Immunomodulation in Cystic Fibrosis: Why and How?

 and
and
Abstract
: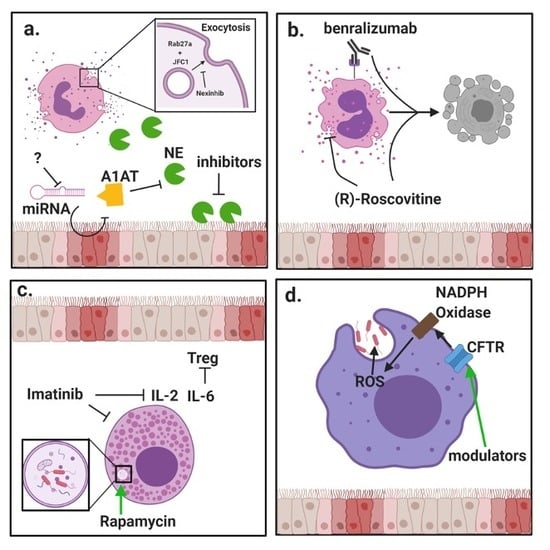
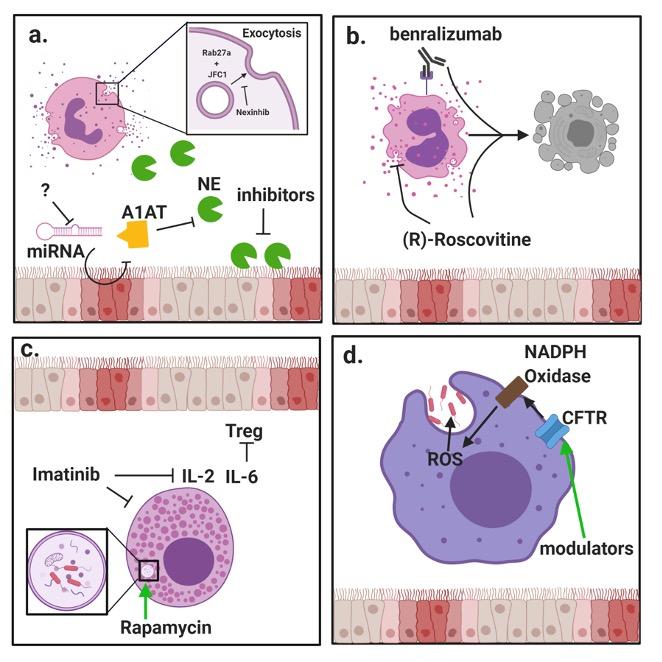{kind=link}
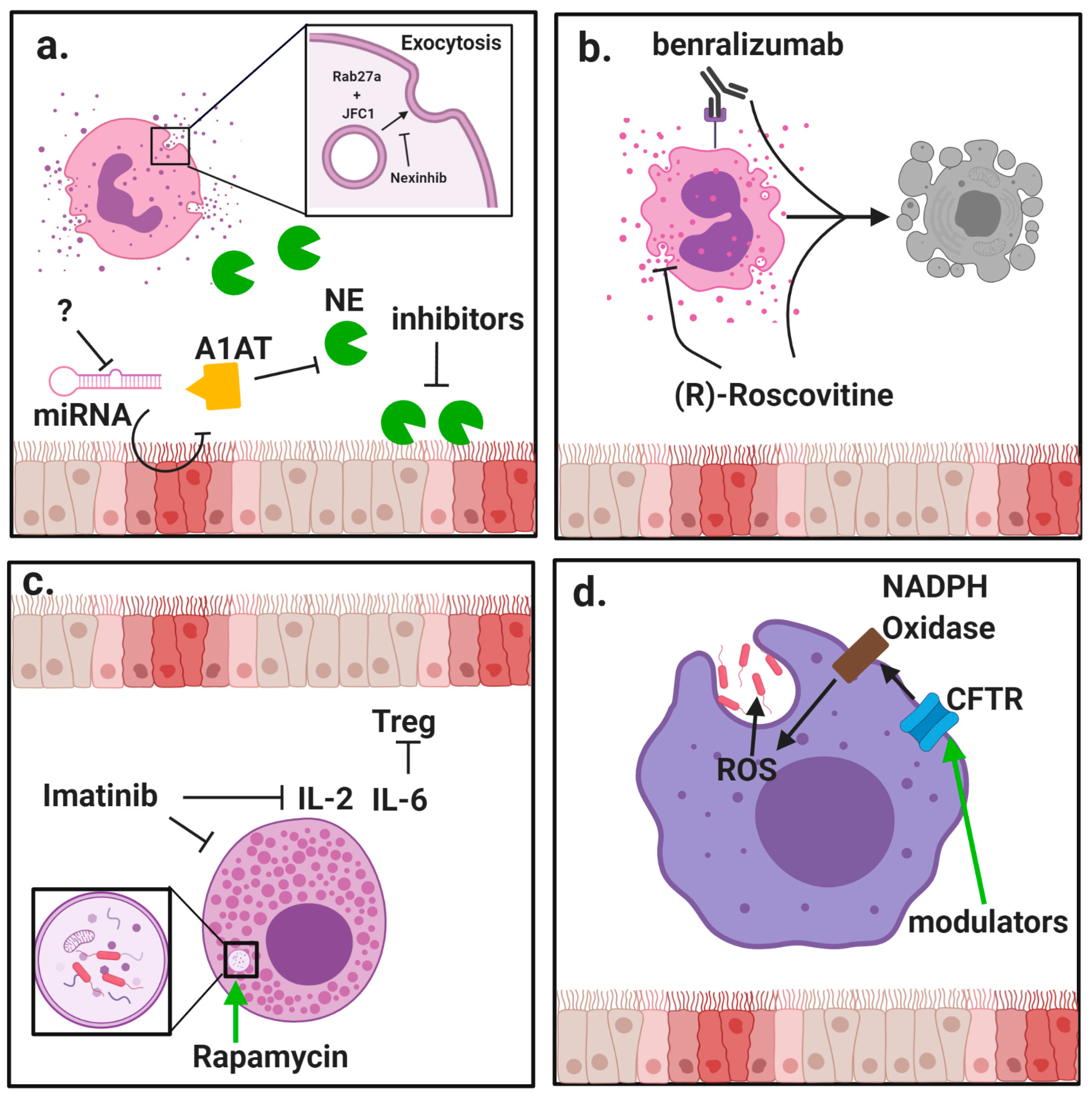{kind=link}
{kind=link}
1. Introduction
2. Targeting Immune Cells in the CF Lung
2.1. Neutrophils
2.2. Eosinophils
2.3. Basophils
2.4. Mast Cells
2.5. Macrophages
2.6. T Cells
2.7. B Cells
3. Protein-Directed Therapies
3.1. Neutrophil Exocytosis
3.2. Neutrophil Elastase
3.3. Matrix Metalloproteinases
3.4. Effect of CFTR Modulators on Macrophages
4. Nucleic Acid-Based Therapies
5. Conclusions
Unmet Need for Immunotherapies
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| A1AT | Alpha-1 antitrypsin |
| ABPA | Allergic bronchopulmonary aspergillosis |
| ASO | Antisense oligonucleotide |
| Arg1 | Arginase-1 |
| CF | Cystic fibrosis |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| cGAS | Cyclic-GMP-AMP synthase |
| CXCL | C-X-C motif chemokine ligand |
| DNAse | Deoxyribonuclease |
| ECP | Eosinophil cationic protein |
| FEV1 | Forced expiratory volume in 1 s |
| IDO | Indoleamine 2,3-dioxygenase |
| IFN | Interferon |
| Ig | Immunoglobulin |
| IL | Interleukin |
| KC | Keratinocyte chemoattractant |
| MMP | Matrix metalloproteinase |
| MPO | Myeloperoxidase |
| NADPH | Nicotinamide adenine nucleotide |
| NE | Neutrophil elastase |
| NOD | Nucleotide-binding oligomerization domain |
| OAS | Oligoadenylate synthetase |
| PRR | Pattern recognition receptor |
| RIG-I | Retinoic acid inducible gene I |
| RNAse | Ribonuclease |
| ROS | Reactive oxygen species |
| STING | Stimulator of interferon genes |
| TGF-β | Transforming growth factor β |
| Th | T helper cells |
| TLR | Toll-like receptor |
| TNFα | Tumor necrosis factor α |
| Tregs | Regulatory T cells |
References
- Berical, A.; Lee, R.E.; Randell, S.H.; Hawkins, F. Challenges Facing Airway Epithelial Cell-Based Therapy for Cystic Fibrosis. Front. Pharmacol. 2019, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Cholon, D.M.; Gentzsch, M. Recent progress in translational cystic fibrosis research using precision medicine strategies. J. Cyst. Fibros. 2018, 17, S52–S60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rose, V.; Molloy, K.; Gohy, S.; Pilette, C.; Greene, C.M. Airway Epithelium Dysfunction in Cystic Fibrosis and COPD. Mediat. Inflamm. 2018, 2018, 1309746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnaby, R.; Koeppen, K.; Nymon, A.; Hampton, T.H.; Berwin, B.; Ashare, A.; Stanton, B.A. Lumacaftor (VX-809) restores the ability of CF macrophages to phagocytose and kill Pseudomonas aeruginosa. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L432–L438. [Google Scholar] [CrossRef] [Green Version]
- Ng, H.P.; Valentine, V.G.; Wang, G. CFTR targeting during activation of human neutrophils. J. Leukoc. Biol. 2016, 100, 1413–1424. [Google Scholar] [CrossRef]
- Painter, R.G.; Valentine, V.G.; Lanson, N.A., Jr.; Leidal, K.; Zhang, Q.; Lombard, G.; Thompson, C.; Viswanathan, A.; Nauseef, W.M.; Wang, G.; et al. CFTR Expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry 2006, 45, 10260–10269. [Google Scholar] [CrossRef] [Green Version]
- Tarique, A.A.; Sly, P.D.; Holt, P.G.; Bosco, A.; Ware, R.S.; Logan, J.; Bell, S.C.; Wainwright, C.E.; Fantino, E. CFTR-dependent defect in alternatively-activated macrophages in cystic fibrosis. J. Cyst. Fibros. 2017, 16, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Shrestha, C.L.; Kopp, B.T. Cystic fibrosis transmembrane conductance regulator (CFTR) modulators have differential effects on cystic fibrosis macrophage function. Sci. Rep. 2018, 8, 17066. [Google Scholar] [CrossRef] [Green Version]
- Polverino, F.; Lu, B.; Quintero, J.R.; Vargas, S.O.; Patel, A.S.; Owen, C.A.; Gerard, N.P.; Gerard, C.; Cernadas, M. CFTR regulates B cell activation and lymphoid follicle development. Respir. Res. 2019, 20, 133. [Google Scholar] [CrossRef] [Green Version]
- Moss, R.B.; Bocian, R.C.; Hsu, Y.P.; Dong, Y.J.; Kemna, M.; Wei, T.; Gardner, P. Reduced IL-10 secretion by CD4+ T lymphocytes expressing mutant cystic fibrosis transmembrane conductance regulator (CFTR). Clin. Exp. Immunol. 1996, 106, 374–388. [Google Scholar] [CrossRef]
- Law, S.M.; Stanfield, S.J.; Hardisty, G.R.; Dransfield, I.; Campbell, C.J.; Gray, R.D. Human cystic fibrosis monocyte derived macrophages display no defect in acidification of phagolysosomes when measured by optical nanosensors. J. Cyst. Fibros. 2019. [Google Scholar] [CrossRef] [PubMed]
- Leuer, L.; Krill, A.; Wilkens, H.; Wagenpfeil, G.; Bischoff, M.; Meier, C.; Bals, R.; Tschernig, T. The Phagocytosis of Blood Leukocytes from Cystic Fibrosis Patients is not Impaired in General. Lung 2020, 198, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Giacalone, V.D.; Margaroli, C.; Mall, M.A.; Tirouvanziam, R. Neutrophil Adaptations upon Recruitment to the Lung: New Concepts and Implications for Homeostasis and Disease. Int. J. Mol. Sci. 2020, 21, 851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- Perrem, L.; Ratjen, F. Anti-inflammatories and mucociliary clearance therapies in the age of CFTR modulators. Pediatr. Pulmonol. 2019, 54 (Suppl. 3), S46–S55. [Google Scholar] [CrossRef] [Green Version]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef]
- Laval, J.; Ralhan, A.; Hartl, D. Neutrophils in cystic fibrosis. Biol. Chem. 2016, 397, 485–496. [Google Scholar] [CrossRef]
- Margaroli, C.; Tirouvanziam, R. Neutrophil plasticity enables the development of pathological microenvironments: Implications for cystic fibrosis airway disease. Mol. Cell. Pediatr. 2016, 3, 38. [Google Scholar] [CrossRef] [Green Version]
- Byrne, A.J.; Mathie, S.A.; Gregory, L.G.; Lloyd, C.M. Pulmonary macrophages: Key players in the innate defence of the airways. Thorax 2015, 70, 1189–1196. [Google Scholar] [CrossRef] [Green Version]
- Bruscia, E.M.; Bonfield, T.L. Cystic Fibrosis Lung Immunity: The Role of the Macrophage. J. Innate Immun. 2016, 8, 550–563. [Google Scholar] [CrossRef]
