Linear Ubiquitin Code: Its Writer, Erasers, Decoders, Inhibitors, and Implications in Disorders


Abstract
:1. Introduction
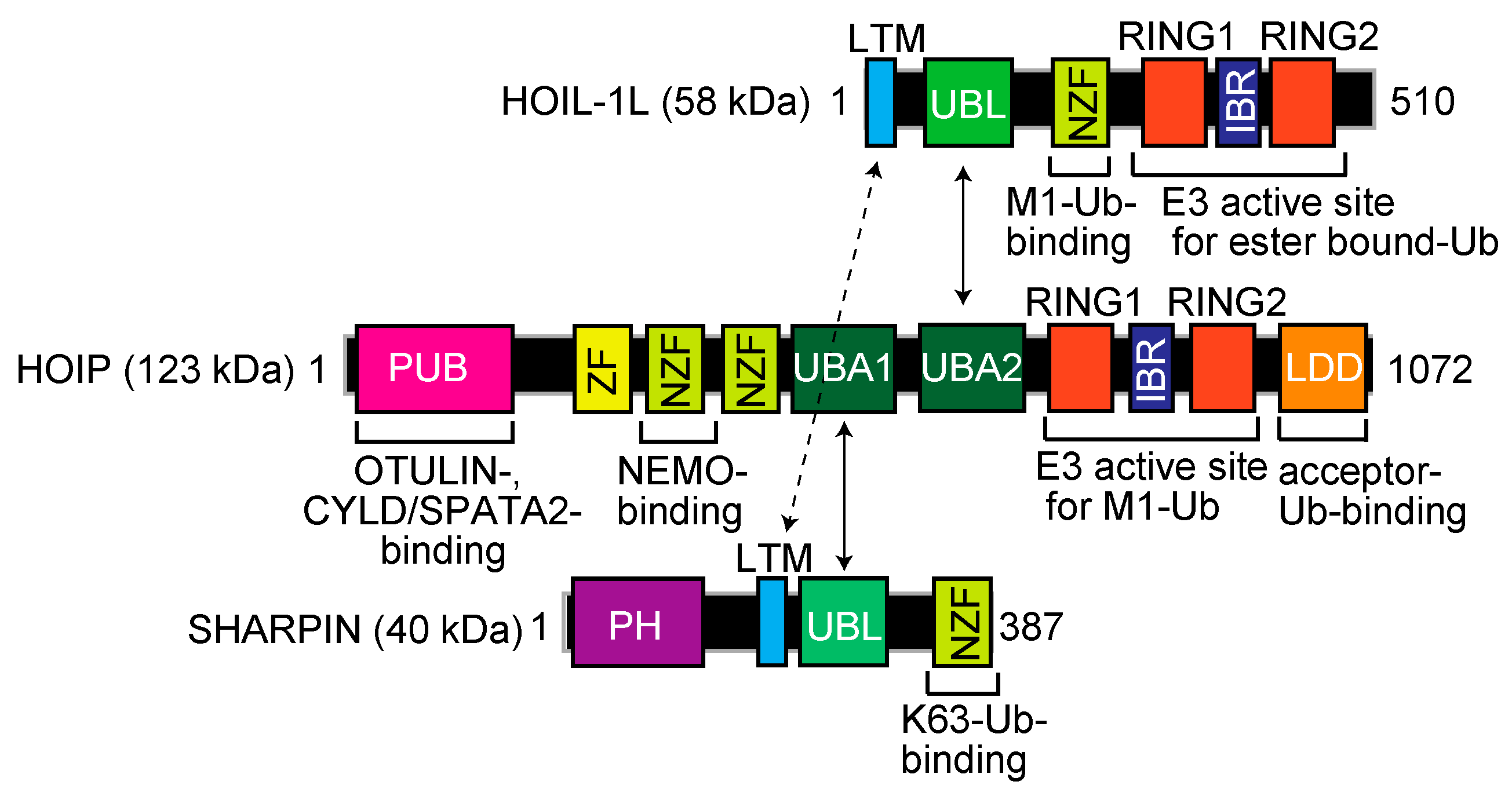
2. LUBAC: The Only Writer of the Linear Ubiquitin Code
2.1. Structure and Catalytic Mechanism of LUBAC
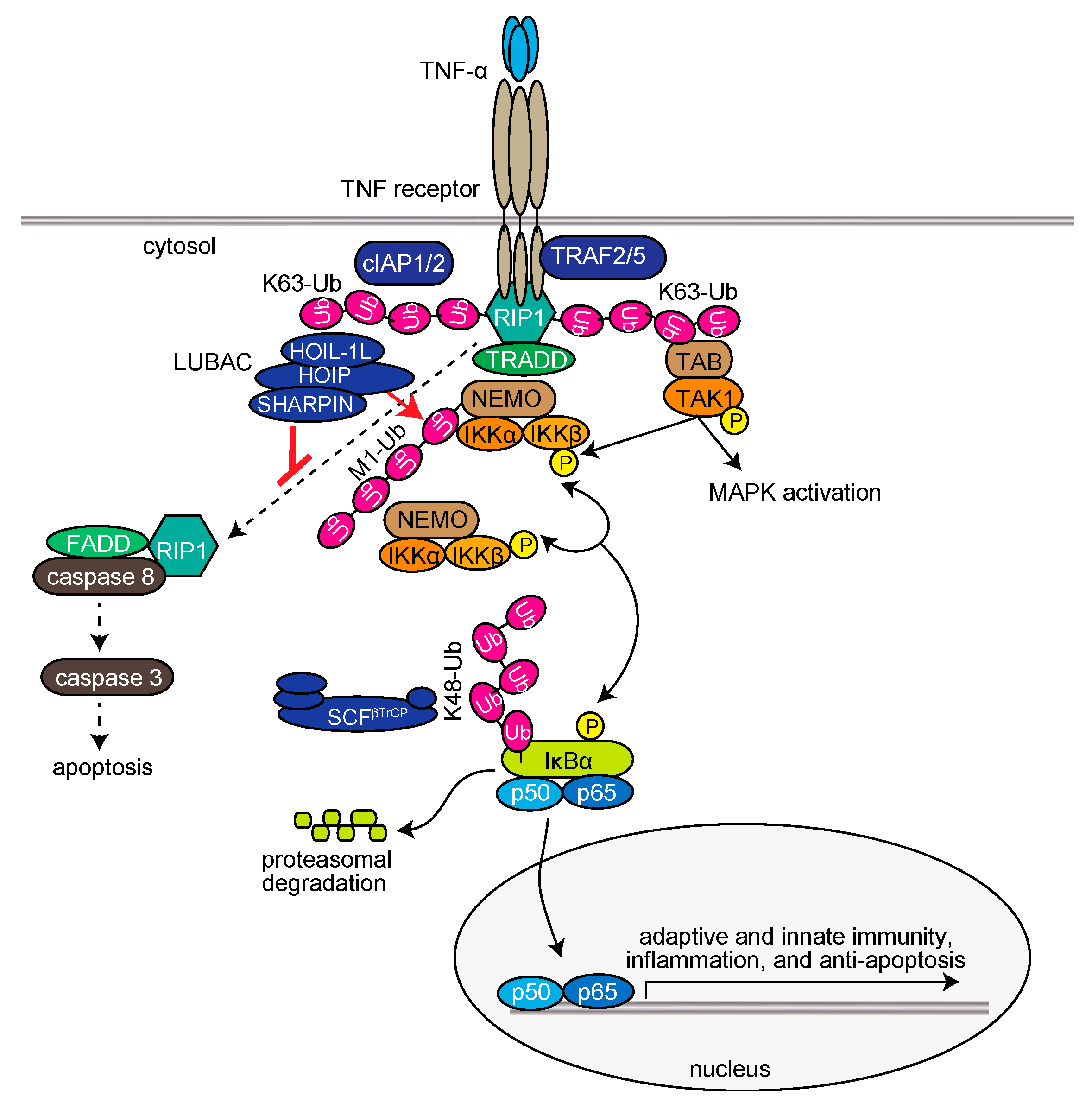
2.2. Cellular Functions of LUBAC
2.2.1. LUBAC in the Inflammatory Cytokine-Induced canonical NF-κB Activation Pathway
2.2.2. LUBAC in Acquired Immune Responses
2.2.3. LUBAC in the Genotoxic Stress Response and Inflammasome Activation
2.2.4. LUBAC-Mediated Regulation of Cell Death
2.2.5. LUBAC-Mediated Regulation of Interferon Signaling
2.2.6. Involvement of Linear Ubiquitination in Selective Autophagy
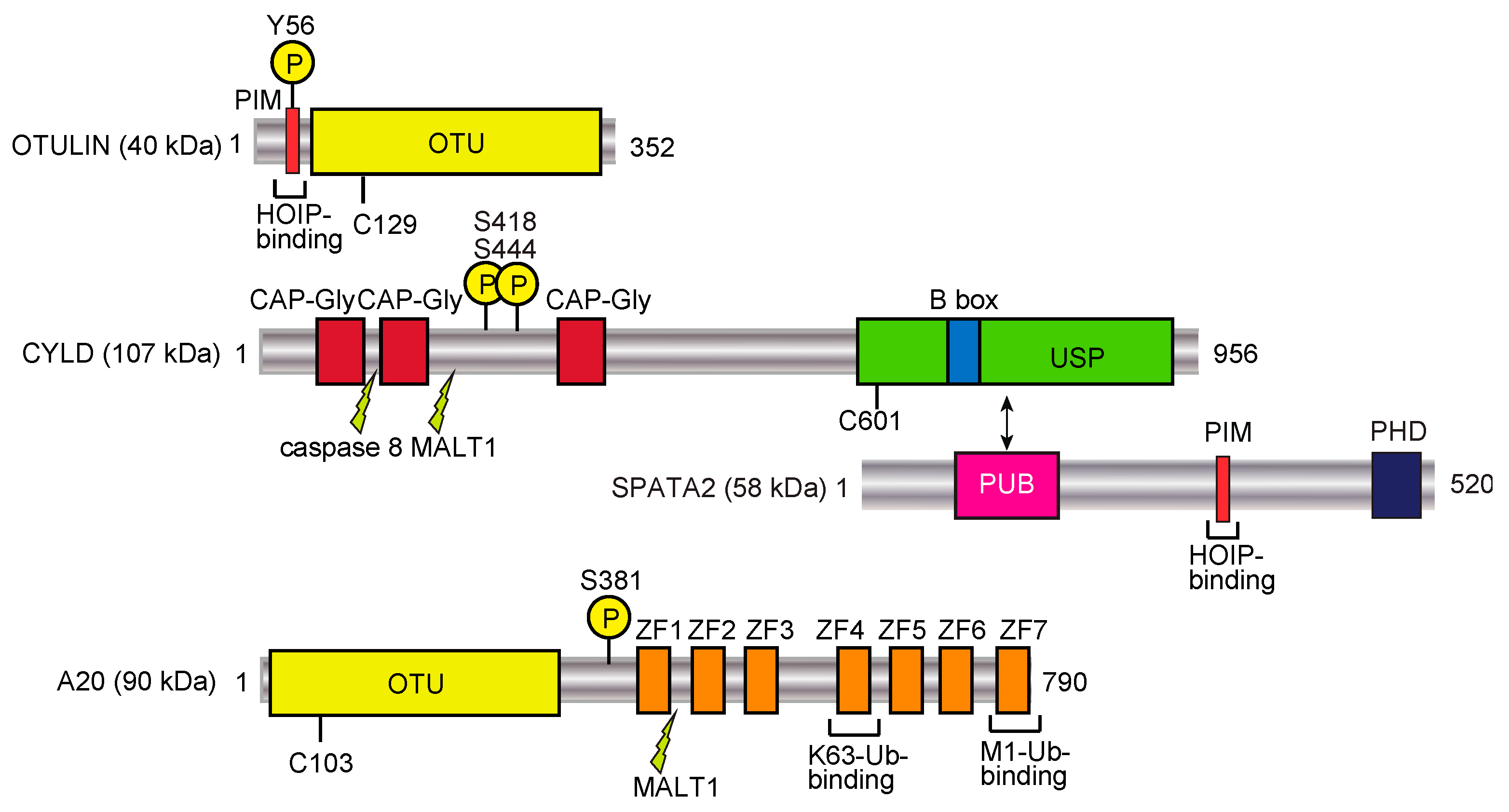
3. Erasers of the Linear Ubiquitin Code
3.1. OTULIN
3.2. CYLD
4. Decoders of the Linear Ubiquitin Code
4.1. UBAN Domain-Containing Proteins: NEMO, OPTN, and ABINs
4.2. A20
5. LUBAC-Related Disorders
5.1. Genetic Deficiency of LUBAC Subunits and Related Diseases
5.2. Enhanced LUBAC Expression and Cancers
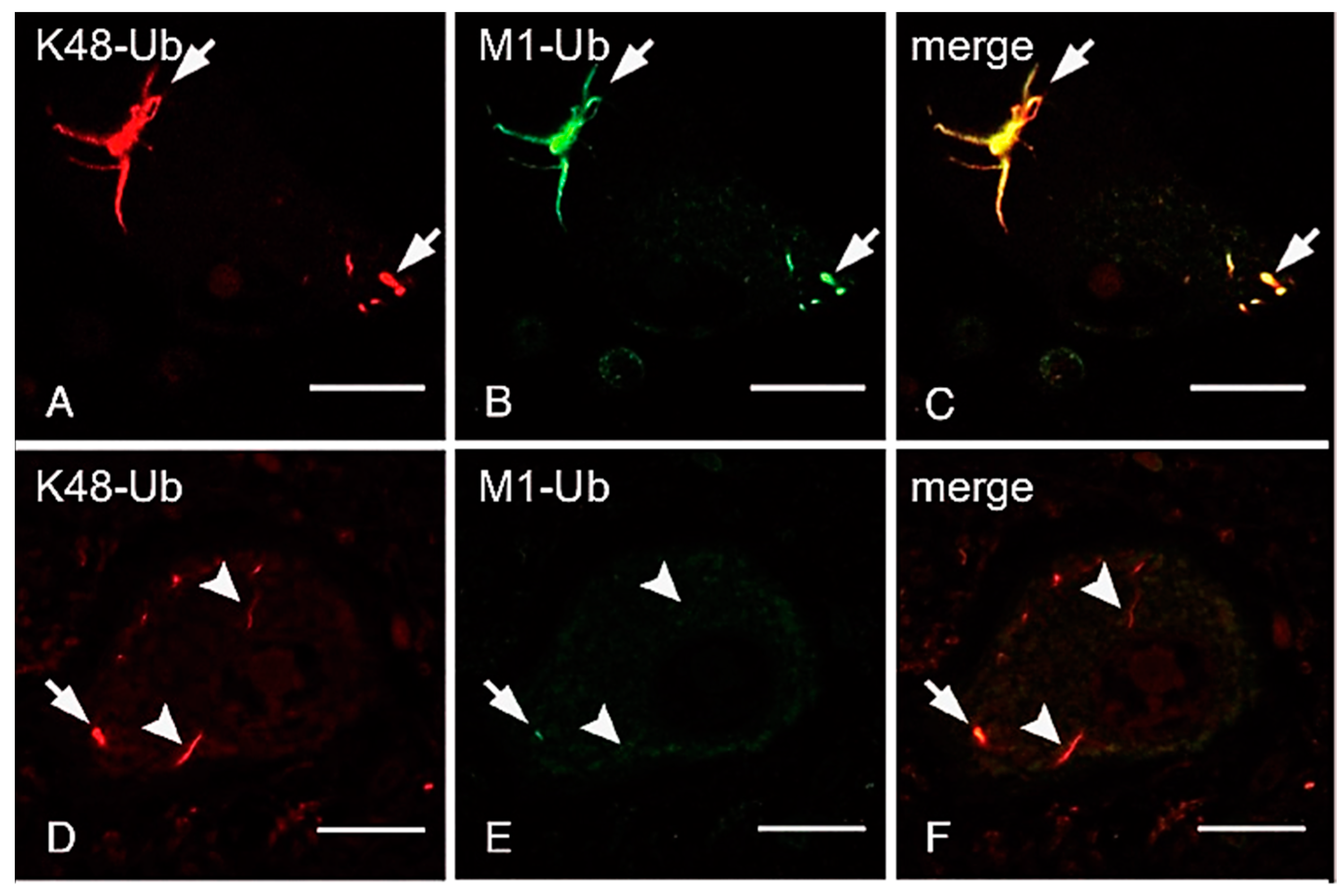
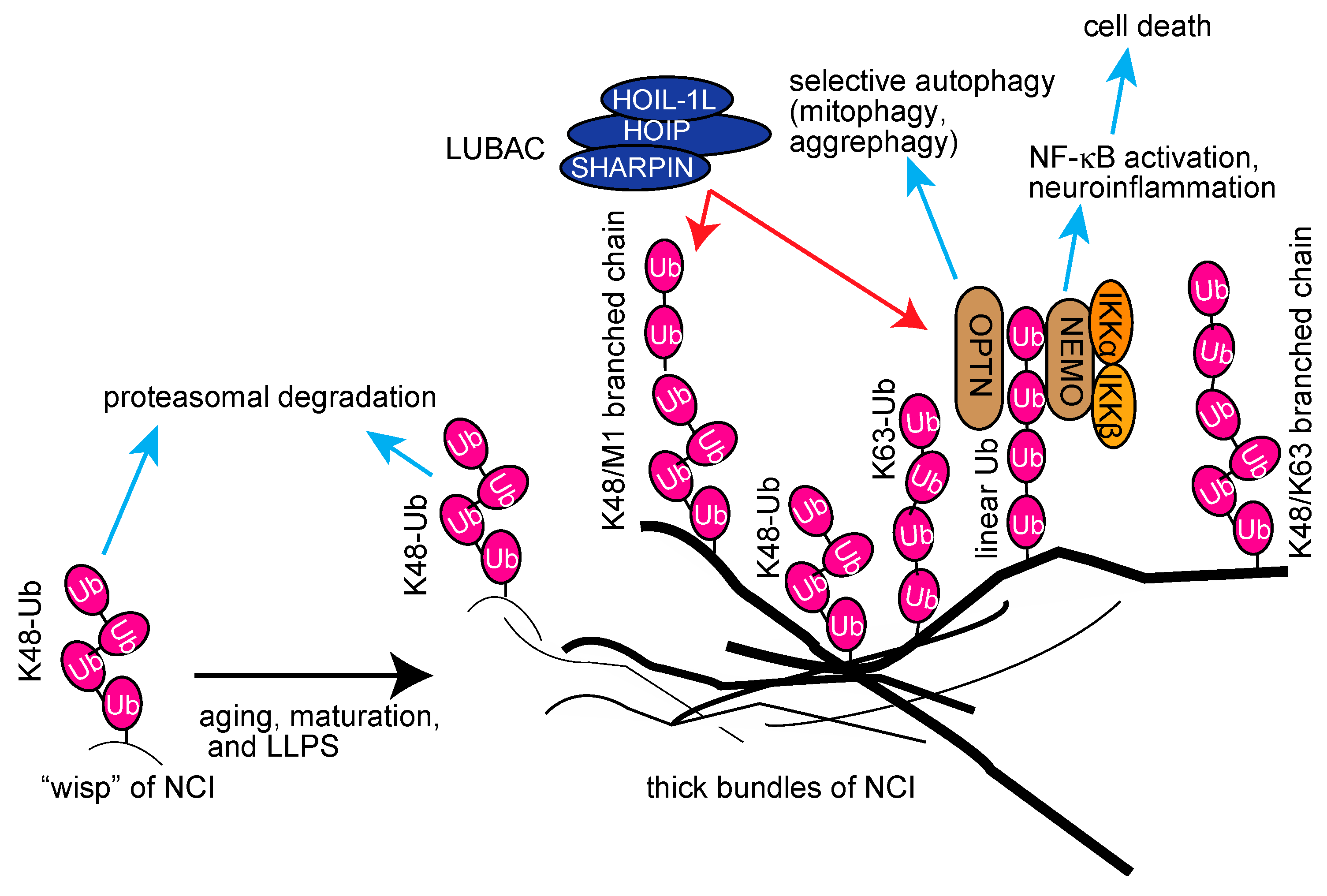
5.3. Linear Ubiquitination in Neurodegenerative Diseases
6. LUBAC Inhibitors
6.1. Chemical Inhibitors of LUBAC
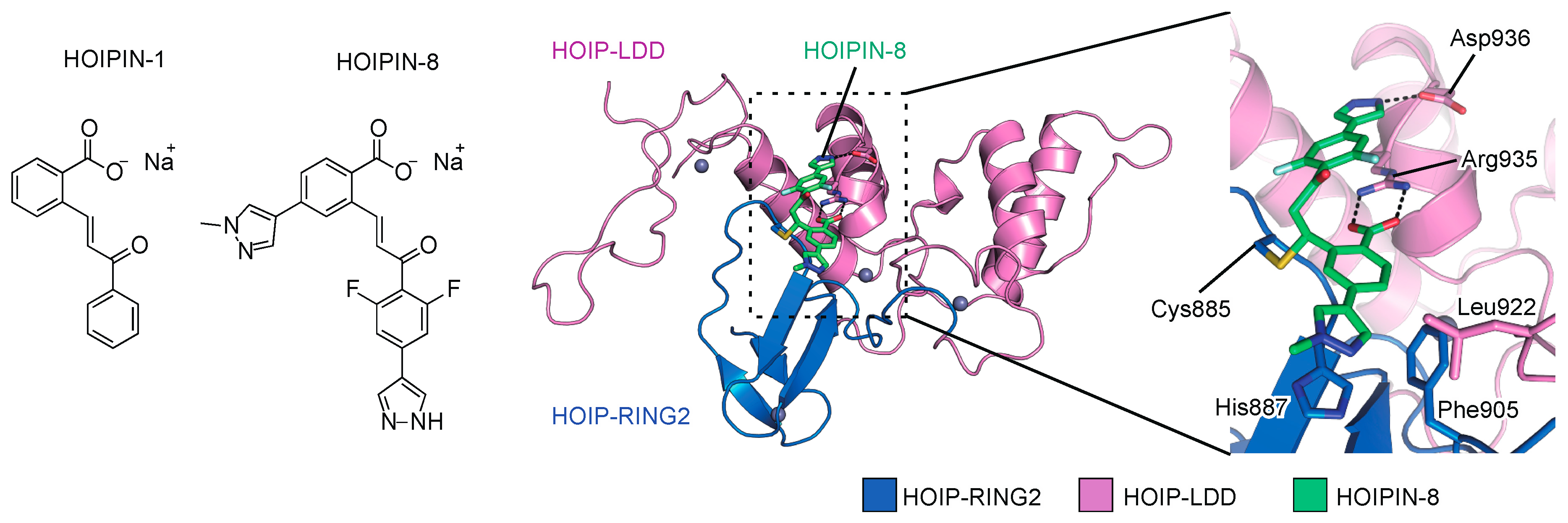
6.2. HOIPINs
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABC-DLBCL | Activated B cell-like diffuse large B cell lymphoma |
| ABIN | A20-binding inhibitor of NF-κB |
| ALS | Amyotrophic lateral sclerosis |
| BCR | B cell receptor |
| DUB | Deubiquitinating enzyme |
| E1 | Ubiquitin-activating enzyme |
| E2 | Ubiquitin-conjugating enzyme |
| E3 | Ubiquitin ligase |
| FTD | Frontotemporal dementia |
