An Immunological Glance on Pancreatic Ductal Adenocarcinoma

 ,
, 
Abstract
:1. Introduction
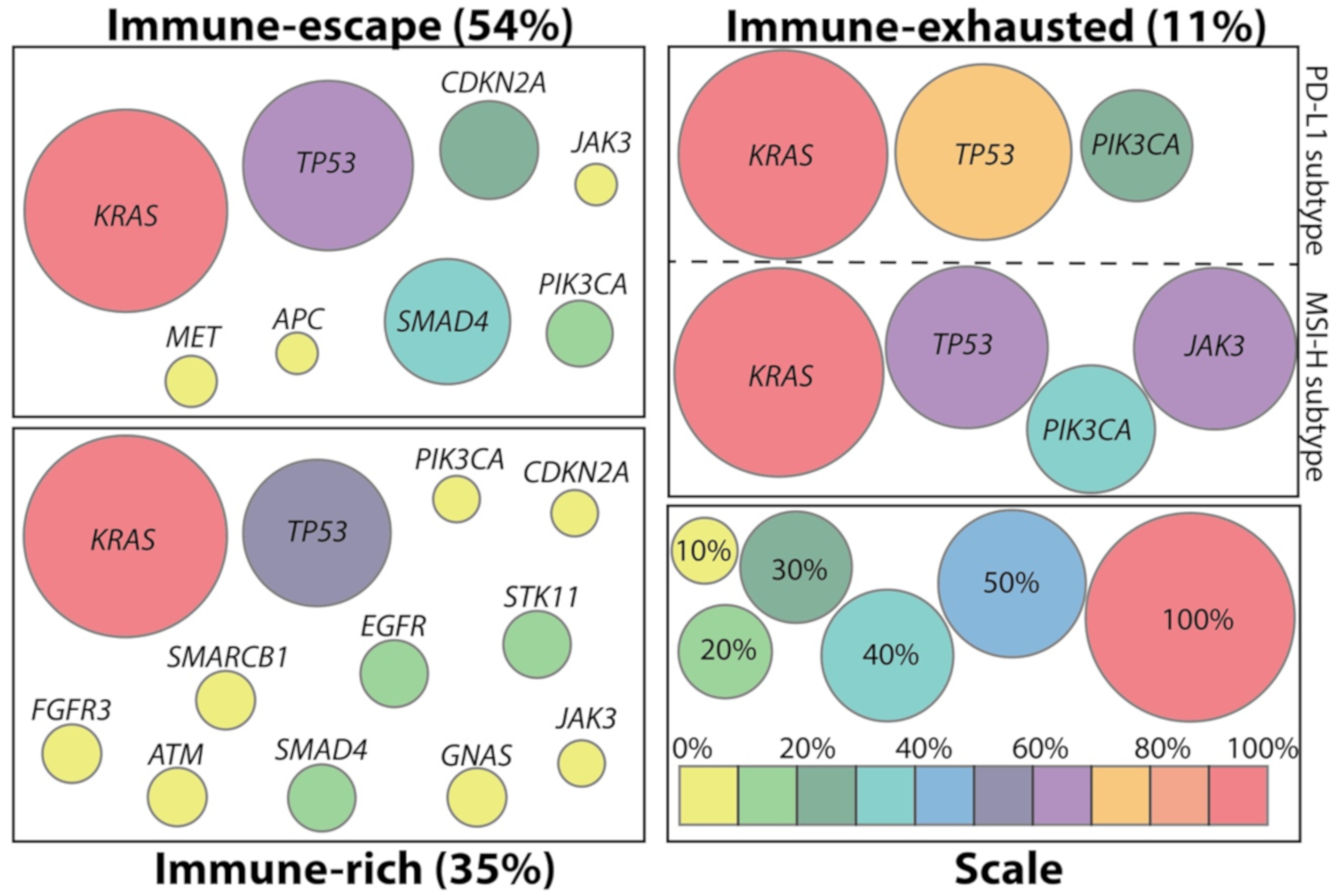
2. Immunologic Subtypes of PDAC
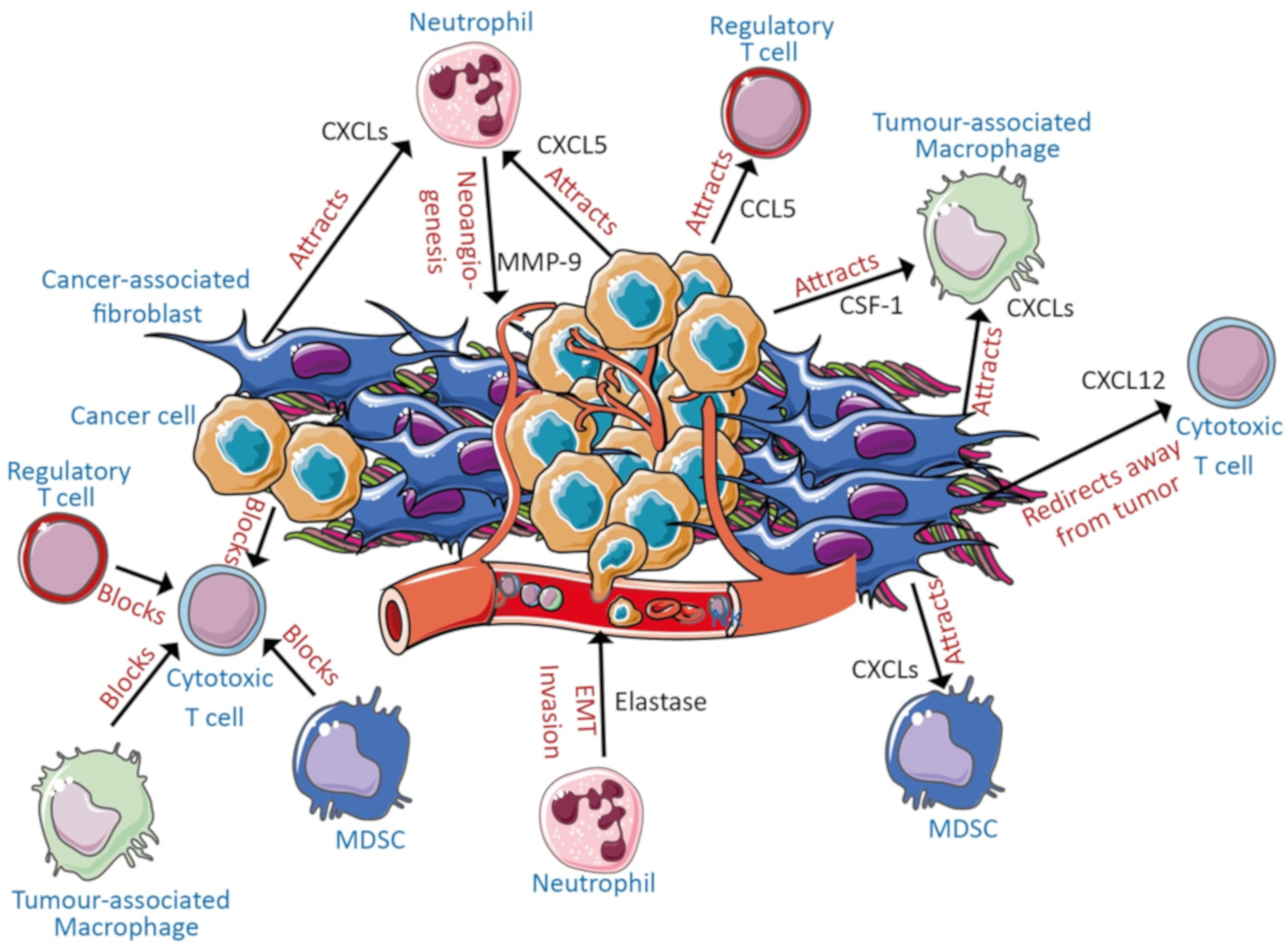
3. Potential Cell Types Involved in Disease Pathogenesis
3.1. Cancer Cells and Stroma
3.2. Myeloid-Derived Suppressor Cells, Neutrophils, and Macrophages
3.3. T Cells
3.4. NK Cells
3.5. B Cells and Mast Cells
4. Translational Models to Study PDAC-Immune System Interactions
5. Clinical Application of Immune Checkpoint Inhibitors and Generation of Neoantigens in PDAC
5.1. Application of Immune Checkpoint Inhibitors in PDAC
5.2. Neoantigens in PDAC
6. Conclusions, Outlook, and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-FU | 5-fluoruracil |
| ACT | adoptive cell therapy |
| ADM | acinar-to-ductal metaplasia |
| apCAF | antigen presenting CAF |
| BiTE | bispecific T cell engager |
| BRCA | breast cancer susceptibility protein |
| CAF | cancer associated fibroblast |
| CAR | chimeric antigen receptor |
| CCL | Chemokine (C-C motif) ligand |
| CSF-1 | colony-stimulating factor-1 |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| CXCL | C-X-C motif binding chemokine |
| CXCR | C-X-C motif chemokine receptor |
| DC | dendritic cell |
| dMMR | deficient mismatch repair |
| EMT | epithelial to mesenchymal transition |
| FDA | Food and Drug Administration |
| FOLFIRINOX | 5-fluoruracil, folinic acid, irinotecan, oxaliplatin |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| iCAF | inflammatory CAF |
| ICI | immune checkpoint inhibitor |
| ICOSL | induced costimulatory ligand |
| IL | interleukin |
| IPMN | intraductal papillary mucinous neoplasm |