- Kim, C.; delaRiva-Velasco, E.; Budhram, A.; Farri, F.; Krich, D.; Nolan, S.S.; Gjonaj, S.; Paul, L.; Dozor, A.J.; Welter, J.J. Incidence and prevalence of common respiratory pathogens before and after implementation of the Cystic Fibrosis Foundation Infection Prevention and Control Guideline. J. Infect. Prev. 2020, 21, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Degiacomi, G.; Sammartino, J.C.; Chiarelli, L.R.; Riabova, O.; Makarov, V.; Pasca, M.R. Mycobacterium abscessus, an Emerging and Worrisome Pathogen among Cystic Fibrosis Patients. Int. J. Mol. Sci. 2019, 20, 5868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingersoll, S.A.; Laval, J.; Forrest, O.A.; Preininger, M.; Brown, M.R.; Arafat, D.; Gibson, G.; Tangpricha, V.; Tirouvanziam, R. Mature cystic fibrosis airway neutrophils suppress T cell function: Evidence for a role of arginase 1 but not programmed death-ligand 1. J. Immunol. 2015, 194, 5520–5528. [Google Scholar] [CrossRef] [PubMed]
- Fritzsching, B.; Zhou-Suckow, Z.; Trojanek, J.B.; Schubert, S.C.; Schatterny, J.; Hirtz, S.; Agrawal, R.; Muley, T.; Kahn, N.; Sticht, C.; et al. Hypoxic epithelial necrosis triggers neutrophilic inflammation via IL-1 receptor signaling in cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2015, 191, 902–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balazs, A.; Mall, M.A. Mucus obstruction and inflammation in early cystic fibrosis lung disease: Emerging role of the IL-1 signaling pathway. Pediatr. Pulmonol. 2019, 54 (Suppl. 3), S5–S12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirouvanziam, R.; Gernez, Y.; Conrad, C.K.; Moss, R.B.; Schrijver, I.; Dunn, C.E.; Davies, Z.A.; Herzenberg, L.A.; Herzenberg, L.A. Profound functional and signaling changes in viable inflammatory neutrophils homing to cystic fibrosis airways. Proc. Natl. Acad. Sci. USA 2008, 105, 4335–4339. [Google Scholar] [CrossRef] [Green Version]
- Makam, M.; Diaz, D.; Laval, J.; Gernez, Y.; Conrad, C.K.; Dunn, C.E.; Davies, Z.A.; Moss, R.B.; Herzenberg, L.A.; Herzenberg, L.A.; et al. Activation of critical, host-induced, metabolic and stress pathways marks neutrophil entry into cystic fibrosis lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 5779–5783. [Google Scholar] [CrossRef] [Green Version]
- Perrem, L.; Ratjen, F. Letting It All Out: Neutrophils in Early Cystic Fibrosis Airway Inflammation. Am. J. Respir. Crit. Care Med. 2019, 199, 816–817. [Google Scholar] [CrossRef]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Bruce, M.C.; Poncz, L.; Klinger, J.D.; Stern, R.C.; Tomashefski, J.F., Jr.; Dearborn, D.G. Biochemical and pathologic evidence for proteolytic destruction of lung connective tissue in cystic fibrosis. Am. Rev. Respir. Dis. 1985, 132, 529–535. [Google Scholar]
- Park, J.A.; He, F.; Martin, L.D.; Li, Y.; Chorley, B.N.; Adler, K.B. Human neutrophil elastase induces hypersecretion of mucin from well-differentiated human bronchial epithelial cells in vitro via a protein kinase C{delta}-mediated mechanism. Am. J. Pathol. 2005, 167, 651–661. [Google Scholar] [CrossRef]
- Le Gars, M.; Descamps, D.; Roussel, D.; Saussereau, E.; Guillot, L.; Ruffin, M.; Tabary, O.; Hong, S.S.; Boulanger, P.; Paulais, M.; et al. Neutrophil elastase degrades cystic fibrosis transmembrane conductance regulator via calpains and disables channel function in vitro and in vivo. Am. J. Respir. Crit. Care Med. 2013, 187, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Sly, P.D.; Gangell, C.L.; Chen, L.; Ware, R.S.; Ranganathan, S.; Mott, L.S.; Murray, C.P.; Stick, S.M.; Investigators, A.C. Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 2013, 368, 1963–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, C.A.; Campbell, M.A.; Sannes, P.L.; Boukedes, S.S.; Campbell, E.J. Cell surface-bound elastase and cathepsin G on human neutrophils: A novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J. Cell. Biol. 1995, 131, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Genschmer, K.R.; Russell, D.W.; Lal, C.; Szul, T.; Bratcher, P.E.; Noerager, B.D.; Abdul Roda, M.; Xu, X.; Rezonzew, G.; Viera, L.; et al. Activated PMN Exosomes: Pathogenic Entities Causing Matrix Destruction and Disease in the Lung. Cell 2019, 176, 113–126.e15. [Google Scholar] [CrossRef] [Green Version]
- Wagner, C.J.; Schultz, C.; Mall, M.A. Neutrophil elastase and matrix metalloproteinase 12 in cystic fibrosis lung disease. Mol. Cell. Pediatr. 2016, 3, 25. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.F.; Hwang, T.L. Neutrophil elastase inhibitors: A patent review and potential applications for inflammatory lung diseases (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 1145–1158. [Google Scholar] [CrossRef]
- Garratt, L.W.; Sutanto, E.N.; Ling, K.M.; Looi, K.; Iosifidis, T.; Martinovich, K.M.; Shaw, N.C.; Buckley, A.G.; Kicic-Starcevich, E.; Lannigan, F.J.; et al. Alpha-1 Antitrypsin Mitigates the Inhibition of Airway Epithelial Cell Repair by Neutrophil Elastase. Am. J. Respir. Cell. Mol. Biol. 2016, 54, 341–349. [Google Scholar] [CrossRef]
- Gaggar, A.; Hector, A.; Bratcher, P.E.; Mall, M.A.; Griese, M.; Hartl, D. The role of matrix metalloproteinases in cystic fibrosis lung disease. Eur. Respir. J. 2011, 38, 721–727. [Google Scholar] [CrossRef]
- van Dalen, C.J.; Whitehouse, M.W.; Winterbourn, C.C.; Kettle, A.J. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem. J. 1997, 327, 487–492. [Google Scholar] [CrossRef]
- Dickerhof, N.; Pearson, J.F.; Hoskin, T.S.; Berry, L.J.; Turner, R.; Sly, P.D.; Kettle, A.J.; Arest, C.F. Oxidative stress in early cystic fibrosis lung disease is exacerbated by airway glutathione deficiency. Free Radic. Biol. Med. 2017, 113, 236–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, J.D.; Margaroli, C.; Horati, H.; Kilgore, M.B.; Veltman, M.; Liu, H.K.; Taurone, A.J.; Peng, L.; Guglani, L.; Uppal, K.; et al. Myeloperoxidase oxidation of methionine associates with early cystic fibrosis lung disease. Eur. Respir. J. 2018, 52, 1801118. [Google Scholar] [CrossRef] [PubMed]