| GCB-DLBCL | Germinal center B cell-like diffuse large B cell lymphoma |
| HECT | Homologous to the E6AP carboxyl terminus |
| HOIL-1L | Heme-oxidized IRP2 ubiquitin ligase-1L |
| HOIP | HOIL-1L-interacting protein |
| IFN | Interferon |
| IKK | IκB kinase |
| IRF | Interferon regulatory factor |
| LDD | Linear ubiquitin chain determining domain |
| LLPS | Liquid-liquid phase separation |
| LSCC | Lung squamous cell carcinoma |
| LTM | LUBAC-tethering motif |
| LUBAC | Linear ubiquitin chain assembly complex |
| NCI | Neuronal cytoplasmic inclusion |
| NEMO | NF-κB-essential modulator |
| NF-κB | Nuclear factor-κB |
| NZF | Npl4-type zinc finger |
| OPTN | Optineurin |
| PAMPs | Pathogen-associated molecular patterns |
| PUB | PNGase/UBA or UBX |
| RBR | RING-in between-RING |
| RING | Really interesting new gene |
| SHARPIN | Shank-associated RH domain interactor |
| TBK1 | TANK-binding kinase 1 |
| TDP-43 | TAR DNA-binding protein 43 |
| TCR | T cell receptor |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| TNFR | TNF receptor |
| UBA | Ubiquitin-associated |
| UBAN | UBD in ABIN proteins and NEMO |
| UBL | Ubiquitin-like |
| ZF | Zinc finger |
References
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- McDowell, G.S.; Philpott, A. Non-canonical ubiquitylation: Mechanisms and consequences. Int. J. Biochem. Cell Biol. 2013, 45, 1833–1842. [Google Scholar] [CrossRef] [Green Version]
- Bhogaraju, S.; Kalayil, S.; Liu, Y.; Bonn, F.; Colby, T.; Matic, I.; Dikic, I. Phosphoribosylation of Ubiquitin Promotes Serine Ubiquitination and Impairs Conventional Ubiquitination. Cell 2016, 167, 1636–1649.e13. [Google Scholar] [CrossRef] [PubMed]