| M-CSF | macrophage colony-stimulating factor |
| MCN | mucinous cystic neoplasm |
| MDSC | myeloid-derived suppressor cell |
| MMP-9 | matrix metalloproteinase-9 |
| MUC16 | Mucin-16 |
| mRNA | messenger ribonucleic acid |
| MSI-H | high microsatellite instability |
| myCAF | myofibroblastic CAF |
| n.a. | not applicable |
| NK cell | natural killer cell |
| OS | overall survival |
| PanIN | pancreatic intraepithelial neoplasm |
| PARP | poly(ADP-ribose)-polymerase |
| PBMC | peripheral blood mononuclear cell |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed cell death 1 ligand 1 |
| PDAC | pancreatic ductal adenocarcinoma |
| PFS | progression-free survival |
| TAM | tumor-associated macrophage |
| TCR | T cell receptor |
| TGF-β | transforming growth factor β |
| TIL | tumor-infiltrating leucocyte |
| TNF-α | tumor necrosis factor α |
| Treg | regulatory T cell |
| α-SMA | α smooth muscle actin |
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef]
- Swayden, M.; Iovanna, J.; Soubeyran, P. Pancreatic cancer chemo-resistance is driven by tumor phenotype rather than tumor genotype. Heliyon 2018, 4, e01055. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Boyiadzis, M.M.; Kirkwood, J.M.; Marshall, J.L.; Pritchard, C.C.; Azad, N.S.; Gulley, J.L. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J. Immunother. Cancer 2018, 6, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Commissioner, O. of the FDA approves first cancer treatment for any solid tumor with a specific genetic feature. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-cancer-treatment-any-solid-tumor-specific-genetic-feature (accessed on 7 May 2020).