- Forrest, O.A.; Chopyk, D.M.; Gernez, Y.; Brown, M.R.; Conrad, C.K.; Moss, R.B.; Tangpricha, V.; Peng, L.; Tirouvanziam, R. Resistin is elevated in cystic fibrosis sputum and correlates negatively with lung function. J. Cyst. Fibros. 2019, 18, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Mejias, J.C.; Forrest, O.A.; Margaroli, C.; Frey Rubio, D.A.; Viera, L.; Li, J.; Xu, X.; Gaggar, A.; Tirouvanziam, R.; Roy, K. Neutrophil-targeted, protease-activated pulmonary drug delivery blocks airway and systemic inflammation. JCI Insight 2019, 4, 131468. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Ramadass, M.; He, J.; Brown, S.J.; Zhang, J.; Abgaryan, L.; Biris, N.; Gavathiotis, E.; Rosen, H.; Catz, S.D. Identification of Neutrophil Exocytosis Inhibitors (Nexinhibs), Small Molecule Inhibitors of Neutrophil Exocytosis and Inflammation: DRUGGABILITY OF THE SMALL GTPase Rab27a. J. Biol. Chem. 2016, 291, 25965–25982. [Google Scholar] [CrossRef] [Green Version]
- Ravin, K.A.; Loy, M. The Eosinophil in Infection. Clin. Rev. Allergy Immunol. 2016, 50, 214–227. [Google Scholar] [CrossRef]
- Bousquet, J.; Jeffery, P.K.; Busse, W.W.; Johnson, M.; Vignola, A.M. Asthma. From bronchoconstriction to airways inflammation and remodeling. Am. J. Respir. Crit. Care Med. 2000, 161, 1720–1745. [Google Scholar] [CrossRef] [Green Version]
- Levine, H.; Cohen-Cymberknoh, M.; Klein, N.; Hoshen, M.; Mussaffi, H.; Stafler, P.; Breuer, O.; Kerem, E.; Blau, H. Reversible airway obstruction in cystic fibrosis: Common, but not associated with characteristics of asthma. J. Cyst. Fibros. 2016, 15, 652–659. [Google Scholar] [CrossRef] [Green Version]
- Koller, D.Y.; Gotz, M.; Eichler, I.; Urbanek, R. Eosinophilic activation in cystic fibrosis. Thorax 1994, 49, 496–499. [Google Scholar] [CrossRef] [Green Version]
- Alyasin, S.; Moghtaderi, M.; Farjadian, S.; Babaei, M.; Teshnizi, S.H. Allergic bronchopulmonary aspergillosis in patients with cystic fibrosis and non-cystic fibrosis bronchiectasis. Electron. Physician 2018, 10, 6273–6278. [Google Scholar] [CrossRef] [Green Version]
- Goralski, J.L.; Lercher, D.M.; Davis, S.D.; Dellon, E.S. Eosinophilic esophagitis in cystic fibrosis: A case series and review of the literature. J. Cyst. Fibros. 2013, 12, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinke, J.W.; Payne, S.C.; Chen, P.G.; Negri, J.; Stelow, E.B.; Borish, L. Etiology of nasal polyps in cystic fibrosis: Not a unimodal disease. Ann. Otol. Rhinol. Laryngol. 2012, 121, 579–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koller, D.Y.; Urbanek, R.; Gotz, M. Increased degranulation of eosinophil and neutrophil granulocytes in cystic fibrosis. Am. J. Respir. Crit. Care Med. 1995, 152, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Lammertyn, E.J.; Vandermeulen, E.; Bellon, H.; Everaerts, S.; Verleden, S.E.; Van Den Eynde, K.; Bracke, K.R.; Brusselle, G.G.; Goeminne, P.C.; Verbeken, E.K.; et al. End-stage cystic fibrosis lung disease is characterised by a diverse inflammatory pattern: An immunohistochemical analysis. Respir. Res. 2017, 18, 10. [Google Scholar] [CrossRef] [Green Version]
- Meijer, L.; Nelson, D.J.; Riazanski, V.; Gabdoulkhakova, A.G.; Hery-Arnaud, G.; Le Berre, R.; Loaec, N.; Oumata, N.; Galons, H.; Nowak, E.; et al. Modulating Innate and Adaptive Immunity by (R)-Roscovitine: Potential Therapeutic Opportunity in Cystic Fibrosis. J. Innate Immun. 2016, 8, 330–349. [Google Scholar] [CrossRef] [Green Version]
- Acharya, K.R.; Ackerman, S.J. Eosinophil granule proteins: Form and function. J. Biol. Chem. 2014, 289, 17406–17415. [Google Scholar] [CrossRef] [Green Version]
- Farahi, N.; Uller, L.; Juss, J.K.; Langton, A.J.; Cowburn, A.S.; Gibson, A.; Foster, M.R.; Farrow, S.N.; Marco-Casanova, P.; Sobolewski, A.; et al. Effects of the cyclin-dependent kinase inhibitor R-roscovitine on eosinophil survival and clearance. Clin. Exp. Allergy 2011, 41, 673–687. [Google Scholar] [CrossRef]
- Pelaia, C.; Calabrese, C.; Vatrella, A.; Busceti, M.T.; Garofalo, E.; Lombardo, N.; Terracciano, R.; Pelaia, G. Benralizumab: From the Basic Mechanism of Action to the Potential Use in the Biological Therapy of Severe Eosinophilic Asthma. Biomed. Res. Int. 2018, 2018, 4839230. [Google Scholar] [CrossRef]
- Pham, T.H.; Damera, G.; Newbold, P.; Ranade, K. Reductions in eosinophil biomarkers by benralizumab in patients with asthma. Respir. Med. 2016, 111, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Gernez, Y.; Dunn, C.E.; Everson, C.; Mitsunaga, E.; Gudiputi, L.; Krasinska, K.; Davies, Z.A.; Herzenberg, L.A.; Tirouvanziam, R.; Moss, R.B. Blood basophils from cystic fibrosis patients with allergic bronchopulmonary aspergillosis are primed and hyper-responsive to stimulation by aspergillus allergens. J. Cyst. Fibros. 2012, 11, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Altman, L.C.; Hill, J.S.; Hairfield, W.M.; Mullarkey, M.F. Effects of corticosteroids on eosinophil chemotaxis and adherence. J. Clin. Investig. 1981, 67, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Kita, H.; Abu-Ghazaleh, R.; Sanderson, C.J.; Gleich, G.J. Effect of steroids on immunoglobulin-induced eosinophil degranulation. J. Allergy Clin. Immunol. 1991, 87, 70–77. [Google Scholar] [CrossRef]
- Cohen-Cymberknoh, M.; Blau, H.; Shoseyov, D.; Mei-Zahav, M.; Efrati, O.; Armoni, S.; Kerem, E. Intravenous monthly pulse methylprednisolone treatment for ABPA in patients with cystic fibrosis. J. Cyst. Fibros. 2009, 8, 253–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Ent, C.K.; Hoekstra, H.; Rijkers, G.T. Successful treatment of allergic bronchopulmonary aspergillosis with recombinant anti-IgE antibody. Thorax 2007, 62, 276–277. [Google Scholar] [CrossRef] [Green Version]
- Lebecque, P.; Leonard, A.; Argaz, M.; Godding, V.; Pilette, C. Omalizumab for exacerbations of allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. BMJ Case Rep. 2009, 2009. [Google Scholar] [CrossRef] [Green Version]
- Zirbes, J.M.; Milla, C.E. Steroid-sparing effect of omalizumab for allergic bronchopulmonary aspergillosis and cystic fibrosis. Pediatr. Pulmonol. 2008, 43, 607–610. [Google Scholar] [CrossRef]