- Eisenhaber, B.; Chumak, N.; Eisenhaber, F.; Hauser, M.T. The ring between ring fingers (RBR) protein family. Genome Biol. 2007, 8, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokunaga, F. Linear ubiquitination-mediated NF-κB regulation and its related disorders. J. Biochem. 2013, 154, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Asaoka, T.; Almagro, J.; Ehrhardt, C.; Tsai, I.; Schleiffer, A.; Deszcz, L.; Junttila, S.; Ringrose, L.; Mechtler, K.; Kavirayani, A.; et al. Linear ubiquitination by LUBEL has a role in Drosophila heat stress response. EMBO Rep. 2016, 17, 1624–1640. [Google Scholar] [CrossRef]
- Yamanaka, K.; Ishikawa, H.; Megumi, Y.; Tokunaga, F.; Kanie, M.; Rouault, T.A.; Morishima, I.; Minato, N.; Ishimori, K.; Iwai, K. Identification of the ubiquitin-protein ligase that recognizes oxidized IRP2. Nat. Cell Biol. 2003, 5, 336–340. [Google Scholar] [CrossRef]
- Kirisako, T.; Kamei, K.; Murata, S.; Kato, M.; Fukumoto, H.; Kanie, M.; Sano, S.; Tokunaga, F.; Tanaka, K.; Iwai, K. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006, 25, 4877–4887. [Google Scholar] [CrossRef]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Nakagawa, T.; Nakahara, M.; Saeki, Y.; Taniguchi, M.; Sakata, S.; Tanaka, K.; Nakano, H.; Iwai, K. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature 2011, 471, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F.; Deribe, Y.L.; Skanland, S.S.; Stieglitz, B.; Grabbe, C.; Franz-Wachtel, M.; van Wijk, S.J.; Goswami, P.; Nagy, V.; Terzic, J.; et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 2011, 471, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Rittinger, K.; Ikeda, F. Linear ubiquitin chains: Enzymes, mechanisms and biology. Open Biol. 2017, 7, 170026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, K.; Fujita, H.; Sasaki, Y. Linear ubiquitin chains: NF-κB signalling, cell death and beyond. Nat. Rev. Mol. Cell Biol. 2014, 15, 503–508. [Google Scholar] [CrossRef]
