The p53–53BP1-Related Survival of A549 and H1299 Human Lung Cancer Cells after Multifractionated Radiotherapy Demonstrated Different Response to Additional Acute X-ray Exposure


 , , and
, , and 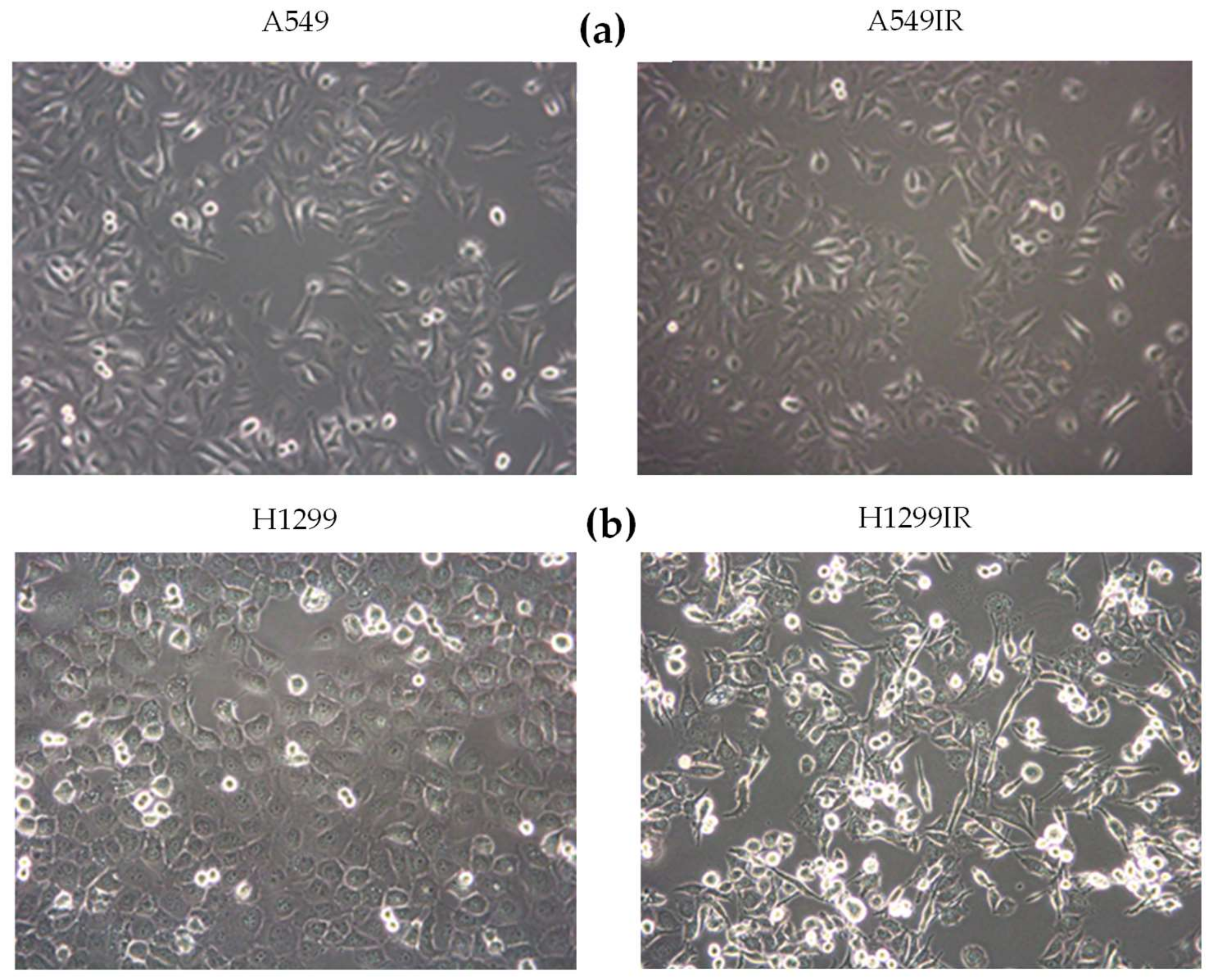{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
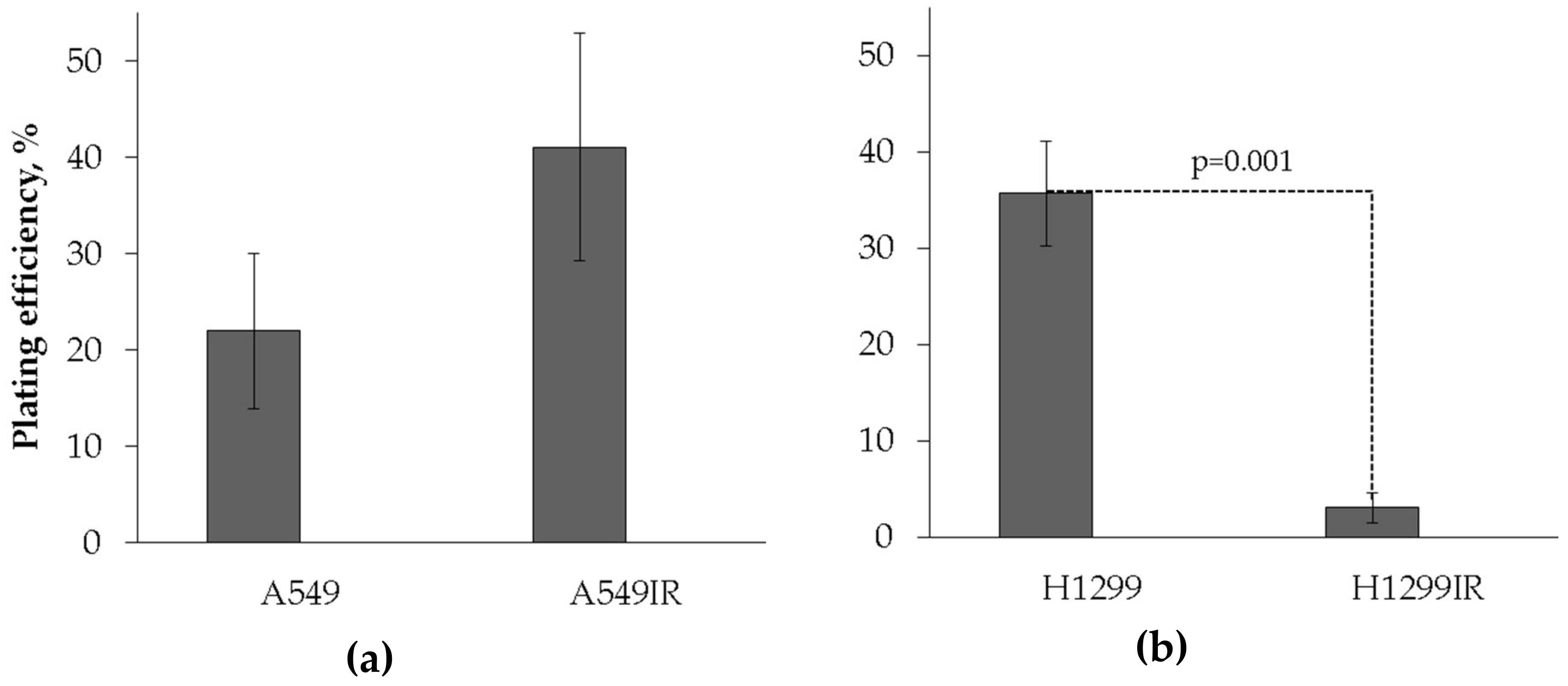
2.1. Establishment and Phenotypic Characterization of A549IR and H1299IR Cells
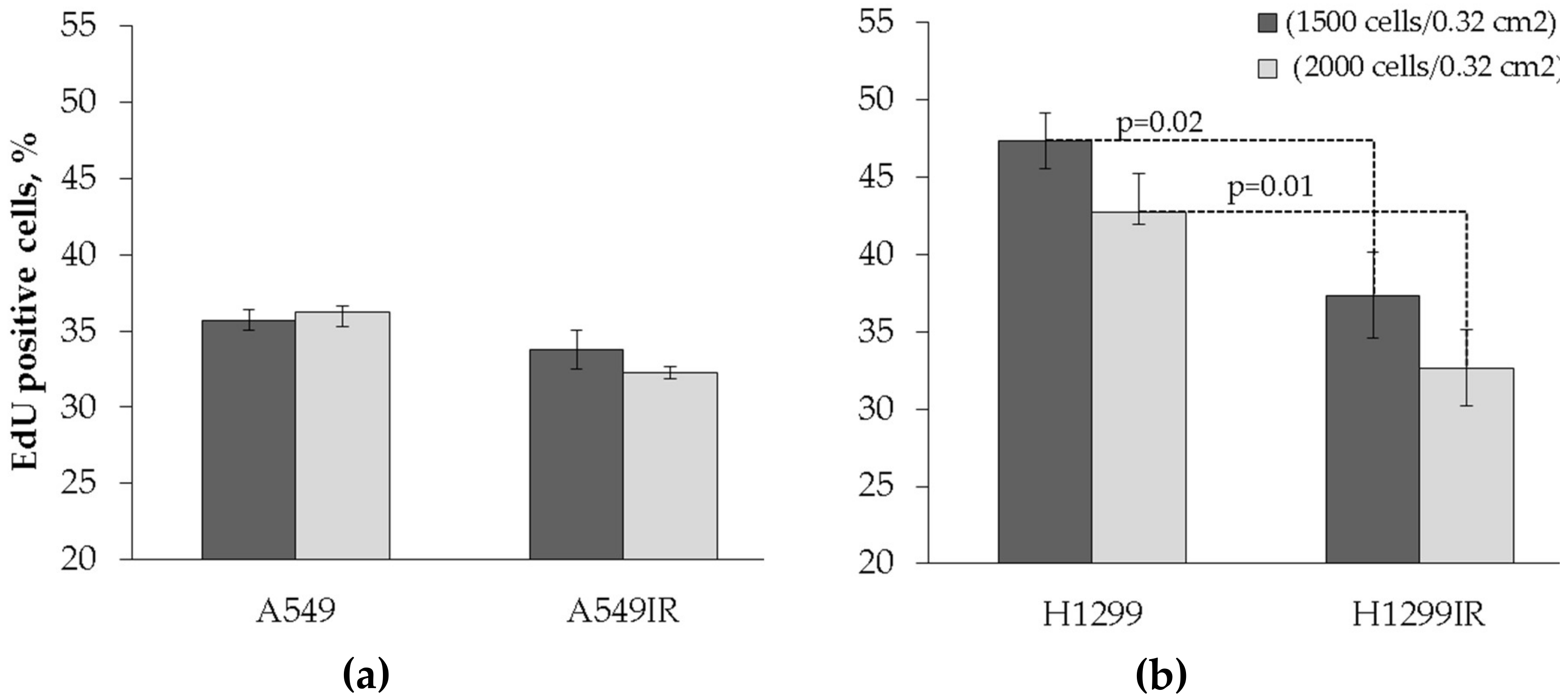
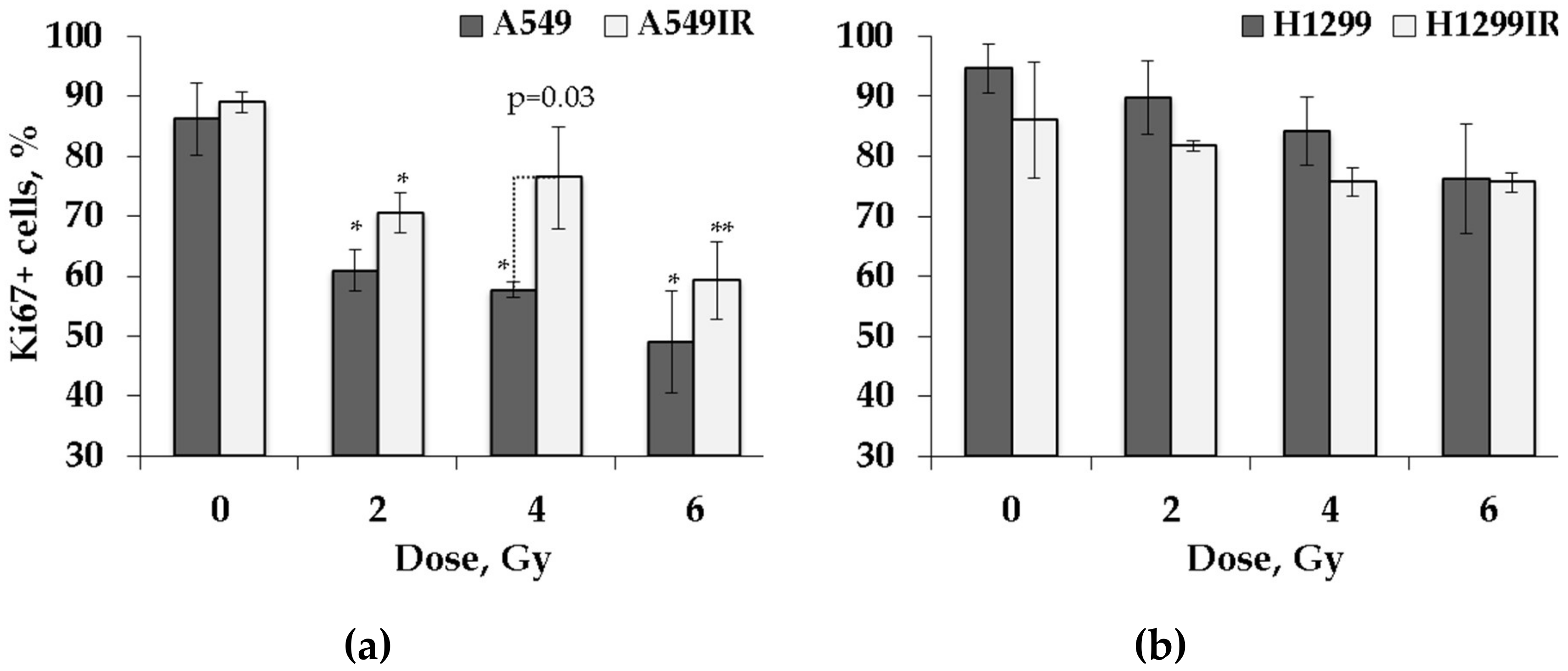
2.2. Proliferative Activity of A549IR and H1299IR Cells Decreased Compared to Parental Cells in a p53-Dependent Manner
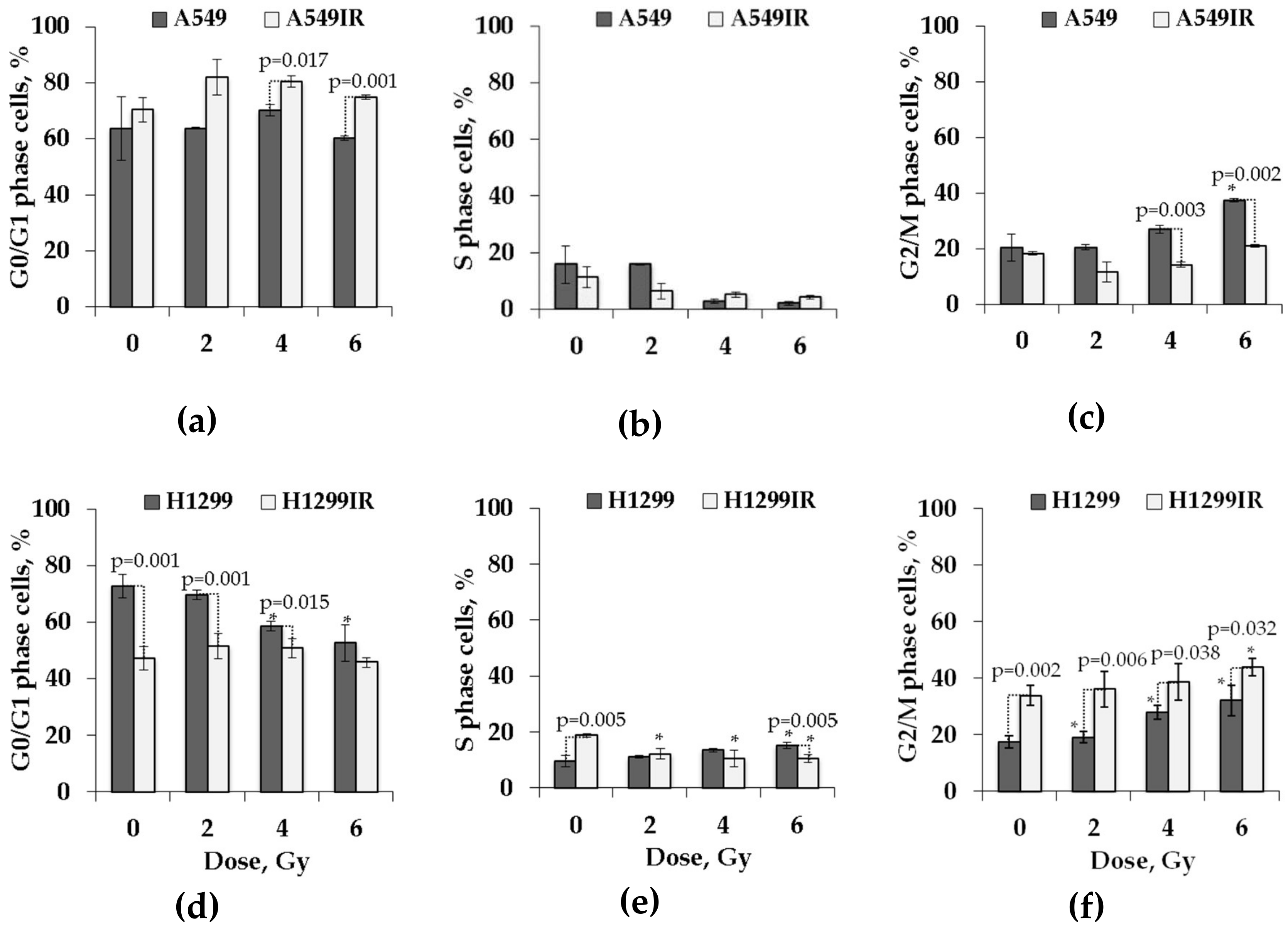
2.3. Functional p53 Contributed to Increased G1 Arrest and Sustained G2 Arrest in Response to Single Doses of Acute X-ray Irradiation
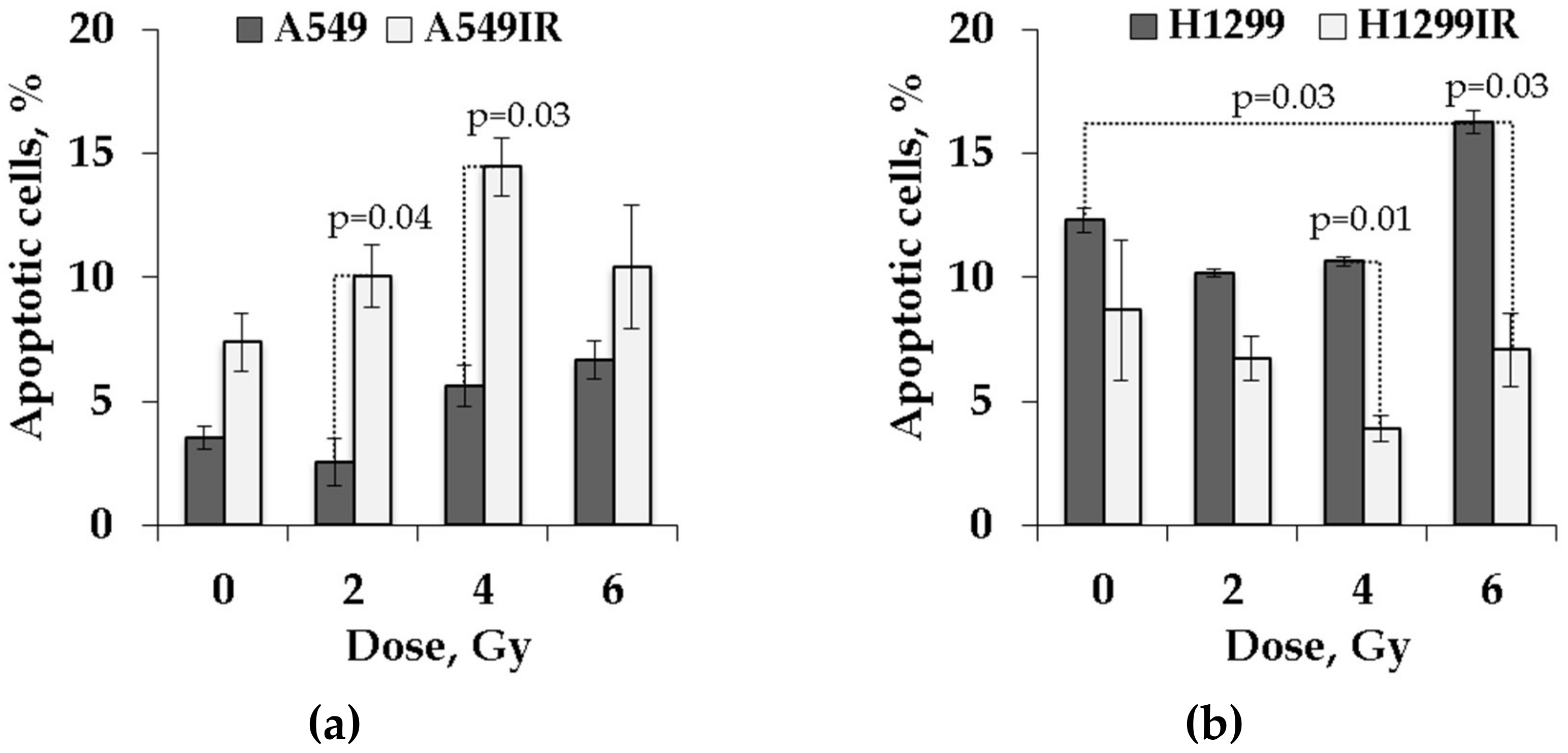
2.4. Functional p53 Subsidized the Level of Apoptosis in Response to Single Doses of Acute X-ray Irradiation
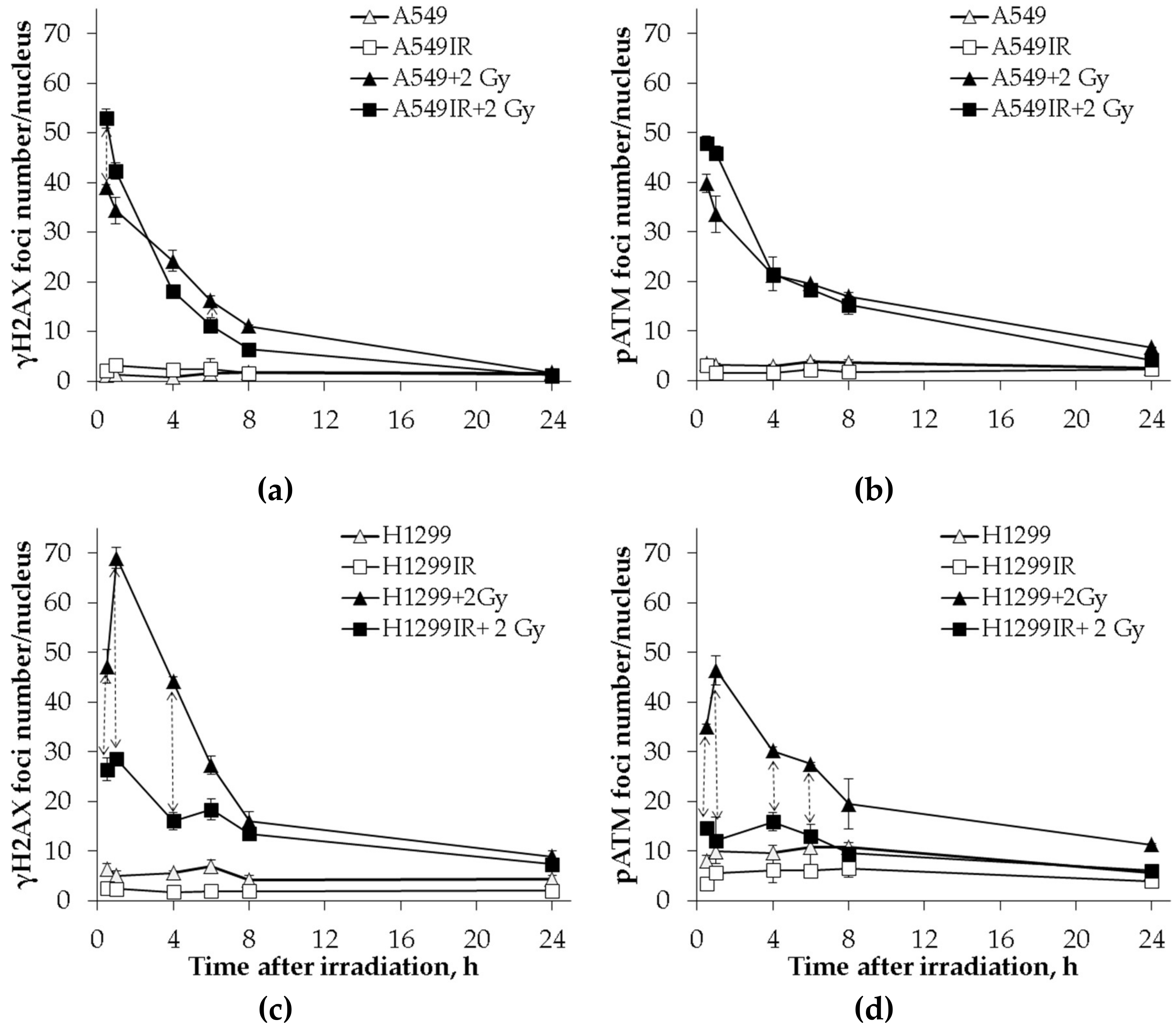
2.5. The p53 Status Influenced the DNA Repair Capacity of Parental Cells and Their IR-Surviving Sublines in Response to Acute Single IR Exposure
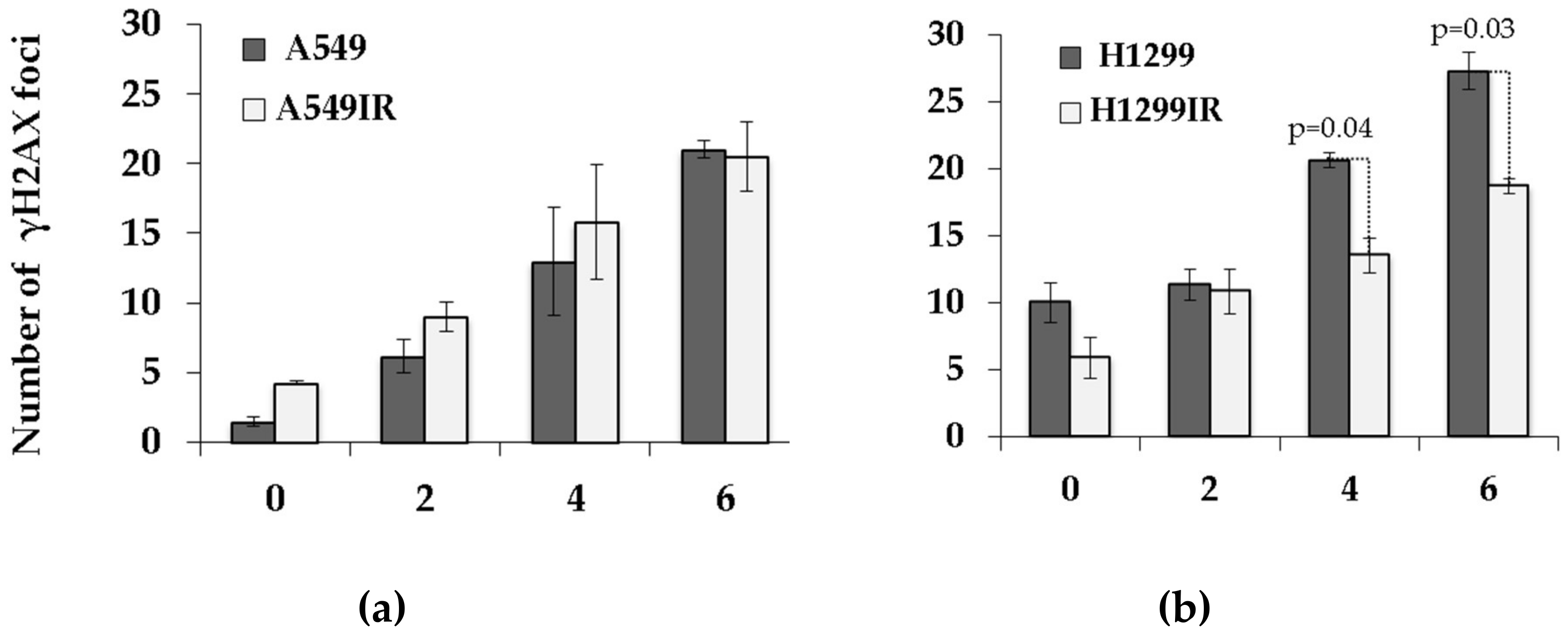
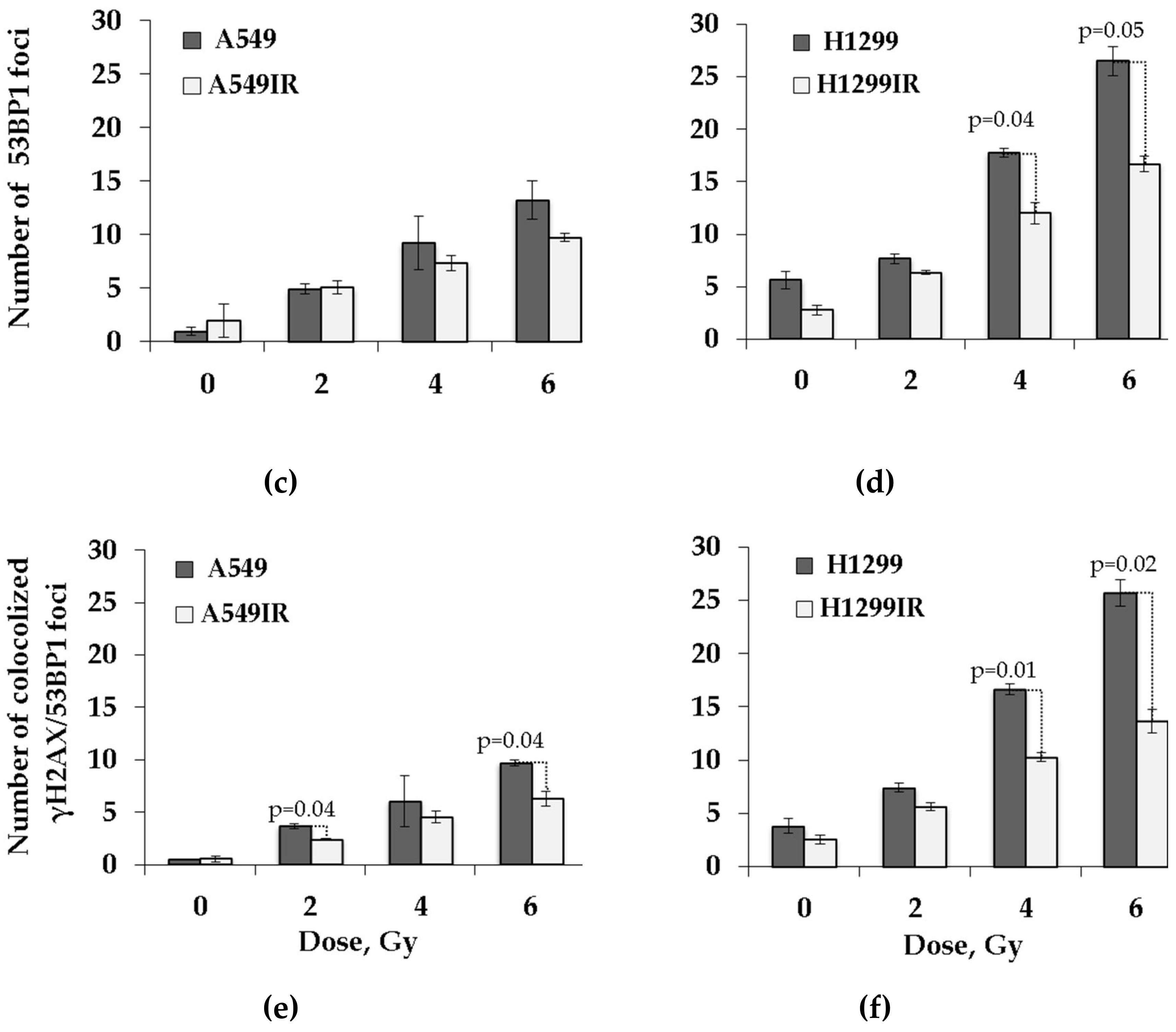
2.6. The Impact of p53 on Residual DNA Repair Foci in Parental Cells and Their IR-Surviving Sublines in Response to Acute Single Doses of IR Exposure
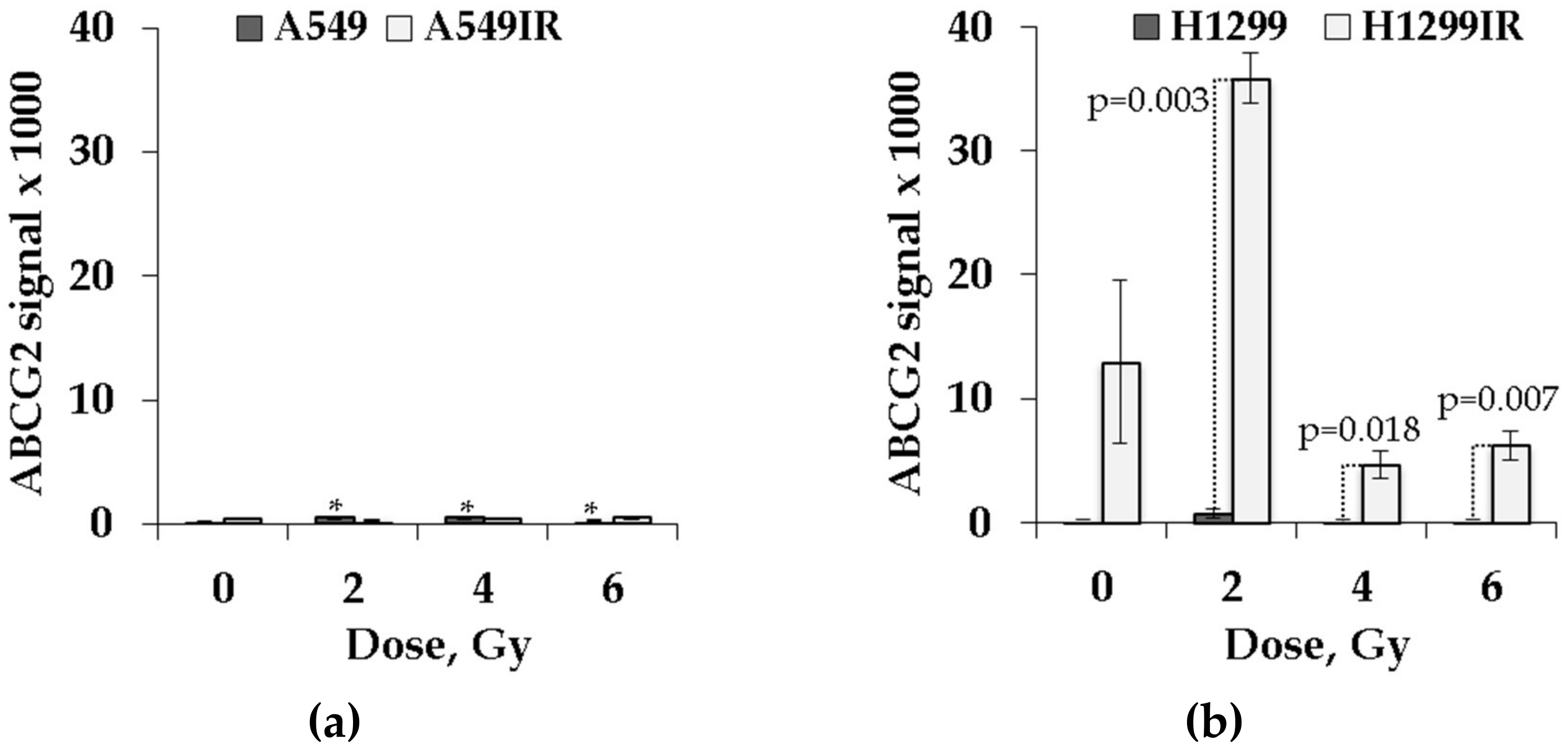
2.7. Expression of ABCG2 in IR-Surviving Sublines and Their Parental Cells Depended on p53 Status in Response to Acute Single Doses of IR Exposure
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Irradiation
4.3. Assessment of Plating Efficiency
4.4. EdU Cell Proliferation Assay
4.5. Immunofluorescence Staining
4.6. Cell Cycle Analysis
4.7. Apoptosis Assay
4.8. Analysis of ABCG2 Expression
4.9. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Moore, S.; Leung, B.; Wu, J.; Ho, C. Population-based analysis of curative therapies in stage II non-small cell lung cancer: The role of radiotherapy in medically inoperable patients. Radiat. Oncol. 