- Kanu, A.; Patel, K. Treatment of allergic bronchopulmonary aspergillosis (ABPA) in CF with anti-IgE antibody (omalizumab). Pediatr. Pulmonol. 2008, 43, 1249–1251. [Google Scholar] [CrossRef]
- Nove-Josserand, R.; Grard, S.; Auzou, L.; Reix, P.; Murris-Espin, M.; Bremont, F.; Mammar, B.; Mely, L.; Hubert, D.; Durieu, I.; et al. Case series of omalizumab for allergic bronchopulmonary aspergillosis in cystic fibrosis patients. Pediatr. Pulmonol. 2017, 52, 190–197. [Google Scholar] [CrossRef]
- Ashkenazi, M.; Sity, S.; Sarouk, I.; Bar Aluma, B.E.; Dagan, A.; Bezalel, Y.; Bentur, L.; De Boeck, K.; Efrati, O. Omalizumab in allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. J. Asthma Allergy 2018, 11, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Keown, K.; Abbott, S.; Kuzeljevic, B.; Rayment, J.H.; Chilvers, M.A.; Yang, C.L. An investigation into biomarkers for the diagnosis of ABPA and aspergillus disease in cystic fibrosis. Pediatr. Pulmonol. 2019, 54, 1787–1793. [Google Scholar] [CrossRef]
- Siracusa, M.C.; Kim, B.S.; Spergel, J.M.; Artis, D. Basophils and allergic inflammation. J. Allergy Clin. Immunol. 2013, 132, 789–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skov, P.S.; Norn, S.; Schiotz, P.O.; Permin, H.; Hoiby, N. Pseudomonas aeruginosa allergy in cystic fibrosis. Involvement of histamine release in the pathogenesis of lung tissue damage. Allergy 1980, 35, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Van Asperen, P.P.; Manglick, P.; Allen, H. Mechanisms of bronchial hyperreactivity in cystic fibrosis. Pediatr. Pulmonol. 1988, 5, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, W.; Saak, A.; Steinkamp, G.; Van der Hardt, H.; Konig, W. Histamine release from basophils in cystic fibrosis. Clin. Exp. Immunol. 1989, 76, 434–439. [Google Scholar] [PubMed]
- Saak, A.; Schonfeld, W.; Konig, W.; von der Hardt, H. [LTB4-metabolism and histamine liberation by granulocytes in the disease pattern of cystic fibrosis (CF)]. Pneumologie 1991, 45, 913–923. [Google Scholar] [PubMed]
- Katelari, A.; Tzanoudaki, M.; Noni, M.; Kanariou, M.; Theodoridou, M.; Kanavakis, E.; Doudounakis, S.E.; Kanaka-Gantenbein, C. The role of basophil activation test in allergic bronchopulmonary aspergillosis and Aspergillus fumigatus sensitization in cystic fibrosis patients. J. Cyst. Fibros. 2016, 15, 587–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Farley, J.M. Effects of first and second generation antihistamines on muscarinic induced mucus gland cell ion transport. BMC Pharmacol. 2005, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Homnick, D.N.; Marks, J.H.; Rubin, B.K. The effect of a first-generation antihistamine on sputum viscoelasticity in cystic fibrosis. J. Aerosol. Med. 2007, 20, 45–49. [Google Scholar] [CrossRef]
- Bielen, K.; ‘S Jongers, B.; Boddaert, J.; Raju, T.K.; Lammens, C.; Malhotra-Kumar, S.; Jorens, P.G.; Goossens, H.; Kumar-Singh, S. Biofilm-Induced Type 2 Innate Immunity in a Cystic Fibrosis Model of Pseudomonas aeruginosa. Front. Cell Infect. Microbiol. 2017, 7, 274. [Google Scholar] [CrossRef] [Green Version]
- da Silva, E.Z.; Jamur, M.C.; Oliver, C. Mast cell function: A new vision of an old cell. J. Histochem. Cytochem. 2014, 62, 698–738. [Google Scholar] [CrossRef]
- Andersson, C.K.; Andersson-Sjoland, A.; Mori, M.; Hallgren, O.; Pardo, A.; Eriksson, L.; Bjermer, L.; Lofdahl, C.G.; Selman, M.; Westergren-Thorsson, G.; et al. Activated MCTC mast cells infiltrate diseased lung areas in cystic fibrosis and idiopathic pulmonary fibrosis. Respir. Res. 2011, 12, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Kishimoto, T. IL-6: Regulator of Treg/Th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Karmouty-Quintana, H.; Melicoff, E.; Le, T.T.; Weng, T.; Chen, N.Y.; Pedroza, M.; Zhou, Y.; Davies, J.; Philip, K.; et al. Blockade of IL-6 Trans signaling attenuates pulmonary fibrosis. J. Immunol. 2014, 193, 3755–3768. [Google Scholar] [CrossRef] [Green Version]
- Moretti, S.; Renga, G.; Oikonomou, V.; Galosi, C.; Pariano, M.; Iannitti, R.G.; Borghi, M.; Puccetti, M.; De Zuani, M.; Pucillo, C.E.; et al. A mast cell-ILC2-Th9 pathway promotes lung inflammation in cystic fibrosis. Nat. Commun. 2017, 8, 14017. [Google Scholar] [CrossRef]
- Henderson, W.R., Jr.; Chi, E.Y. Degranulation of cystic fibrosis nasal polyp mast cells. J. Pathol. 1992, 166, 395–404. [Google Scholar] [CrossRef]
- Junkins, R.D.; Shen, A.; Rosen, K.; McCormick, C.; Lin, T.J. Autophagy enhances bacterial clearance during P. aeruginosa lung infection. PLoS ONE 2013, 8, e72263. [Google Scholar] [CrossRef] [Green Version]
- Gomez, C.; Carsin, A.; Gouitaa, M.; Reynaud-Gaubert, M.; Dubus, J.C.; Mege, J.L.; Ranque, S.; Vitte, J. Mast cell tryptase changes with Aspergillus fumigatus—Host crosstalk in cystic fibrosis patients. J. Cyst. Fibros. 2018, 17, 631–635. [Google Scholar] [CrossRef] [Green Version]
- Piliponsky, A.M.; Acharya, M.; Shubin, N.J. Mast Cells in Viral, Bacterial, and Fungal Infection Immunity. Int. J. Mol. Sci. 2019, 20, 2851. [Google Scholar] [CrossRef] [Green Version]
- Leveque, M.; Le Trionnaire, S.; Del Porto, P.; Martin-Chouly, C. The impact of impaired macrophage functions in cystic fibrosis disease progression. J. Cyst. Fibros. 2017, 16, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Hartl, D.; Latzin, P.; Hordijk, P.; Marcos, V.; Rudolph, C.; Woischnik, M.; Krauss-Etschmann, S.; Koller, B.; Reinhardt, D.; Roscher, A.A.; et al. Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat. Med. 2007, 13, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Simonin-Le Jeune, K.; Le Jeune, A.; Jouneau, S.; Belleguic, C.; Roux, P.F.; Jaguin, M.; Dimanche-Boitre, M.T.; Lecureur, V.; Leclercq, C.; Desrues, B.; et al. Impaired functions of macrophage from cystic fibrosis patients: CD11b, TLR-5 decrease and sCD14, inflammatory cytokines increase. PLoS ONE 2013, 8, e75667. [Google Scholar] [CrossRef] [Green Version]