- Fujita, H.; Tokunaga, A.; Shimizu, S.; Whiting, A.L.; Aguilar-Alonso, F.; Takagi, K.; Walinda, E.; Sasaki, Y.; Shimokawa, T.; Mizushima, T.; et al. Cooperative Domain Formation by Homologous Motifs in HOIL-1L and SHARPIN Plays A Crucial Role in LUBAC Stabilization. Cell Rep. 2018, 23, 1192–1204. [Google Scholar] [CrossRef]
- Wenzel, D.M.; Lissounov, A.; Brzovic, P.S.; Klevit, R.E. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 2011, 474, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Dove, K.K.; Stieglitz, B.; Duncan, E.D.; Rittinger, K.; Klevit, R.E. Molecular insights into RBR E3 ligase ubiquitin transfer mechanisms. EMBO Rep. 2016, 17, 1221–1235. [Google Scholar] [CrossRef] [Green Version]
- Stieglitz, B.; Morris-Davies, A.C.; Koliopoulos, M.G.; Christodoulou, E.; Rittinger, K. LUBAC synthesizes linear ubiquitin chains via a thioester intermediate. EMBO Rep. 2012, 13, 840–846. [Google Scholar] [CrossRef]
- Stieglitz, B.; Rana, R.R.; Koliopoulos, M.G.; Morris-Davies, A.C.; Schaeffer, V.; Christodoulou, E.; Howell, S.; Brown, N.R.; Dikic, I.; Rittinger, K. Structural basis for ligase-specific conjugation of linear ubiquitin chains by HOIP. Nature 2013, 503, 422–426. [Google Scholar] [CrossRef]
- Smit, J.J.; Monteferrario, D.; Noordermeer, S.M.; van Dijk, W.J.; van der Reijden, B.A.; Sixma, T.K. The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. Embo J. 2012, 31, 3833–3844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechtenberg, B.C.; Rajput, A.; Sanishvili, R.; Dobaczewska, M.K.; Ware, C.F.; Mace, P.D.; Riedl, S.J. Structure of a HOIP/E2~ubiquitin