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Shima, H.; Tsurita, G.; Wada, S.; Hirohashi, Y.; Yasui, H.; Hayashi, H.; Miyakoshi, T.; Watanabe, K.; Murai, A.; Asanuma, H.; et al. Randomized phase II trial of survivin 2B peptide vaccination for patients with HLA-A24-positive pancreatic adenocarcinoma. Cancer Sci. 2019, 110, 2378–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Russell, R.; Perkhofer, L.; Liebau, S.; Lin, Q.; Lechel, A.; Feld, F.M.; Hessmann, E.; Gaedcke, J.; Güthle, M.; Zenke, M.; et al. Loss of ATM accelerates pancreatic cancer formation and epithelial-mesenchymal transition. Nat. Commun. 2015, 6, 7677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkhofer, L.; Schmitt, A.; Romero Carrasco, M.C.; Ihle, M.; Hampp, S.; Ruess, D.A.; Hessmann, E.; Russell, R.; Lechel, A.; Azoitei, N.; et al. ATM Deficiency Generating Genomic Instability Sensitizes Pancreatic Ductal Adenocarcinoma Cells to Therapy-Induced DNA Damage. Cancer Res. 2017, 77, 5576–5590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wartenberg, M.; Cibin, S.; Zlobec, I.; Vassella, E.; Eppenberger-Castori, S.; Terracciano, L.; Eichmann, M.D.; Worni, M.; Gloor, B.; Perren, A.; et al. Integrated Genomic and Immunophenotypic Classification of Pancreatic Cancer Reveals Three Distinct Subtypes with Prognostic/Predictive Significance. Clin. Cancer Res. 2018, 24, 4444–4454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lang, M.; Zhao, T.; Feng, X.; Zheng, C.; Huang, C.; Hao, J.; Dong, J.; Luo, L.; Li, X.; et al. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3+Treg cells in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 3048–3058. [Google Scholar] [CrossRef]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Candido, J.B.; Morton, J.P.; Bailey, P.; Campbell, A.D.; Karim, S.A.; Jamieson, T.; Lapienyte, L.; Gopinathan, A.; Clark, W.; McGhee, E.J.; et al. CSF1R+ Macrophages Sustain Pancreatic Tumor Growth through T Cell Suppression and Maintenance of Key Gene Programs that Define the Squamous Subtype. Cell Rep. 2018, 23, 1448–1460. [Google Scholar] [CrossRef] [Green Version]
- Basso, D.; Gnatta, E.; Padoan, A.; Fogar, P.; Furlanello, S.; Aita, A.; Bozzato, D.; Zambon, C.-F.; Arrigoni, G.; Frasson, C.; et al. PDAC-derived exosomes enrich the microenvironment in MDSCs in a SMAD4-dependent manner through a new calcium related axis. Oncotarget 2017, 8, 84928–84944. [Google Scholar] [CrossRef] [Green Version]
- Penny, H.L.; Sieow, J.L.; Adriani, G.; Yeap, W.H.; See Chi Ee, P.; San Luis, B.; Lee, B.; Lee, T.; Mak, S.Y.; Ho, Y.S.; et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology 2016, 5, e1191731. [Google Scholar] [CrossRef]
- Pu, N.; Zhao, G.; Gao, S.; Cui, Y.; Xu, Y.; Lv, Y.; Nuerxiati, A.; Wu, W. Neutralizing TGF-β promotes anti-tumor immunity of dendritic cells against pancreatic cancer by regulating T lymphocytes. Cent. Eur. J. Immunol. 2018, 43, 123–131. [Google Scholar] [CrossRef]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, B.; Hu, Q.; Qin, Y.; Xu, W.; Liu, W.; Yu, X.; Xu, J. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics 2018, 8, 5072–5087. [Google Scholar] [CrossRef] [PubMed]
- Kadaba, R.; Birke, H.; Wang, J.; Hooper, S.; Andl, C.D.; Di Maggio, F.; Soylu, E.; Ghallab, M.; Bor, D.; Froeling, F.E.; et al. Imbalance of desmoplastic stromal cell numbers drives aggressive cancer processes. J. Pathol. 2013, 230, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuta, K.; Masamune, A.; Watanabe, T.; Ariga, H.; Itoh, H.; Hamada, S.; Satoh, K.; Egawa, S.; Unno, M.; Shimosegawa, T. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2010, 403, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Rucki, A.A.; Foley, K.; Zhang, P.; Xiao, Q.; Kleponis, J.; Wu, A.A.; Sharma, R.; Mo, G.; Liu, A.; Van Eyk, J.; et al. Heterogeneous Stromal Signaling within the Tumor Microenvironment Controls the Metastasis of Pancreatic Cancer. Cancer Res. 2017, 77, 41–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175. [Google Scholar] [CrossRef]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [Green Version]