- Lara-Reyna, S.; Scambler, T.; Holbrook, J.; Wong, C.; Jarosz-Griffiths, H.H.; Martinon, F.; Savic, S.; Peckham, D.; McDermott, M.F. Metabolic Reprograming of Cystic Fibrosis Macrophages via the IRE1alpha Arm of the Unfolded Protein Response Results in Exacerbated Inflammation. Front. Immunol. 2019, 10, 1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regamey, N.; Tsartsali, L.; Hilliard, T.N.; Fuchs, O.; Tan, H.L.; Zhu, J.; Qiu, Y.S.; Alton, E.W.; Jeffery, P.K.; Bush, A.; et al. Distinct patterns of inflammation in the airway lumen and bronchial mucosa of children with cystic fibrosis. Thorax 2012, 67, 164–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasemann, H.; Schwiertz, R.; Matthiesen, S.; Racke, K.; Ratjen, F. Increased arginase activity in cystic fibrosis airways. Am. J. Respir. Crit. Care Med. 2005, 172, 1523–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munder, M.; Schneider, H.; Luckner, C.; Giese, T.; Langhans, C.D.; Fuentes, J.M.; Kropf, P.; Mueller, I.; Kolb, A.; Modolell, M.; et al. Suppression of T-cell functions by human granulocyte arginase. Blood 2006, 108, 1627–1634. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.E.; Prober, C.G.; Manson, B.; Corey, M.; Levison, H. Association of respiratory viral infections with pulmonary deterioration in patients with cystic fibrosis. N. Engl. J. Med. 1984, 311, 1653–1658. [Google Scholar] [CrossRef]
- Akazawa, Y.; Kubo, M.; Zhang, R.; Matsumoto, K.; Yan, F.; Setiawan, H.; Takahashi, H.; Fujikura, Y.; Ogino, K. Inhibition of arginase ameliorates experimental ulcerative colitis in mice. Free Radic. Res. 2013, 47, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Pera, T.; Zuidhof, A.B.; Smit, M.; Menzen, M.H.; Klein, T.; Flik, G.; Zaagsma, J.; Meurs, H.; Maarsingh, H. Arginase inhibition prevents inflammation and remodeling in a guinea pig model of chronic obstructive pulmonary disease. J. Pharmacol. Exp. Ther. 2014, 349, 229–238. [Google Scholar] [CrossRef]
- Pincikova, T.; Paquin-Proulx, D.; Moll, M.; Flodstrom-Tullberg, M.; Hjelte, L.; Sandberg, J.K. Severely Impaired Control of Bacterial Infections in a Patient with Cystic Fibrosis Defective in Mucosal-Associated Invariant T Cells. Chest 2018, 153, e93–e96. [Google Scholar] [CrossRef] [Green Version]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Anil, N.; Singh, M. CD4(+)CD25(high) FOXP3(+) regulatory T cells correlate with FEV1 in North Indian children with cystic fibrosis. Immunol. Investig. 2014, 43, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Hector, A.; Schafer, H.; Poschel, S.; Fischer, A.; Fritzsching, B.; Ralhan, A.; Carevic, M.; Oz, H.; Zundel, S.; Hogardt, M.; et al. Regulatory T-cell impairment in cystic fibrosis patients with chronic pseudomonas infection. Am. J. Respir. Crit. Care Med. 2015, 191, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Iannitti, R.G.; Carvalho, A.; Cunha, C.; De Luca, A.; Giovannini, G.; Casagrande, A.; Zelante, T.; Vacca, C.; Fallarino, F.; Puccetti, P.; et al. Th17/Treg imbalance in murine cystic fibrosis is linked to indoleamine 2,3-dioxygenase deficiency but corrected by kynurenines. Am. J. Respir. Crit. Care Med. 2013, 187, 609–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubin, P.J.; McAllister, F.; Kolls, J.K. Is cystic fibrosis a TH17 disease? Inflamm. Res. 2007, 56, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Dubin, P.J.; Kolls, J.K. IL-17 in cystic fibrosis: More than just Th17 cells. Am. J. Respir. Crit. Care Med. 2011, 184, 155–157. [Google Scholar] [CrossRef]
- Tan, H.L.; Regamey, N.; Brown, S.; Bush, A.; Lloyd, C.M.; Davies, J.C. The Th17 pathway in cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2011, 184, 252–258. [Google Scholar] [CrossRef]
- Sorensen, R.U.; Ruuskanen, O.; Miller, K.; Stern, R.C. B-lymphocyte function in cystic fibrosis. Eur. J. Respir. Dis. 1983, 64, 524–533. [Google Scholar]
- Hubeau, C.; Lorenzato, M.; Couetil, J.P.; Hubert, D.; Dusser, D.; Puchelle, E.; Gaillard, D. Quantitative analysis of inflammatory cells infiltrating the cystic fibrosis airway mucosa. Clin. Exp. Immunol. 2001, 124, 69–76. [Google Scholar] [CrossRef]
- Emilie, D.; Crevon, M.C.; Chicheportiche, R.; Auffredou, M.T.; Barot-Ciorbaru, R.; Lenoir, G.; Dayer, J.M.; Galanaud, P. Cystic fibrosis patients’ B-lymphocyte response is resistant to the in vitro enhancing effect of corticosteroids. Eur. J. Clin. Investig. 1990, 20, 620–626. [Google Scholar] [CrossRef]
- Ben-Ari, J.; Gozal, D.; Dorio, R.J.; Bowman, C.M.; Reiff, A.; Walker, S.M. Superantigens and cystic fibrosis: Resistance of presenting cells to dexamethasone. Clin. Diagn. Lab. Immunol. 2000, 7, 553–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frija-Masson, J.; Martin, C.; Regard, L.; Lothe, M.N.; Touqui, L.; Durand, A.; Lucas, B.; Damotte, D.; Alifano, M.; Fajac, I.; et al. Bacteria-driven peribronchial lymphoid neogenesis in bronchiectasis and cystic fibrosis. Eur. Respir. J. 2017, 49, 1601873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regard, L.; Martin, C.; Zemoura, L.; Geolier, V.; Sage, E.; Burgel, P.R. Peribronchial tertiary lymphoid structures persist after rituximab therapy in patients with cystic fibrosis. J. Clin. Pathol. 2018, 71, 752–753. [Google Scholar] [CrossRef]
- Gohy, S.T.; Ladjemi, M.Z.; Pilette, C. Lung lymphoid neogenesis in cystic fibrosis: A model of adaptive responses to bacteria? Eur. Respir. J. 2017, 49, 1700380. [Google Scholar] [CrossRef] [Green Version]
- Fleige, H.; Ravens, S.; Moschovakis, G.L.; Bolter, J.; Willenzon, S.; Sutter, G.; Haussler, S.; Kalinke, U.; Prinz, I.; Forster, R. IL-17-induced CXCL12 recruits B cells and induces follicle formation in BALT in the absence of differentiated FDCs. J. Exp. Med. 2014, 211, 643–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legler, D.F.; Loetscher, M.; Roos, R.S.; Clark-Lewis, I.; Baggiolini, M.; Moser, B. B cell-attracting chemokine 1, a human CXC chemokine expressed in lymphoid tissues, selectively attracts B lymphocytes via BLR1/CXCR5. J. Exp. Med. 1998, 187, 655–660. [Google Scholar] [CrossRef]