complex reveals RBR E3 ligase mechanism and regulation. Nature 2016, 529, 546–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelsall, I.R.; Zhang, J.; Knebel, A.; Arthur, J.S.C.; Cohen, P. The E3 ligase HOIL-1 catalyses ester bond formation between ubiquitin and components of the Myddosome in mammalian cells. Proc. Natl. Acad. Sci. USA 2019, 116, 13293–13298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, M.D.; Buchberger, A.; Bycroft, M. The PUB domain functions as a p97 binding module in human peptide N-glycanase. J. Biol. Chem. 2006, 281, 25502–25508. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, V.; Akutsu, M.; Olma, M.H.; Gomes, L.C.; Kawasaki, M.; Dikic, I. Binding of OTULIN to the PUB domain of HOIP controls NF-κB signaling. Mol. Cell 2014, 54, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.R.; Leske, D.; Hrdinka, M.; Bagola, K.; Fiil, B.K.; McLaughlin, S.H.; Wagstaff, J.; Volkmar, N.; Christianson, J.C.; Kessler, B.M.; et al. SPATA2 Links CYLD to LUBAC, Activates CYLD, and Controls LUBAC Signaling. Mol. Cell 2016, 63, 990–1005. [Google Scholar] [CrossRef] [Green Version]
- Kupka, S.; De Miguel, D.; Draber, P.; Martino, L.; Surinova, S.; Rittinger, K.; Walczak, H. SPATA2-Mediated Binding of CYLD to HOIP Enables CYLD Recruitment to Signaling Complexes. Cell Rep. 2016, 16, 2271–2280. [Google Scholar] [CrossRef] [Green Version]
- Schlicher, L.; Wissler, M.; Preiss, F.; Brauns-Schubert, P.; Jakob, C.; Dumit, V.; Borner, C.; Dengjel, J.; Maurer, U. SPATA2 promotes CYLD activity and regulates TNF-induced NF-κB signaling and cell death. EMBO Rep. 2016, 17, 1485–1497. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.A.; Satpathy, S.; Beli, P.; Choudhary, C. SPATA2 links CYLD to the TNF-alpha receptor signaling complex and modulates the receptor signaling outcomes. EMBO J. 2016, 35, 1868–1884. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Fujita, H.; Yoshikawa, A.; Yamashita, M.; Yamagata, A.; Kaiser, S.E.; Iwai, K.; Fukai, S. Specific recognition of linear ubiquitin chains by the Npl4 zinc finger (NZF) domain of the HOIL-1L subunit of the linear ubiquitin chain assembly complex. Proc. Natl. Acad. Sci. USA 2011, 108, 20520–20525. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Fujita, H.; Sasaki, Y.; Tsuruyama, T.; Fukuda, K.; Iwai, K. Differential Involvement of the Npl4 Zinc Finger Domains of SHARPIN and HOIL-1L in Linear Ubiquitin Chain Assembly Complex-Mediated Cell Death Protection. Mol. Cell. Biol. 2016, 36, 1569–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, H.; Rahighi, S.; Akita, M.; Kato, R.; Sasaki, Y.; Wakatsuki, S.; Iwai, K. Mechanism underlying IkappaB kinase activation mediated by the linear ubiquitin chain assembly complex. Mol. Cell. Biol. 2014, 34, 1322–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, Y.; Sano, S.; Nakahara, M.; Murata, S.; Kometani, K.; Aiba, Y.; Sakamoto, S.; Watanabe, Y.; Tanaka, K.; Kurosaki, T.; et al. Defective immune responses in mice lacking LUBAC-mediated linear ubiquitination in B cells. Embo J. 2013, 32, 2463–2476. [Google Scholar] [CrossRef] [Green Version]
- de Almagro, M.C.; Goncharov, T.; Newton, K.; Vucic, D. Cellular IAP proteins and LUBAC differentially regulate necrosome-associated RIP1 ubiquitination. Cell Death Dis. 2015, 6, e1800. [Google Scholar] [CrossRef]
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef]
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat. Cell Biol. 2009, 11, 123–132. [Google Scholar] [CrossRef]