2020, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeller, B.; Balagamwala, E.H.; Chen, A.; Creach, K.M.; Giaccone, G.; Koshy, M.; Zaky, S.; Rodrigues, G. Palliative thoracic radiation therapy for non-small cell lung cancer: 2018 Update of an American Society for Radiation Oncology (ASTRO) Evidence-Based Guideline. Pract. Radiat. Oncol. 2018, 8, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.; Dahele, M.; Lagerwaard, F.J.; Senan, S. A critical review of recent developments in radiotherapy for non-small cell lung cancer. Radiat. Oncol. 2016, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, B.L.; Goyal, N.G.; Weiss, D.M.; Westbrook, T.D.; Maddocks, K.J.; Byrd, J.C.; Johnson, A.J. Cells, cytokines, chemokines, and cancer stress: A biobehavioral study of patients with chronic lymphocytic leukemia. Cancer 2018, 124, 3240–3248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Graham, P.; Hao, J.; Ni, J.; Deng, J.; Bucci, J.; Malouf, D.; Gillatt, D.; Li, Y. Cancer stem cells and signaling pathways in radioresistance. Oncotarget 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2017, 15, 81–94. [Google Scholar] [CrossRef]
- Lim, Z.-F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Lovly, C.M.; Salama, A.K.S.; Salgia, R. Tumor Heterogeneity and Therapeutic Resistance. Am. Soc. Clin. Oncol. Educ. Book 2016, 36, e585–e593. [Google Scholar] [CrossRef]
- Rich, J.N. Cancer stem cells. Medicine 2016, 95, S2–S7. [Google Scholar] [CrossRef]
- Puhr, M.; Hoefer, J.; Schäfer, G.; Erb, H.H.H.; Oh, S.J.; Klocker, H.; Heidegger, I.; Neuwirt, H.; Culig, Z. Epithelial-to-Mesenchymal Transition Leads to Docetaxel Resistance in Prostate Cancer and Is Mediated by Reduced Expression of miR-200c and miR-205. Am. J. Pathol. 2012, 181, 2188–2201. [Google Scholar] [CrossRef] [Green Version]
- Rycaj, K.; Tang, D.G. Cancer stem cells and radioresistance. Int. J. Radiat. Biol. 2014, 90, 615–621. [Google Scholar] [CrossRef] [Green Version]
- Zaider, M.; Hanin, L. Tumor control probability in radiation treatment. Med Phys. 2011, 38, 574–583. [Google Scholar] [CrossRef]