- Taylor, P.C.; Williams, R.O. Combination cytokine blockade: The way forward in therapy for rheumatoid arthritis? Arthritis Rheumatol. 2015, 67, 14–16. [Google Scholar] [CrossRef]
- Estabrooks, S.; Brodsky, J.L. Regulation of CFTR Biogenesis by the Proteostatic Network and Pharmacological Modulators. Int. J. Mol. Sci. 2020, 21, 452. [Google Scholar] [CrossRef] [Green Version]
- Bear, C.E. A Therapy for Most with Cystic Fibrosis. Cell 2020, 180, 211. [Google Scholar] [CrossRef]
- Habib, A.R.; Kajbafzadeh, M.; Desai, S.; Yang, C.L.; Skolnik, K.; Quon, B.S. A Systematic Review of the Clinical Efficacy and Safety of CFTR Modulators in Cystic Fibrosis. Sci. Rep. 2019, 9, 7234. [Google Scholar] [CrossRef]
- Sergeev, V.; Chou, F.Y.; Lam, G.Y.; Hamilton, C.M.; Wilcox, P.G.; Quon, B.S. The Extrapulmonary Effects of Cystic Fibrosis Transmembrane Conductance Regulator Modulators in Cystic Fibrosis. Ann. Am. Thorac. Soc. 2020, 17, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, C.; Tindall, J.M.; Hong, J.S.; Sorscher, E.J. Making precision medicine personal for cystic fibrosis. Science 2019, 365, 220–221. [Google Scholar] [CrossRef] [PubMed]
- Amico, G.; Brandas, C.; Moran, O.; Baroni, D. Unravelling the Regions of Mutant F508del-CFTR More Susceptible to the Action of Four Cystic Fibrosis Correctors. Int. J. Mol. Sci. 2019, 20, 5463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianotti, A.; Capurro, V.; Delpiano, L.; Mielczarek, M.; Garcia-Valverde, M.; Carreira-Barral, I.; Ludovico, A.; Fiore, M.; Baroni, D.; Moran, O.; et al. Small Molecule Anion Carriers Correct Abnormal Airway Surface Liquid Properties in Cystic Fibrosis Airway Epithelia. Int. J. Mol. Sci. 2020, 21, 1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraglia, K.A.; Chorghade, R.S.; Kim, B.R.; Tang, X.X.; Shah, V.S.; Grillo, A.S.; Daniels, P.N.; Cioffi, A.G.; Karp, P.H.; Zhu, L.; et al. Small-molecule ion channels increase host defences in cystic fibrosis airway epithelia. Nature 2019, 567, 405–408. [Google Scholar] [CrossRef]
- McElvaney, O.J.; Wade, P.; Murphy, M.; Reeves, E.P.; McElvaney, N.G. Targeting airway inflammation in cystic fibrosis. Expert Rev. Respir. Med. 2019, 13, 1041–1055. [Google Scholar] [CrossRef]
- Reihill, J.A.; Walker, B.; Hamilton, R.A.; Ferguson, T.E.; Elborn, J.S.; Stutts, M.J.; Harvey, B.J.; Saint-Criq, V.; Hendrick, S.M.; Martin, S.L. Inhibition of Protease-Epithelial Sodium Channel Signaling Improves Mucociliary Function in Cystic Fibrosis Airways. Am. J. Respir. Crit. Care Med. 2016, 194, 701–710. [Google Scholar] [CrossRef]
- Laval, J.; Touhami, J.; Herzenberg, L.A.; Conrad, C.; Taylor, N.; Battini, J.L.; Sitbon, M.; Tirouvanziam, R. Metabolic adaptation of neutrophils in cystic fibrosis airways involves distinct shifts in nutrient transporter expression. J. Immunol. 2013, 190, 6043–6050. [Google Scholar] [CrossRef] [Green Version]
- McElvaney, O.J.; Zaslona, Z.; Becker-Flegler, K.; Palsson-McDermott, E.M.; Boland, F.; Gunaratnam, C.; Gulbins, E.; O’Neill, L.A.; Reeves, E.P.; McElvaney, N.G. Specific Inhibition of the NLRP3 Inflammasome as an Antiinflammatory Strategy in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 1381–1391. [Google Scholar] [CrossRef]
- Margaroli, C.; Garratt, L.W.; Horati, H.; Dittrich, A.S.; Rosenow, T.; Montgomery, S.T.; Frey, D.L.; Brown, M.R.; Schultz, C.; Guglani, L.; et al. Elastase Exocytosis by Airway Neutrophils Is Associated with Early Lung Damage in Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 873–881. [Google Scholar] [CrossRef] [Green Version]
- Garratt, L.W.; Sutanto, E.N.; Ling, K.M.; Looi, K.; Iosifidis, T.; Martinovich, K.M.; Shaw, N.C.; Kicic-Starcevich, E.; Knight, D.A.; Ranganathan, S.; et al. Matrix metalloproteinase activation by free neutrophil elastase contributes to bronchiectasis progression in early cystic fibrosis. Eur. Respir. J. 2015, 46, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, E.P.; Banville, N.; Ryan, D.M.; O’Reilly, N.; Bergin, D.A.; Pohl, K.; Molloy, K.; McElvaney, O.J.; Alsaleh, K.; Aljorfi, A.; et al. Intracellular secretory leukoprotease inhibitor modulates inositol 1,4,5-triphosphate generation and exerts an anti-inflammatory effect on neutrophils of individuals with cystic fibrosis and chronic obstructive pulmonary disease. Biomed. Res. Int. 2013, 2013, 560141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, K.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Molloy, K.; Jundi, B.; Bergin, D.A.; McCarthy, C.; McElvaney, O.J.; et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood 2014, 124, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Forrest, O.A.; Ingersoll, S.A.; Preininger, M.K.; Laval, J.; Limoli, D.H.; Brown, M.R.; Lee, F.E.; Bedi, B.; Sadikot, R.T.; Goldberg, J.B.; et al. Frontline Science: Pathological conditioning of human neutrophils recruited to the airway milieu in cystic fibrosis. J. Leukoc. Biol. 2018, 104, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Greene, C.M.; McElvaney, N.G. Targeting neutrophil elastase in cystic fibrosis. Expert Opin. Ther. Targets 2008, 12, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Morla, S.; Sankaranarayanan, N.V.; Afosah, D.K.; Kumar, M.; Kummarapurugu, A.B.; Voynow, J.A.; Desai, U.R. On the Process of Discovering Leads That Target the Heparin-Binding Site of Neutrophil Elastase in the Sputum of Cystic Fibrosis Patients. J. Med. Chem. 2019, 62, 5501–5511. [Google Scholar] [CrossRef]
- Dittrich, A.S.; Kuhbandner, I.; Gehrig, S.; Rickert-Zacharias, V.; Twigg, M.; Wege, S.; Taggart, C.C.; Herth, F.; Schultz, C.; Mall, M.A. Elastase activity on sputum neutrophils correlates with severity of lung disease in cystic fibrosis. Eur. Respir. J. 2018, 51, 1701910. [Google Scholar] [CrossRef] [Green Version]
- Newsome, S.J.; Daniel, R.M.; Carr, S.B.; Bilton, D.; Keogh, R.H. Investigating the effects of long-term dornase alfa use on lung function using registry data. J. Cyst. Fibros. 2019, 18, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Podolska, M.J.; Mahajan, A.; Hahn, J.; Knopf, J.; Maueroder, C.; Petru, L.; Ullmann, M.; Schett, G.; Leppkes, M.; Herrmann, M.; et al. Treatment with DNases rescues hidden neutrophil elastase from aggregated NETs. J. Leukoc. Biol. 2019, 106, 1359–1366. [Google Scholar] [CrossRef] [Green Version]