- Sano, M.; Ijichi, H.; Takahashi, R.; Miyabayashi, K.; Fujiwara, H.; Yamada, T.; Kato, H.; Nakatsuka, T.; Tanaka, Y.; Tateishi, K.; et al. Blocking CXCLs-CXCR2 axis in tumor-stromal interactions contributes to survival in a mouse model of pancreatic ductal adenocarcinoma through reduced cell invasion/migration and a shift of immune-inflammatory microenvironment. Oncogenesis 2019, 8, 8. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [Green Version]
- Nagathihalli, N.S.; Castellanos, J.A.; VanSaun, M.N.; Dai, X.; Ambrose, M.; Guo, Q.; Xiong, Y.; Merchant, N.B. Pancreatic stellate cell secreted IL-6 stimulates STAT3 dependent invasiveness of pancreatic intraepithelial neoplasia and cancer cells. Oncotarget 2016, 7, 65982–65992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, E.S.; Vail, P.; Balaji, U.; Ngo, H.; Botros, I.W.; Makarov, V.; Riaz, N.; Balachandran, V.; Leach, S.; Thompson, D.M.; et al. Stratification of Pancreatic Ductal Adenocarcinoma: Combinatorial Genetic, Stromal, and Immunologic Markers. Clin. Cancer Res. 2017, 23, 4429–4440. [Google Scholar] [CrossRef] [Green Version]
- Miksch, R.C.; Schoenberg, M.B.; Weniger, M.; Bösch, F.; Ormanns, S.; Mayer, B.; Werner, J.; Bazhin, A.V.; D’Haese, J.G. Prognostic Impact of Tumor-Infiltrating Lymphocytes and Neutrophils on Survival of Patients with Upfront Resection of Pancreatic Cancer. Cancers (Basel) 2019, 11, 39. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, U.M.; Langhoff, E.; Goni, E.; Costello, E.; Greenhalf, W.; Halloran, C.; Ormanns, S.; Kruger, S.; Boeck, S.; Ribback, S.; et al. Immune Cell and Stromal Signature Associated With Progression-Free Survival of Patients With Resected Pancreatic Ductal Adenocarcinoma. Gastroenterology 2018, 155, 1625–1639. [Google Scholar] [CrossRef] [Green Version]
- Steins, A.; van Mackelenbergh, M.G.; van der Zalm, A.P.; Klaassen, R.; Serrels, B.; Goris, S.G.; Kocher, H.M.; Waasdorp, C.; de Jong, J.H.; Tekin, C.; et al. High-grade mesenchymal pancreatic ductal adenocarcinoma drives stromal deactivation through CSF-1. EMBO Rep. 2020, 21, e48780. [Google Scholar] [CrossRef]
- Trovato, R.; Fiore, A.; Sartori, S.; Canè, S.; Giugno, R.; Cascione, L.; Paiella, S.; Salvia, R.; De Sanctis, F.; Poffe, O.; et al. Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J. Immunother. Cancer 2019, 7, 255. [Google Scholar] [CrossRef]
- Takeuchi, S.; Baghdadi, M.; Tsuchikawa, T.; Wada, H.; Nakamura, T.; Abe, H.; Nakanishi, S.; Usui, Y.; Higuchi, K.; Takahashi, M.; et al. Chemotherapy-Derived Inflammatory Responses Accelerate the Formation of Immunosuppressive Myeloid Cells in the Tissue Microenvironment of Human Pancreatic Cancer. Cancer Res. 2015, 75, 2629–2640. [Google Scholar] [CrossRef] [Green Version]
- Bausch, D.; Pausch, T.; Krauss, T.; Hopt, U.T.; Fernandez-del-Castillo, C.; Warshaw, A.L.; Thayer, S.P.; Keck, T. Neutrophil granulocyte derived MMP-9 is a VEGF independent functional component of the angiogenic switch in pancreatic ductal adenocarcinoma. Angiogenesis 2011, 14, 235–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaida, M.M.; Steffen, T.G.; Günther, F.; Tschaharganeh, D.F.; Felix, K.; Bergmann, F.; Schirmacher, P.; Hänsch, G.M. Polymorphonuclear neutrophils promote dyshesion of tumor cells and elastase-mediated degradation of E-cadherin in pancreatic tumors. Eur. J. Immunol. 2012, 42, 3369–3380. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Steffen, T.; Giese, T.; Giese, N.; Longerich, T.; Schirmacher, P.; Hänsch, G.M.; Gaida, M.M. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma and pancreatic tumor cell lines: The role of neutrophils and neutrophil-derived elastase. Clin. Dev. Immunol. 2012, 2012, 720768. [Google Scholar] [CrossRef] [PubMed]