- McDermott, N.; Meunier, A.; Mooney, B.; Nortey, G.; Hernandez, C.; Hurley, S.; Lynam-Lennon, N.; Barsoom, S.H.; Bowman, K.J.; Marples, B.; et al. Fractionated radiation exposure amplifies the radioresistant nature of prostate cancer cells. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.; Turnbull, A.K.; Ward, C.; Meehan, J.; Martínez-Pérez, C.; Bonello, M.; Pang, L.Y.; Langdon, S.P.; Kunkler, I.H.; Murray, A.; et al. Development and characterisation of acquired radioresistant breast cancer cell lines. Radiat. Oncol. 2019, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, F.; Popat, S. Combining targeted agents and hypo- and hyper-fractionated radiotherapy in NSCLC. J. Thorac. Dis. 2014, 6, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Falkson, C.B.; Vella, E.T.; Yu, E.; El-Mallah, M.; Mackenzie, R.; Ellis, P.M.; Ung, Y.C. Radiotherapy With Curative Intent in Patients With Early-stage, Medically Inoperable, Non-Small-cell Lung Cancer: A Systematic Review. Clin. Lung Cancer 2017, 18, 105–121.e105. [Google Scholar] [CrossRef]
- Milano, M.T.; Kong, F.-M.S.; Movsas, B. Stereotactic body radiotherapy as salvage treatment for recurrence of non-small cell lung cancer after prior surgery or radiotherapy. Transl. Lung Cancer Res. 2018, 8, 78–87. [Google Scholar] [CrossRef]
- Prezzano, K.M.; Ma, S.J.; Hermann, G.M.; Rivers, C.I.; Gomez-Suescun, J.A.; Singh, A.K. Stereotactic body radiation therapy for non-small cell lung cancer: A review. World J. Clin. Oncol. 2019, 10, 14–27. [Google Scholar] [CrossRef]
- Kumar, S.S.; McGarry, R.C. Management of local recurrences and regional failure in early stage non-small cell lung cancer after stereotactic body radiation therapy. Transl. Lung Cancer Res. 2019, 8, S213–S221. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.; Murray, D. Ionizing Radiation-Induced Responses in Human Cells with Differing TP53 Status. Int. J. Mol. Sci. 2013, 14, 22409–22435. [Google Scholar] [CrossRef] [Green Version]