- Staudinger, B.J.; Muller, J.F.; Halldorsson, S.; Boles, B.; Angermeyer, A.; Nguyen, D.; Rosen, H.; Baldursson, O.; Gottfreethsson, M.; Guethmundsson, G.H.; et al. Conditions associated with the cystic fibrosis defect promote chronic Pseudomonas aeruginosa infection. Am. J. Respir. Crit. Care Med. 2014, 189, 812–824. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Yanagihara, K.; Araki, N.; Harada, Y.; Morinaga, Y.; Izumikawa, K.; Kakeya, H.; Yamamoto, Y.; Hasegawa, H.; Kohno, S.; et al. In vivo efficacy of KRP-109, a novel elastase inhibitor, in a murine model of severe pneumococcal pneumonia. Pulm. Pharmacol. Ther. 2011, 24, 660–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chillappagari, S.; Muller, C.; Mahavadi, P.; Guenther, A.; Nahrlich, L.; Rosenblum, J.; Rubin, B.K.; Henke, M.O. A small molecule neutrophil elastase inhibitor, KRP-109, inhibits cystic fibrosis mucin degradation. J. Cyst. Fibros. 2016, 15, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Barth, P.; Bruijnzeel, P.; Wach, A.; Sellier Kessler, O.; Hooftman, L.; Zimmermann, J.; Naue, N.; Huber, B.; Heimbeck, I.; Kappeler, D.; et al. Single dose escalation studies with inhaled POL6014, a potent novel selective reversible inhibitor of human neutrophil elastase, in healthy volunteers and subjects with cystic fibrosis. J. Cyst. Fibros. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Q.; Woehl, J.L.; Kitamura, S.; Santos-Martins, D.; Smedley, C.J.; Li, G.; Forli, S.; Moses, J.E.; Wolan, D.W.; Sharpless, K.B. SuFEx-enabled, agnostic discovery of covalent inhibitors of human neutrophil elastase. Proc. Natl. Acad. Sci. USA 2019, 116, 18808–18814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, A.M.D.; Glasgow, A.M.A.; Humphreys, H.; Greene, C.M. Alpha-1 Antitrypsin—A Target for MicroRNA-Based Therapeutic Development for Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, 836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggar, A.; Li, Y.; Weathington, N.; Winkler, M.; Kong, M.; Jackson, P.; Blalock, J.E.; Clancy, J.P. Matrix metalloprotease-9 dysregulation in lower airway secretions of cystic fibrosis patients. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L96–L104. [Google Scholar] [CrossRef] [Green Version]
- Gaggar, A.; Jackson, P.L.; Noerager, B.D.; O’Reilly, P.J.; McQuaid, D.B.; Rowe, S.M.; Clancy, J.P.; Blalock, J.E. A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J. Immunol. 2008, 180, 5662–5669. [Google Scholar] [CrossRef] [Green Version]
- Gaggar, A.; Weathington, N. Bioactive extracellular matrix fragments in lung health and disease. J. Clin. Investig. 2016, 126, 3176–3184. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.; Pilkington, R.; Callaghan, M.; McClean, S. Activation of MMP-9 by human lung epithelial cells in response to the cystic fibrosis-associated pathogen Burkholderia cenocepacia reduced wound healing in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L575–L586. [Google Scholar] [CrossRef] [Green Version]
- Averna, M.; Bavestrello, M.; Cresta, F.; Pedrazzi, M.; De Tullio, R.; Minicucci, L.; Sparatore, B.; Salamino, F.; Pontremoli, S.; Melloni, E. Abnormal activation of calpain and protein kinase Calpha promotes a constitutive release of matrix metalloproteinase 9 in peripheral blood mononuclear cells from cystic fibrosis patients. Arch. Biochem. Biophys. 2016, 604, 103–112. [Google Scholar] [CrossRef]
- Xu, X.; Abdalla, T.; Bratcher, P.E.; Jackson, P.L.; Sabbatini, G.; Wells, J.M.; Lou, X.Y.; Quinn, R.; Blalock, J.E.; Clancy, J.P.; et al. Doxycycline improves clinical outcomes during cystic fibrosis exacerbations. Eur. Respir. J. 2017, 49, 1601102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentschel, J.; Jager, M.; Beiersdorf, N.; Fischer, N.; Doht, F.; Michl, R.K.; Lehmann, T.; Markert, U.R.; Boer, K.; Keller, P.M.; et al. Dynamics of soluble and cellular inflammatory markers in nasal lavage obtained from cystic fibrosis patients during intravenous antibiotic treatment. BMC Pulm. Med. 2014, 14, 82. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Maretti-Mira, A.C.; Wang, L.; DeLeve, L.D. Liver-Selective MMP-9 Inhibition in the Rat Eliminates Ischemia-Reperfusion Injury and Accelerates Liver Regeneration. Hepatology 2019, 69, 314–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McColley, S.A.; Stellmach, V.; Boas, S.R.; Jain, M.; Crawford, S.E. Serum vascular endothelial growth factor is elevated in cystic fibrosis and decreases with treatment of acute pulmonary exacerbation. Am. J. Respir. Crit. Care Med. 2000, 161, 1877–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettit, R.S.; Fellner, C. CFTR Modulators for the Treatment of Cystic Fibrosis. Pharm. Ther. 2014, 39, 500–511. [Google Scholar]
- Leveque, M.; Penna, A.; Le Trionnaire, S.; Belleguic, C.; Desrues, B.; Brinchault, G.; Jouneau, S.; Lagadic-Gossmann, D.; Martin-Chouly, C. Phagocytosis depends on TRPV2-mediated calcium influx and requires TRPV2 in lipids rafts: Alteration in macrophages from patients with cystic fibrosis. Sci. Rep. 2018, 8, 4310. [Google Scholar] [CrossRef]
- Assani, K.; Shrestha, C.L.; Robledo-Avila, F.; Rajaram, M.V.; Partida-Sanchez, S.; Schlesinger, L.S.; Kopp, B.T. Human Cystic Fibrosis Macrophages Have Defective Calcium-Dependent Protein Kinase C Activation of the NADPH Oxidase, an Effect Augmented by Burkholderia cenocepacia. J. Immunol. 2017, 198, 1985–1994. [Google Scholar] [CrossRef] [Green Version]
- Bernut, A.; Dupont, C.; Ogryzko, N.V.; Neyret, A.; Herrmann, J.L.; Floto, R.A.; Renshaw, S.A.; Kremer, L. CFTR Protects against Mycobacterium abscessus Infection by Fine-Tuning Host Oxidative Defenses. Cell Rep. 2019, 26, 1828–1840.e4. [Google Scholar] [CrossRef] [Green Version]
