of gut microbiota and suppression of intestinal carcinogenesis. Oncogenesis 2016, 5, e238. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Pietrocola, F.; Kroemer, G. Nutrition, inflammation and cancer. Nat. Immunol. 2017, 18, 843–850. [Google Scholar] [CrossRef]
- Wu, W.; Sun, M.; Chen, F.; Cao, A.T.; Liu, H.; Zhao, Y.; Huang, X.; Xiao, Y.; Yao, S.; Zhao, Q.; et al. Microbiota metabolite short-chain fatty acid acetate promotes intestinal IgA response to microbiota which is mediated by GPR43. Mucosal Immunol. 2017, 10, 946–956. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, N.; Yuan, R.; Prestwood, T.R.; Penny, H.L.; DiMaio, M.A.; Reticker-Flynn, N.E.; Krois, C.R.; Kenkel, J.A.; Pham, T.D.; Carmi, Y.; et al. Normalizing Microbiota-Induced Retinoic Acid Deficiency Stimulates Protective CD8 + T Cell-Mediated Immunity in Colorectal Cancer. Immunity 2016, 45, 641–655. [Google Scholar] [CrossRef] [Green Version]
- Lyu, Y.; Wu, L.; Wang, F.; Shen, X.; Lin, D. Carotenoid supplementation and retinoic acid in immunoglobulin A regulation of the gut microbiota dysbiosis. Exp. Biol Med. (Maywood) 2018, 243, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Okai, S.; Usui, F.; Ohta, M.; Mori, H.; Kurokawa, K.; Matsumoto, S.; Kato, T.; Miyauchi, E.; Ohno, H.; Shinkura, R. Intestinal IgA as a modulator of the gut microbiota. Gut Microbes 2017, 8, 486–492. [Google Scholar] [CrossRef] [Green Version]
- Peterson, D.A.; McNulty, N.P.; Guruge, J.L.; Gordon, J.I. IgA Response to Symbiotic Bacteria as a Mediator of Gut Homeostasis. Cell Host Microbe 2007, 2, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, A.; Vogelzang, A.; Maruya, M.; Miyajima, M.; Murata, M.; Son, A.; Kuwahara, T.; Tsuruyama, T.; Yamada, S.; Matsuura, M.; et al. IgA regulates the composition and metabolic function of gut microbiota by promoting symbiosis between bacteria. J. Exp. Med. 2018, 215, 2019–2034. [Google Scholar] [CrossRef] [Green Version]
- Statovci, D.; Aguilera, M.; MacSharry, J.; Melgar, S. The Impact of Western Diet and Nutrients on the Microbiota and Immune Response at Mucosal Interfaces. Front. Immunol. 2017, 8, 838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agus, A.; Denizot, J.; Thévenot, J.; Martinez-Medina, M.; Massier, S.; Sauvanet, P.; Bernalier-Donadille, A.; Denis, S.; Hofman, P.; Bonnet, R.; et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive, E. coli infection and intestinal inflammation. Sci Rep. 2016, 6, 19032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadaka, A.O.; Ojo, B.A.; Adewale, O.B.; Esho, T.; Pretorius, A. Effect of dietary components on miRNA and colorectal carcinogenesis. Cancer Cell Int. 2018, 18, 130. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Dong, T.S.; Dalal, S.R.; Wu, F.; Bissonnette, M.; Kwon, J.H.; Chang, E.B. The Microbe-Derived Short Chain Fatty Acid Butyrate Targets miRNA-Dependent p21 Gene Expression in Human Colon Cancer. PLoS ONE 2011, 6, e16221. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Liu, L.; Chang, E.B.; Wang, J.-Y.; Raufman, J.-P. Butyrate inhibits pro-proliferative miR-92a by diminishing c-Myc-induced miR-17-92a cluster transcription in human colon cancer cells. Mol. Cancer 2015, 14, 180. [Google Scholar] [CrossRef] [Green Version]
- Donohoe, D.R.; Holley, D.; Collins, L.B.; Montgomery, S.A.; Whitmore, A.C.; Hillhouse, A.; Curry, K.P.; Renner, S.W.; Greenwalt, A.; Ryan, E.P.; et al. A Gnotobiotic Mouse Model Demonstrates That Dietary Fiber Protects against Colorectal Tumorigenesis in a Microbiota- and Butyrate-Dependent Manner. Cancer Discov. 2014, 4, 1387–1397. [Google Scholar] [CrossRef] [Green Version]
- Marks, P.A.; Richon, V.M.; Rifkind, R.A. Histone Deacetylase Inhibitors: Inducers of Differentiation or Apoptosis of Transformed Cells. J. Natl. Cancer 2000, 92, 1210–1216. [Google Scholar] [CrossRef]
- Bordonaro, M.; Lazarova, D.L.; Sartorelli, A.C. Butyrate and Wnt signaling: A possible solution to the puzzle of dietary fiber and colon cancer risk? Cell Cycle 2008, 7, 1178–1183. [Google Scholar] [CrossRef]
- Hu, Y.; Le Leu, R.K.; Christophersen, C.T.; Somashekar, R.; Conlon, M.A.; Meng, X.Q.; Winter, J.M.; Woodman, R.J.; McKinnon, R.; Young, G.P. Manipulation of the gut microbiota using resistant starch is associated with protection against colitis-associated colorectal cancer in rats. Carcin 2016, 37, 366–375. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Chen, Y.; Jiang, H.; Robbins, G.T.; Nie, D. G-protein-coupled receptor for short-chain fatty acids suppresses colon cancer. Int. J. Cancer 2011, 128, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Luu, M.; Weigand, K.; Wedi, F.; Breidenbend, C.; Leister, H.; Pautz, S.; Adhikary, T.; Visekruna, A. Regulation of the effector function of CD8+ T cells by gut microbiota-derived metabolite butyrate. Sci Rep. 2018, 8, 14430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcheva, A.; Irrazabal, T.; Robertson, S.J.; Streutker, C.; Maughan, H.; Rubino, S.; Moriyama, E.H.; Copeland, J.K.; Surendra, A.; Kumar, S.; et al. Gut Microbial Metabolism Drives Transformation of Msh2-Deficient Colon Epithelial Cells. Cell 2014, 158, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Lazarova, D.L.; Bordonaro, M.; Carbone, R.; Sartorelli, A.C. Linear relationship between Wnt activity levels and apoptosis in colorectal carcinoma cells exposed to butyrate. Int. J. Cancer 2004, 110, 523–531. [Google Scholar] [CrossRef]