- Buch, K.; Peters, T.; Nawroth, T.; Sänger, M.; Schmidberger, H.; Langguth, P. Determination of cell survival after irradiation via clonogenic assay versus multiple MTT Assay - A comparative study. Radiat. Oncol. 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM Phosphorylates Histone H2AX in Response to DNA Double-strand Breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, R.; Krautloher, A.; Ramírez-Fernández, A.; Nicke, A. P2X7 Interactions and Signaling – Making Head or Tail of It. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, S.; Romin, Y.; Barlas, A.; Petrovic, L.M.; Turkekul, M.; Fan, N.; Xu, K.; Garcia, A.R.; Monette, S.; Klimstra, D.S.; et al. Evaluation of YO-PRO-1 as an early marker of apoptosis following radiofrequency ablation of colon cancer liver metastases. Cytotechnology 2013, 66, 259–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ota, S.; Ishii, G.; Goto, K.; Kubota, K.; Kim, Y.H.; Kojika, M.; Murata, Y.; Yamazaki, M.; Nishiwaki, Y.; Eguchi, K.; et al. Immunohistochemical expression of BCRP and ERCC1 in biopsy specimen predicts survival in advanced non-small-cell lung cancer treated with cisplatin-based chemotherapy. Lung Cancer 2009, 64, 98–104. [Google Scholar] [CrossRef]
- Burger, H.; Foekens, J.A.; Look, M.P.; Meijer-van Gelder, M.E.; Klijn, J.G.; Wiemer, E.A.; Stoter, G.; Nooter, K. RNA expression of breast cancer resistance protein, lung resistance-related protein, multidrug resistance-associated proteins 1 and 2, and multidrug resistance gene 1 in breast cancer: Correlation with chemotherapeutic response. Clin Cancer Res 2003, 9, 827–836. [Google Scholar]
- Zhou, S.; Schuetz, J.D.; Bunting, K.D.; Colapietro, A.-M.; Sampath, J.; Morris, J.J.; Lagutina, I.; Grosveld, G.C.; Osawa, M.; Nakauchi, H.; et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 2001, 7, 1028–1034. [Google Scholar] [CrossRef]
- Sato, K.; Shimokawa, T.; Imai, T. Difference in Acquired Radioresistance Induction Between Repeated Photon and Particle Irradiation. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Alshaimaa, A.; Maria, M.V.; Daniel, D.; Herbert, R.; Jozsef, D.; Ira-Ida, S. Epithelial-mesenchymal crosstalk induces radioresistance in HNSCC cells. Oncotarget 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Casal, R.; Bhattacharya, C.; Ganesh, N.; Bailey, L.; Basse, P.; Gibson, M.; Epperly, M.; Levina, V. Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial-mesenchymal transition phenotypes. Mol. Cancer 2013, 12. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Pei, F.; Yang, F.; Li, L.; Amin, A.; Liu, S.; Buchan, J.; Cho, W. Role of Autophagy and Apoptosis in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, A.; Minaguchi, T.; Fujieda, K.; Hosokawa, Y.; Nishida, K.; Shikama, A.; Tasaka, N.; Sakurai, M.; Ochi, H.; Satoh, T. Abnormal accumulation of p53 predicts radioresistance leading to poor survival in patients with endometrial carcinoma. Oncol. Lett. 2019. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.L.; Earle, J.D.; Gumerlock, P.H. Functional p53 increases prostate cancer cell survival after exposure to fractionated doses of ionizing radiation. Cancer Res 2003, 63, 7190–7196. [Google Scholar] [PubMed]
- Kuwahara, Y.; Li, L.; Baba, T.; Nakagawa, H.; Shimura, T.; Yamamoto, Y.; Ohkubo, Y.; Fukumoto, M. Clinically relevant radioresistant cells efficiently repair DNA double-strand breaks induced by X-rays. Cancer Sci. 2009, 100, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Babayan, N.; Grigoryan, B.; Khondkaryan, L.; Tadevosyan, G.; Sarkisyan, N.; Grigoryan, R.; Apresyan, L.; Aroutiounian, R.; Vorobyeva, N.; Pustovalova, M.; et al. Laser-Driven Ultrashort Pulsed Electron Beam Radiation at Doses of 0.5 and 1.0 Gy Induces Apoptosis in Human Fibroblasts. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorokin, M.; Kholodenko, R.; Grekhova, A.; Suntsova, M.; Pustovalova, M.; Vorobyeva, N.; Kholodenko, I.; Malakhova, G.; Garazha, A.; Nedoluzhko, A.; et al. Acquired resistance to tyrosine kinase inhibitors may be linked with the decreased sensitivity to X-ray irradiation. Oncotarget 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Pustovalova, M.; Astrelina, T.A.; Grekhova, A.; Vorobyeva, N.; Tsvetkova, A.; Blokhina, T.; Nikitina, V.; Suchkova, Y.; Usupzhanova, D.; Brunchukov, V.; et al. Residual γH2AX foci induced by low dose x-ray radiation in bone marrow mesenchymal stem cells do not cause accelerated senescence in the progeny of irradiated cells. Aging 2017, 9, 2397–2410. [Google Scholar] [CrossRef] [Green Version]