- Mayer, P.; Dinkic, C.; Jesenofsky, R.; Klauss, M.; Schirmacher, P.; Dapunt, U.; Hackert, T.; Uhle, F.; Hänsch, G.M.; Gaida, M.M. Changes in the microarchitecture of the pancreatic cancer stroma are linked to neutrophil-dependent reprogramming of stellate cells and reflected by diffusion-weighted magnetic resonance imaging. Theranostics 2018, 8, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Yin, Z.; Ma, T.; Huang, B.; Lin, L.; Zhou, Y.; Yan, J.; Zou, Y.; Chen, S. Macrophage-derived exosomal microRNA-501-3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3-mediated TGF-β signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 310. [Google Scholar] [CrossRef] [Green Version]
- Jackute, J.; Zemaitis, M.; Pranys, D.; Sitkauskiene, B.; Miliauskas, S.; Vaitkiene, S.; Sakalauskas, R. Distribution of M1 and M2 macrophages in tumor islets and stroma in relation to prognosis of non-small cell lung cancer. BMC Immunol. 2018, 19, 3. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.-Y.; Döppler, H.; Necela, B.; Krishna, M.; Crawford, H.C.; Raimondo, M.; Storz, P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-κB and MMPs. J. Cell Biol. 2013, 202, 563–577. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Döppler, H.; Necela, B.; Edenfield, B.; Zhang, L.; Dawson, D.W.; Storz, P. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2015, 5, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Komura, T.; Sakai, Y.; Harada, K.; Kawaguchi, K.; Takabatake, H.; Kitagawa, H.; Wada, T.; Honda, M.; Ohta, T.; Nakanuma, Y.; et al. Inflammatory features of pancreatic cancer highlighted by monocytes/macrophages and CD4+ T cells with clinical impact. Cancer Sci. 2015, 106, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Goebel, L.; Grage-Griebenow, E.; Gorys, A.; Helm, O.; Genrich, G.; Lenk, L.; Wesch, D.; Ungefroren, H.; Freitag-Wolf, S.; Sipos, B.; et al. CD4+ T cells potently induce epithelial-mesenchymal-transition in premalignant and malignant pancreatic ductal epithelial cells-novel implications of CD4+ T cells in pancreatic cancer development. Oncoimmunology 2015, 4, e1000083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.-Q.; Liu, L.; Xu, H.-X.; Wu, C.-T.; Xiang, J.-F.; Xu, J.; Liu, C.; Long, J.; Ni, Q.-X.; Yu, X.-J. Infiltrating immune cells and gene mutations in pancreatic ductal adenocarcinoma. Br. J. Surg. 2016, 103, 1189–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helm, O.; Mennrich, R.; Petrick, D.; Goebel, L.; Freitag-Wolf, S.; Röder, C.; Kalthoff, H.; Röcken, C.; Sipos, B.; Kabelitz, D.; et al. Comparative characterization of stroma cells and ductal epithelium in chronic pancreatitis and pancreatic ductal adenocarcinoma. PLoS ONE 2014, 9, e94357. [Google Scholar] [CrossRef] [Green Version]
- Shevchenko, I.; Karakhanova, S.; Soltek, S.; Link, J.; Bayry, J.; Werner, J.; Umansky, V.; Bazhin, A.V. Low-dose gemcitabine depletes regulatory T cells and improves survival in the orthotopic Panc02 model of pancreatic cancer. Int. J. Cancer 2013, 133, 98–107. [Google Scholar] [CrossRef]
- Lim, S.A.; Kim, J.; Jeon, S.; Shin, M.H.; Kwon, J.; Kim, T.-J.; Im, K.; Han, Y.; Kwon, W.; Kim, S.-W.; et al. Defective Localization With Impaired Tumor Cytotoxicity Contributes to the Immune Escape of NK Cells in Pancreatic Cancer Patients. Front. Immunol. 2019, 10, 496. [Google Scholar] [CrossRef] [Green Version]
- Wartenberg, M.; Zlobec, I.; Perren, A.; Koelzer, V.H.; Gloor, B.; Lugli, A.; Karamitopoulou, E. Accumulation of FOXP3+T-cells in the tumor microenvironment is associated with an epithelial-mesenchymal-transition-type tumor budding phenotype and is an independent prognostic factor in surgically resected pancreatic ductal adenocarcinoma. Oncotarget 2015, 6, 4190–4201. [Google Scholar] [CrossRef] [Green Version]
- Bengsch, F.; Knoblock, D.M.; Liu, A.; McAllister, F.; Beatty, G.L. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol. Immunother. 2017, 66, 1609–1617. [Google Scholar] [CrossRef]
- Kenkel, J.A.; Tseng, W.W.; Davidson, M.G.; Tolentino, L.L.; Choi, O.; Bhattacharya, N.; Seeley, E.S.; Winer, D.A.; Reticker-Flynn, N.E.; Engleman, E.G. An Immunosuppressive Dendritic Cell Subset Accumulates at Secondary Sites and Promotes Metastasis in Pancreatic Cancer. Cancer Res. 2017, 77, 4158–4170. [Google Scholar] [CrossRef] [Green Version]