- Baxter, N.T.; Zackular, J.P.; Chen, G.Y.; Schloss, P.D. Structure of the gut microbiome following colonization with human feces determines colonic tumor burden. Microbiome 2014, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Linden, D.R. Hydrogen Sulfide Signaling in the Gastrointestinal Tract. Antioxidants Redox Signal. 2013, 20, 818–830. [Google Scholar] [CrossRef]
- Windey, K.; De Preter, V.; Verbeke, K. Relevance of protein fermentation to gut health. Mol. Nutr. Food Res. 2012, 56, 184–196. [Google Scholar] [CrossRef]
- Liu, J.; Lkhagva, E.; Chung, H.-J.; Kim, H.-J.; Hong, S.-T. The Pharmabiotic Approach to Treat Hyperammonemia. Nutrients 2018, 10, 140. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteria/Clade | Mode of Action | Cancer |
|---|---|---|
| Helicobacter pylori | Disruption of stomach and colonic epithelial integrity creates a niche in stomach suitable for further pathogenic bacterial invasion [40]. | Stomach and colorectal. |
| Fusobacterium nucleatum | Suspension of disintegration of β-catenin signalling, increased expression of TLR4 activation of p21- activated kinase and cyclin D1 [41], increased inflammatory gene expression, and suppression of antitumour NKT cells via effector molecules FadA and Fap2 [42,43]. | Stomach, colorectal, oral, and lung. |
| Bacteroides fragilis | Reactive-oxygen-species (ROS) generation leading to DNA damage, colon-epithelial-barrier disruption, and depletion of mucous membrane, causing increased inflammation [44,45,46]. | Stomach, colorectal, and lung. |
| Pathogenic Escherichia coli | Toxin colibactin indirectly induces release of growth factors in tumour microenvironment; cytotoxic necrotizing factor (CNF)-mediated disruption of host cell DNA repair mechanism [47,48]. | Stomach and lung. |
| Salmonella sp. | Stabilises and prevents degradation of β-catenin by deubiquitinase activity of its AvrA protein [49,50,51]. | Stomach, colorectal, gall-bladder, and lung. |
| Peptostreptococcus anaerobius | Increases expression of SREB2 gene via ROS, causing increased cholesterol biosynthesis in colon [52]. | Colorectal. |
| Citrobacter rodentium | Loss of cell polarity, depletion of epithelial barrier, and increased inflammation [53]. | Colorectal. |
| Mycobacterium tuberculosis, Streptococcus viridans, Haemophilus influenza, Streptococcus pnuemoniae, Staphyloccocus | Involved in various chronic inflammatory lung disorders like asthma, cystic fibrosis, and chronic obstructive pulmonary disease; potential for accelerating tumourigenesis via inflammatory cytokines like tumour necrosis factor [54,55,56]. | Lung. |
| Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans | Reach pancreas from oral cavity through blood circulation and act as secondary drivers of cancer; impair host innate immunity, leading to increased colonisation by other bacteria, leading to chronic inflammation of pancreas causing accelerated tumourigenesis [57,58,59]. | Pancreatic. |
| Proteobacteria, Betaproteobacteria, Firmicutes, Alcaligenaceae, Burkholderiales | Alter metabolism and oestrogen recycling, and exert pressure on immune system [38]. | Breast. |
| P. gingivalis and Tannerella forsythia | Cause overexpression of inflammatory cytokines; gingipain K produced by P. gingivalis paralyses immune cells, and induce indirect overexpression of glucose-transporter (GLUT-1 and GLUT-4) genes that help in faster tumour-cell proliferation [60,61,62,63]. | Oesophageal. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rastogi, Y.R.; Saini, A.K.; Thakur, V.K.; Saini, R.V. New Insights into Molecular Links Between Microbiota and Gastrointestinal Cancers: A Literature Review. Int. J. Mol. Sci. 2020, 21, 3212. https://doi.org/10.3390/ijms21093212
Rastogi YR, Saini AK, Thakur VK, Saini RV. New Insights into Molecular Links Between Microbiota and Gastrointestinal Cancers: A Literature Review. International Journal of Molecular Sciences. 2020; 21(9):3212. https://doi.org/10.3390/ijms21093212
Chicago/Turabian StyleRastogi, Yash Raj, Adesh K. Saini, Vijay Kumar Thakur, and Reena V. Saini. 2020. "New Insights into Molecular Links Between Microbiota and Gastrointestinal Cancers: A Literature Review" International Journal of Molecular Sciences 21, no. 9: 3212. https://doi.org/10.3390/ijms21093212
APA StyleRastogi, Y. R., Saini, A. K., Thakur, V. K., & Saini, R. V. (2020). New Insights into Molecular Links Between Microbiota and Gastrointestinal Cancers: A Literature Review. International Journal of Molecular Sciences, 21(9), 3212. https://doi.org/10.3390/ijms21093212