- Ulyanenko, S.; Pustovalova, M.; Koryakin, S.; Beketov, E.; Lychagin, A.; Ulyanenko, L.; Kaprin, A.; Grekhova, A.; Ozerova, A.M.; Ozerov, I.V.; et al. Formation of γH2AX and pATM Foci in Human Mesenchymal Stem Cells Exposed to Low Dose-Rate Gamma-Radiation. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Osipov, A.N.; Pustovalova, M.; Grekhova, A.; Eremin, P.; Vorobyova, N.; Pulin, A.; Zhavoronkov, A.; Roumiantsev, S.; Klokov, D.Y.; Eremin, I. Low doses of X-rays induce prolonged and ATM-independent persistence of γH2AX foci in human gingival mesenchymal stem cells. Oncotarget 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. -H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef]
- Xu, B.; Kim, S.-T.; Lim, D.-S.; Kastan, M.B. Two Molecularly Distinct G2/M Checkpoints Are Induced by Ionizing Irradiation. Mol. Cell. Biol. 2002, 22, 1049–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Löbrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. Embo J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Yang, C.-X.; Mei, Z.-J.; Chen, J.; Zhang, S.-M.; Sun, S.-X.; Zhou, F.-X.; Zhou, Y.-F.; Xie, C.-H. Involvement of Cdc25c in Cell Cycle Alteration of a Radioresistant Lung Cancer Cell Line Established with Fractionated Ionizing Radiation. Asian Pac. J. Cancer Prev. 2013, 14, 5725–5730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löbrich, M.; Jeggo, P. A Process of Resection-Dependent Nonhomologous End Joining Involving the Goddess Artemis. Trends Biochem. Sci. 2017, 42, 690–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djuzenova, C.S.; Elsner, I.; Katzer, A.; Worschech, E.; Distel, L.V.; Flentje, M.; Polat, B. Radiosensitivity in breast cancer assessed by the histone γ-H2AX and 53BP1 foci. Radiat. Oncol. 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyaev, I.Y. Radiation-induced DNA repair foci: Spatio-temporal aspects of formation, application for assessment of radiosensitivity and biological dosimetry. Mutat. Res. /Rev. Mutat. Res. 2010, 704, 132–141. [Google Scholar] [CrossRef]