- Sakellariou-Thompson, D.; Forget, M.-A.; Creasy, C.; Bernard, V.; Zhao, L.; Kim, Y.U.; Hurd, M.W.; Uraoka, N.; Parra, E.R.; Kang, Y.; et al. 4-1BB Agonist Focuses CD8+ Tumor-Infiltrating T-Cell Growth into a Distinct Repertoire Capable of Tumor Recognition in Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 7263–7275. [Google Scholar] [CrossRef] [Green Version]
- Bräunlein, E.; Krackhardt, A.M. Tools to define the melanoma-associated immunopeptidome. Immunology 2017, 152, 536–544. [Google Scholar] [CrossRef]
- Quaranta, V.; Rainer, C.; Nielsen, S.R.; Raymant, M.L.; Ahmed, M.S.; Engle, D.D.; Taylor, A.; Murray, T.; Campbell, F.; Palmer, D.H.; et al. Macrophage-Derived Granulin Drives Resistance to Immune Checkpoint Inhibition in Metastatic Pancreatic Cancer. Cancer Res. 2018, 78, 4253–4269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.-F.; Lu, Y.; Cheng, H.; Shi, S.; Xu, J.; Long, J.; Liu, L.; Liu, C.; Yu, X. Abnormal distribution of peripheral lymphocyte subsets induced by PDAC modulates overall survival. Pancreatology 2014, 14, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Basso, D.; Fogar, P.; Falconi, M.; Fadi, E.; Sperti, C.; Frasson, C.; Greco, E.; Tamburrino, D.; Teolato, S.; Moz, S.; et al. Pancreatic tumors and immature immunosuppressive myeloid cells in blood and spleen: Role of inhibitory co-stimulatory molecules PDL1 and CTLA4. An in vivo and in vitro study. PLoS ONE 2013, 8, e54824. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhou, Z.; Cheng, Q.; Wang, H.; Cao, H.; Xu, Q.; Tuo, Y.; Jiang, L.; Zou, Y.; Ren, H.; et al. Acceleration of pancreatic tumorigenesis under immunosuppressive microenvironment induced by Reg3g overexpression. Cell Death Dis. 2017, 8, e3033. [Google Scholar] [CrossRef] [Green Version]
- Gürlevik, E.; Fleischmann-Mundt, B.; Brooks, J.; Demir, I.E.; Steiger, K.; Ribback, S.; Yevsa, T.; Woller, N.; Kloos, A.; Ostroumov, D.; et al. Administration of Gemcitabine After Pancreatic Tumor Resection in Mice Induces an Antitumor Immune Response Mediated by Natural Killer Cells. Gastroenterology 2016, 151, 338–350. [Google Scholar] [CrossRef] [Green Version]
- Brooks, J.; Fleischmann-Mundt, B.; Woller, N.; Niemann, J.; Ribback, S.; Peters, K.; Demir, I.E.; Armbrecht, N.; Ceyhan, G.O.; Manns, M.P.; et al. Perioperative, Spatiotemporally Coordinated Activation of T and NK Cells Prevents Recurrence of Pancreatic Cancer. Cancer Res. 2018, 78, 475–488. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Schlößer, H.A.; Wang, Z.; Qin, J.; Li, J.; Popp, F.; Popp, M.C.; Alakus, H.; Chon, S.-H.; Hansen, H.P.; et al. Tumor-Derived Extracellular Vesicles Inhibit Natural Killer Cell Function in Pancreatic Cancer. Cancers (Basel) 2019, 11, 874. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Huang, M.; Xie, F.; Zhou, H.; Yang, J.; Huang, Q. Gemcitabine inhibits immune escape of pancreatic cancer by down regulating the soluble ULBP2 protein. Oncotarget 2016, 7, 70092–70099. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.F.; Cui, J.W. The Role of Tumor-Infiltrating B Cells in Tumor Immunity. J. Oncol. 2019, 2019, 2592419. [Google Scholar] [CrossRef]
- Castino, G.F.; Cortese, N.; Capretti, G.; Serio, S.; Di Caro, G.; Mineri, R.; Magrini, E.; Grizzi, F.; Cappello, P.; Novelli, F.; et al. Spatial distribution of B cells predicts prognosis in human pancreatic adenocarcinoma. Oncoimmunology 2016, 5, e1085147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcelli, L.; Iacobazzi, R.M.; Di Fonte, R.; Serratì, S.; Intini, A.; Solimando, A.G.; Brunetti, O.; Calabrese, A.; Leonetti, F.; Azzariti, A.; et al. CAFs and TGF-β Signaling Activation by Mast Cells Contribute to Resistance to Gemcitabine/Nabpaclitaxel in Pancreatic Cancer. Cancers (Basel) 2019, 11, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Hwang, R.F.; Logsdon, C.D.; Ullrich, S.E. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res. 2013, 73, 3927–3937. [Google Scholar] [CrossRef] [Green Version]