- Christopher Boyd, A.; Guo, S.; Huang, L.; Kerem, B.; Oren, Y.S.; Walker, A.J.; Hart, S.L. New approaches to genetic therapies for cystic fibrosis. J. Cyst. Fibros. 2020, 19 (Suppl. 1), S54–S59. [Google Scholar] [CrossRef] [Green Version]
- Pranke, I.; Golec, A.; Hinzpeter, A.; Edelman, A.; Sermet-Gaudelus, I. Emerging Therapeutic Approaches for Cystic Fibrosis. From Gene Editing to Personalized Medicine. Front. Pharmacol. 2019, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S.; Guo, S. Nucleic Acid Therapies for Cystic Fibrosis. Nucleic Acid Ther. 2018, 28, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vencken, S.F.; Greene, C.M. Toll-Like Receptors in Cystic Fibrosis: Impact of Dysfunctional microRNA on Innate Immune Responses in the Cystic Fibrosis Lung. J. Innate Immun. 2016, 8, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Alton, E.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R.; et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015, 3, 684–691. [Google Scholar] [CrossRef] [Green Version]
- Magrone, T.; Jirillo, E. The Tolerant Immune System: Biological Significance and Clinical Implications of T Cell Tolerance. Endocr. Metab. Immune Disord. Drug Targets 2019, 19, 580–593. [Google Scholar] [CrossRef]
- Miller, C.M.; Harris, E.N. Antisense Oligonucleotides: Treatment Strategies and Cellular Internalization. RNA Dis. 2016, 3, e1393. [Google Scholar]
- Crooke, S.T. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic Acid Ther. 2017, 27, 70–77. [Google Scholar] [CrossRef]
- Havens, M.A.; Duelli, D.M.; Hastings, M.L. Targeting RNA splicing for disease therapy. Wiley Interdiscip. Rev. RNA 2013, 4, 247–266. [Google Scholar] [CrossRef]
- Rader, D.J.; Kastelein, J.J. Lomitapide and mipomersen: Two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation 2014, 129, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Pires, V.B.; Simoes, R.; Mamchaoui, K.; Carvalho, C.; Carmo-Fonseca, M. Short (16-mer) locked nucleic acid splice-switching oligonucleotides restore dystrophin production in Duchenne Muscular Dystrophy myotubes. PLoS ONE 2017, 12, e0181065. [Google Scholar] [CrossRef] [Green Version]
- Drevinek, P.; Pressler, T.; Cipolli, M.; De Boeck, K.; Schwarz, C.; Bouisset, F.; Boff, M.; Henig, N.; Paquette-Lamontagne, N.; Montgomery, S.; et al. Antisense oligonucleotide eluforsen is safe and improves respiratory symptoms in F508DEL cystic fibrosis. J. Cyst. Fibros. 2020, 19, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Freund, I.; Eigenbrod, T.; Helm, M.; Dalpke, A.H. RNA Modifications Modulate Activation of Innate Toll-Like Receptors. Genes 2019, 10, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mankan, A.K.; Schmidt, T.; Chauhan, D.; Goldeck, M.; Honing, K.; Gaidt, M.; Kubarenko, A.V.; Andreeva, L.; Hopfner, K.P.; Hornung, V. Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J. 2014, 33, 2937–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigby, R.E.; Webb, L.M.; Mackenzie, K.J.; Li, Y.; Leitch, A.; Reijns, M.A.; Lundie, R.J.; Revuelta, A.; Davidson, D.J.; Diebold, S.; et al. RNA:DNA hybrids are a novel molecular pattern sensed by TLR9. EMBO J. 2014, 33, 542–558. [Google Scholar] [CrossRef] [PubMed]
- Urban, E.; Noe, C.R. Structural modifications of antisense oligonucleotides. Farmaco 2003, 58, 243–258. [Google Scholar] [CrossRef]
- Haque, A.; Dewerth, A.; Antony, J.S.; Riethmuller, J.; Schweizer, G.R.; Weinmann, P.; Latifi, N.; Yasar, H.; Pedemonte, N.; Sondo, E.; et al. Chemically modified hCFTR mRNAs recuperate lung function in a mouse model of cystic fibrosis. Sci. Rep. 2018, 8, 16776. [Google Scholar] [CrossRef] [Green Version]
- Bangel-Ruland, N.; Tomczak, K.; Fernandez Fernandez, E.; Leier, G.; Leciejewski, B.; Rudolph, C.; Rosenecker, J.; Weber, W.M. Cystic fibrosis transmembrane conductance regulator-mRNA delivery: A novel alternative for cystic fibrosis gene therapy. J. Gene Med. 2013, 15, 414–426. [Google Scholar] [CrossRef]
- Robinson, E.; MacDonald, K.D.; Slaughter, K.; McKinney, M.; Patel, S.; Sun, C.; Sahay, G. Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis. Mol. Ther. 2018, 26, 2034–2046. [Google Scholar] [CrossRef] [Green Version]
- Kariko, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Durbin, A.F.; Wang, C.; Marcotrigiano, J.; Gehrke, L. RNAs Containing Modified Nucleotides Fail To Trigger RIG-I Conformational Changes for Innate Immune Signaling. mBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Zhang, Z.; Xue, M.; Zhao, B.S.; Harder, O.; Li, A.; Liang, X.; Gao, T.Z.; Xu, Y.; Zhou, J.; et al. N(6)-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat. Microbiol. 2020, 5, 584–598. [Google Scholar] [CrossRef]
- Anderson, B.R.; Muramatsu, H.; Jha, B.K.; Silverman, R.H.; Weissman, D.; Kariko, K. Nucleoside modifications in RNA limit activation of 2′-5′-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res. 2011, 39, 9329–9338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuyx, S.; De Boeck, K. Treating the Underlying Cystic Fibrosis Transmembrane Conductance Regulator Defect in Patients with Cystic Fibrosis. Semin. Respir. Crit. Care Med. 2019, 40, 762–774. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giacalone, V.D.; Dobosh, B.S.; Gaggar, A.; Tirouvanziam, R.; Margaroli, C. Immunomodulation in Cystic Fibrosis: Why and How? Int. J. Mol. Sci. 2020, 21, 3331. https://doi.org/10.3390/ijms21093331
Giacalone VD, Dobosh BS, Gaggar A, Tirouvanziam R, Margaroli C. Immunomodulation in Cystic Fibrosis: Why and How? International Journal of Molecular Sciences. 2020; 21(9):3331. https://doi.org/10.3390/ijms21093331
Chicago/Turabian StyleGiacalone, Vincent D., Brian S. Dobosh, Amit Gaggar, Rabindra Tirouvanziam, and Camilla Margaroli. 2020. "Immunomodulation in Cystic Fibrosis: Why and How?" International Journal of Molecular Sciences 21, no. 9: 3331. https://doi.org/10.3390/ijms21093331
APA StyleGiacalone, V. D., Dobosh, B. S., Gaggar, A., Tirouvanziam, R., & Margaroli, C. (2020). Immunomodulation in Cystic Fibrosis: Why and How? International Journal of Molecular Sciences, 21(9), 3331. https://doi.org/10.3390/ijms21093331