- Cuella-Martin, R.; Oliveira, C.; Lockstone, H.E.; Snellenberg, S.; Grolmusova, N.; Chapman, J.R. 53BP1 Integrates DNA Repair and p53-Dependent Cell Fate Decisions via Distinct Mechanisms. Mol. Cell 2016, 64, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Zaugg, K.; Mak, T.W.; Elledge, S.J. A Role for the Deubiquitinating Enzyme USP28 in Control of the DNA-Damage Response. Cell 2006, 126, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Kilic, S.; Lezaja, A.; Gatti, M.; Bianco, E.; Michelena, J.; Imhof, R.; Altmeyer, M. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. Embo J. 2019, 38. [Google Scholar] [CrossRef]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Guan, H.; Liu, X.D.; Xie, D.F.; Wang, Y.; Ma, T.; Huang, B.; Zhou, P.K. p53 positively regulates the expression of cancer stem cell marker CD133 in HCT116 colon cancer cells. Oncol. Lett. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, H.; Spike, B.T.; Wahl, G.M.; Levine, A.J. Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proc. Natl. Acad. Sci. 2010, 107, 22745–22750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shien, K.; Toyooka, S.; Ichimura, K.; Soh, J.; Furukawa, M.; Maki, Y.; Muraoka, T.; Tanaka, N.; Ueno, T.; Asano, H.; et al. Prognostic impact of cancer stem cell-related markers in non-small cell lung cancer patients treated with induction chemoradiotherapy. Lung Cancer 2012, 77, 162–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, W.; Zhang, J.T. Human ABCG2: Structure, function, and its role in multidrug resistance. Int. J. Biochem. Mol. Biol. 2012, 3, 1–27. [Google Scholar] [PubMed]
- Bunting, K.D. ABC Transporters as Phenotypic Markers and Functional Regulators of Stem Cells. Stem Cells 2002, 20, 11–20. [Google Scholar] [CrossRef]
- Wang, L.; Huang, X.; Zheng, X.; Wang, X.; Li, S.; Zhang, L.; Yang, Z.; Xia, Z. Enrichment of Prostate Cancer Stem-Like Cells from Human Prostate Cancer Cell Lines by Culture in Serum-Free Medium and Chemoradiotherapy. Int. J. Biol. Sci. 2013, 9, 472–479. [Google Scholar] [CrossRef]
- Ding, R.; Jin, S.; Pabon, K.; Scotto, K.W. A role for ABCG2 beyond drug transport: Regulation of autophagy. Autophagy 2016, 12, 737–751. [Google Scholar] [CrossRef] [Green Version]