- Goto, E.; Tokunaga, F. Decreased linear ubiquitination of NEMO and FADD on apoptosis with caspase-mediated cleavage of HOIP. Biochem. Biophys. Res. Commun. 2017, 485, 152–159. [Google Scholar] [CrossRef]
- Kensche, T.; Tokunaga, F.; Ikeda, F.; Goto, E.; Iwai, K.; Dikic, I. Analysis of nuclear factor-kappaB (NF-κB) essential modulator (NEMO) binding to linear and lysine-linked ubiquitin chains and its role in the activation of NF-κB. J. Biol. Chem. 2012, 287, 23626–23634. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.Y.; Lim, J.M.; Cho, H.; Kim, E.; Kim, Y.; Oh, H.K.; Yang, W.S.; Roh, K.H.; Park, H.W.; Mo, J.S.; et al. MST1 Negatively Regulates TNFalpha-Induced NF-κB Signaling through Modulating LUBAC Activity. Mol. Cell 2019, 73, 1138–1149. [Google Scholar] [CrossRef] [Green Version]
- Meschede, J.; Sadic, M.; Furthmann, N.; Miedema, T.; Sehr, D.A.; Muller-Rischart, A.K.; Bader, V.; Berlemann, L.A.; Pilsl, A.; Schlierf, A.; et al. The parkin-coregulated gene product PACRG promotes TNF signaling by stabilizing LUBAC. Sci. Signal. 2020, 13, eaav1256. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, N.; Tinevez, J.Y.; Crowell, E.F.; Boisson, B.; Henriques, R.; Mhlanga, M.; Agou, F.; Israel, A.; Laplantine, E. TNF and IL-1 exhibit distinct ubiquitin requirements for inducing NEMO-IKK supramolecular structures. J. Cell Biol. 2014, 204, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, C.H.; Ordureau, A.; Strickson, S.; Arthur, J.S.; Pedrioli, P.G.; Komander, D.; Cohen, P. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc. Natl. Acad. Sci. USA 2013, 110, 15247–15252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meininger, I.; Krappmann, D. Lymphocyte signaling and activation by the CARMA1-BCL10-MALT1 signalosome. Biol. Chem. 2016, 397, 1315–1333. [Google Scholar] [CrossRef]