- Tjomsland, V.; Spångeus, A.; Sandström, P.; Borch, K.; Messmer, D.; Larsson, M. Semi mature blood dendritic cells exist in patients with ductal pancreatic adenocarcinoma owing to inflammatory factors released from the tumor. PLoS ONE 2010, 5, e13441. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Komar, C.A.; Bengsch, F.; Graham, K.; Beatty, G.L. Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-Kras(G12D/+); LSL-Trp53(R172H/+); Pdx-1-Cre), Its Variants, and Their Application in Immuno-oncology Drug Discovery. Curr. Protoc. Pharmacol. 2016, 73, 14–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, D.Z.; Ma, Y.; Ji, B.; Wang, H.; Deng, D.; Liu, Y.; Abbruzzese, J.L.; Liu, Y.; Logsdon, C.D.; Hwu, P. Mast cells in tumor microenvironment promotes the in vivo growth of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2011, 17, 7015–7023. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.-I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [Green Version]
- Frappart, P.-O.; Walter, K.; Gout, J.; Beutel, A.K.; Morawe, M.; Arnold, F.; Breunig, M.; Barth, T.F.; Marienfeld, R.; Schulte, L.; et al. Pancreatic cancer-derived organoids - a disease modeling tool to predict drug response. United Eur. Gastroenterol. J. 2020, 2050640620905183. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 335. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, A.C.; Yarchoan, M.; Durham, J.N.; Yusko, E.C.; Rytlewski, J.A.; Robins, H.S.; Laheru, D.A.; Le, D.T.; Lutz, E.R.; Jaffee, E.M. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Kamath, S.D.; Kalyan, A.; Kircher, S.; Nimeiri, H.; Fought, A.J.; Benson, A.; Mulcahy, M. Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A Phase Ib Study. Oncologist 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahalingam, D.; Wilkinson, G.A.; Eng, K.H.; Fields, P.; Raber, P.; Moseley, J.L.; Cheetham, K.; Coffey, M.; Nuovo, G.; Kalinski, P.; et al. Pembrolizumab in Combination with the Oncolytic Virus Pelareorep and Chemotherapy in Patients with Advanced Pancreatic Adenocarcinoma: A Phase Ib Study. Clin. Cancer Res. 2020, 26, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macherla, S.; Laks, S.; Naqash, A.R.; Bulumulle, A.; Zervos, E.; Muzaffar, M. Emerging Role of Immune Checkpoint Blockade in Pancreatic Cancer. Int. J. Mol. Sci. 2018, 19, E3505. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Itoh, F.; Nakamura, H.; Fukushima, H.; Sasaki, S.; Perucho, M.; Imai, K. Genetic and clinical features of human pancreatic ductal adenocarcinomas with widespread microsatellite instability. Cancer Res. 2001, 61, 3139–3144. [Google Scholar]
- Nakata, B.; Wang, Y.Q.; Yashiro, M.; Nishioka, N.; Tanaka, H.; Ohira, M.; Ishikawa, T.; Nishino, H.; Hirakawa, K. Prognostic value of microsatellite instability in resectable pancreatic cancer. Clin. Cancer Res. 2002, 8, 2536–2540. [Google Scholar]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Ott, P.A.; Bang, Y.-J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.-C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated With Pembrolizumab Across 20 Cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.-Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.-C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019. [Google Scholar] [CrossRef]
- Hong, D.; Rasco, D.; Veeder, M.; Luke, J.J.; Chandler, J.; Balmanoukian, A.; George, T.J.; Munster, P.; Berlin, J.D.; Gutierrez, M.; et al. A Phase 1b/2 Study of the Bruton Tyrosine Kinase Inhibitor Ibrutinib and the PD-L1 Inhibitor Durvalumab in Patients with Pretreated Solid Tumors. Oncology 2019, 97, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Forget, M.-A.; Lucas, F.A.S.; Alvarez, H.A.; Haymaker, C.; Chattopadhyay, C.; Kim, S.-H.; Ekmekcioglu, S.; Grimm, E.A.; et al. Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma. Sci. Rep. 2016, 6, 35848. [Google Scholar] [CrossRef] [PubMed]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.-R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Dotti, G.; Yee, C.; Goff, S.L. An Update on Adoptive T-Cell Therapy and Neoantigen Vaccines. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, e70–e78. [Google Scholar] [CrossRef]