- Khoo, K.H.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef]
- Lehmann, S.; Bykov, V.J.N.; Ali, D.; Andrén, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in Vivo: A First-in-Human Study With p53-Targeting Compound APR-246 in Refractory Hematologic Malignancies and Prostate Cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef]
- Stathis, A.; Bertoni, F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018, 8, 24–36. [Google Scholar] [CrossRef] [Green Version]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pustovalova, M.; Alhaddad, L.; Smetanina, N.; Chigasova, A.; Blokhina, T.; Chuprov-Netochin, R.; Osipov, A.N.; Leonov, S. The p53–53BP1-Related Survival of A549 and H1299 Human Lung Cancer Cells after Multifractionated Radiotherapy Demonstrated Different Response to Additional Acute X-ray Exposure. Int. J. Mol. Sci. 2020, 21, 3342. https://doi.org/10.3390/ijms21093342
Pustovalova M, Alhaddad L, Smetanina N, Chigasova A, Blokhina T, Chuprov-Netochin R, Osipov AN, Leonov S. The p53–53BP1-Related Survival of A549 and H1299 Human Lung Cancer Cells after Multifractionated Radiotherapy Demonstrated Different Response to Additional Acute X-ray Exposure. International Journal of Molecular Sciences. 2020; 21(9):3342. https://doi.org/10.3390/ijms21093342
Chicago/Turabian StylePustovalova, Margarita, Lina Alhaddad, Nadezhda Smetanina, Anna Chigasova, Taisia Blokhina, Roman Chuprov-Netochin, Andreyan N. Osipov, and Sergey Leonov. 2020. "The p53–53BP1-Related Survival of A549 and H1299 Human Lung Cancer Cells after Multifractionated Radiotherapy Demonstrated Different Response to Additional Acute X-ray Exposure" International Journal of Molecular Sciences 21, no. 9: 3342. https://doi.org/10.3390/ijms21093342
APA StylePustovalova, M., Alhaddad, L., Smetanina, N., Chigasova, A., Blokhina, T., Chuprov-Netochin, R., Osipov, A. N., & Leonov, S. (2020). The p53–53BP1-Related Survival of A549 and H1299 Human Lung Cancer Cells after Multifractionated Radiotherapy Demonstrated Different Response to Additional Acute X-ray Exposure. International Journal of Molecular Sciences, 21(9), 3342. https://doi.org/10.3390/ijms21093342