- FDA. Research, C. for D.E. and FDA Approves Tisagenlecleucel for Adults with Relapsed or Refractory Large B-Cell Lymphoma; FDA: Silver Spring, MD, USA, 2019. [Google Scholar]
- Akce, M.; Zaidi, M.Y.; Waller, E.K.; El-Rayes, B.F.; Lesinski, G.B. The Potential of CAR T Cell Therapy in Pancreatic Cancer. Front. Immunol. 2018, 9, 2166. [Google Scholar] [CrossRef]
- Fan, J.Q.; Wang, M.F.; Chen, H.L.; Shang, D.; Das, J.K.; Song, J. Current Advances and Outlooks in Immunotherapy for Pancreatic Ductal Adenocarcinoma. Mol. Cancer 2020, 19, 32. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Yang, G.; Luo, W.; Cao, Z.; Liu, Y.; Qiu, J.; Chen, G.; You, L.; Zhao, F.; Zheng, L.; et al. Novel therapeutic strategies and perspectives for metastatic pancreatic cancer: vaccine therapy is more than just a theory. Cancer Cell Int. 2020, 20, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Przepiorka, D.; Ko, C.-W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.-J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| ICI Class | Drugs | Patients/Disease | Treatment | Clinical Trial | Status |
|---|---|---|---|---|---|
| PD-1 blocker, CTLA-4 blocker | Pembrolizumab and/or ipilimumab | Patients with hepatocarcinoma, lung cancer, melanoma, renal cancer, head and neck cancer, pancreatic cancer, ovarian cancer, colorectal cancer, cervical cancer, breast cancer | Vein, artery, or intra-tumour infusion of checkpoint inhibitor | NCT03755739 | Recruiting |
| ICI Class | Co-Drugs | Treatment | Patients/Disease | Median Overall Survival | Reference |
|---|---|---|---|---|---|
| CTLA-4 | GVAX | Ipilimumab ± GVAX | Previously treated, advanced PDAC (n = 15 for iplimumab; n = 15 for ipilimumab ± GVAX) | Ipilimumab: 3.6 months; Ipilimumab ± GVAX: 5.7 months | [83] |
| CTLA-4, PD-1 | GVAX, Listeria monocytogenes | Ipilimumab ± GVAX; GVAX+ mesothelin expressing Listeria monocytogenes± nivolumab | Metastatic PDAC (n = 25 for ipilimumab ± GVAX; n = 32 for GVAX+ mesothelin expressing Listeria monocytogenes± nivolumab) | n.a. for specific therapy | [84] |
| CTLA-4 | Gemcitabine | Ipilimumab + gemcitabine | Advanced or metastatic PDAC (n = 21) | 6.9 months | [85] |
| PD-1 | Oncolytic virus (pelareorep), chemotherapy (gemcitabine or irinotecan or leucovorin and 5-FU followed by 5-FU | Pembrolizumab + pelareorep + chemotherapy | Advanced PDAC (n = 11) | 3.1 months | [86] |
| PD-1 | Pembrolizumab | Previously treated, advanced non-colorectal cancer with DNA mismatch repair or high microsatellite instability (n = 233; pancreatic cancer n = 22) | 4.0 months | [90] | |
| PD-1 | Pembrolizumab | PD-L1 positive advanced solid tumors (n = 475; pancreatic cancer n = 24) | 3.9 months | [91] | |
| PD-L1 CTLA-4 | Durvalumab ± temelimumab | Metastatic pancreatic cancer (n = 65) | n.a. | [92] | |
| PD-L1 | Bruton tyrosine kinase inhibitor ibrutinib | Ibrutinib + durvalumab | Previously treated, advanced PDAC, breast cancer, non-small cell lung cancer (n = 122, pancreatic cancer n = 49) | 4.2 months | [93] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melzer, M.K.; Arnold, F.; Stifter, K.; Zengerling, F.; Azoitei, N.; Seufferlein, T.; Bolenz, C.; Kleger, A. An Immunological Glance on Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 3345. https://doi.org/10.3390/ijms21093345
Melzer MK, Arnold F, Stifter K, Zengerling F, Azoitei N, Seufferlein T, Bolenz C, Kleger A. An Immunological Glance on Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences. 2020; 21(9):3345. https://doi.org/10.3390/ijms21093345
Chicago/Turabian StyleMelzer, Michael Karl, Frank Arnold, Katja Stifter, Friedemann Zengerling, Ninel Azoitei, Thomas Seufferlein, Christian Bolenz, and Alexander Kleger. 2020. "An Immunological Glance on Pancreatic Ductal Adenocarcinoma" International Journal of Molecular Sciences 21, no. 9: 3345. https://doi.org/10.3390/ijms21093345
APA StyleMelzer, M. K., Arnold, F., Stifter, K., Zengerling, F., Azoitei, N., Seufferlein, T., Bolenz, C., & Kleger, A. (2020). An Immunological Glance on Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences, 21(9), 3345. https://doi.org/10.3390/ijms21093345