- Teh, C.E.; Lalaoui, N.; Jain, R.; Policheni, A.N.; Heinlein, M.; Alvarez-Diaz, S.; Sheridan, J.M.; Rieser, E.; Deuser, S.; Darding, M.; et al. Linear ubiquitin chain assembly complex coordinates late thymic T-cell differentiation and regulatory T-cell homeostasis. Nat. Commun. 2016, 7, 13353. [Google Scholar] [CrossRef]
- Klein, T.; Fung, S.Y.; Renner, F.; Blank, M.A.; Dufour, A.; Kang, S.; Bolger-Munro, M.; Scurll, J.M.; Priatel, J.J.; Schweigler, P.; et al. The paracaspase MALT1 cleaves HOIL1 reducing linear ubiquitination by LUBAC to dampen lymphocyte NF-κB signalling. Nat. Commun. 2015, 6, 8777. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.K.; Yang, C.; Chan, W.; Wang, Z.; Deibel, K.E.; Pomerantz, J.L. Molecular Determinants of Scaffold-induced Linear Ubiquitinylation of B Cell Lymphoma/Leukemia 10 (Bcl10) during T Cell Receptor and Oncogenic Caspase Recruitment Domain-containing Protein 11 (CARD11) Signaling. J. Biol. Chem. 2016, 291, 25921–25936. [Google Scholar] [CrossRef] [Green Version]
- Dubois, S.M.; Alexia, C.; Wu, Y.; Leclair, H.M.; Leveau, C.; Schol, E.; Fest, T.; Tarte, K.; Chen, Z.J.; Gavard, J.; et al. A catalytic-independent role for the LUBAC in NF-κB activation upon antigen receptor engagement and in lymphoma cells. Blood 2014, 123, 2199–2203. [Google Scholar] [CrossRef]
- McCool, K.W.; Miyamoto, S. DNA damage-dependent NF-κB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326. [Google Scholar] [CrossRef]
- Niu, J.; Shi, Y.; Iwai, K.; Wu, Z.H. LUBAC regulates NF-κB activation upon genotoxic stress by promoting linear ubiquitination of NEMO. Embo J. 2011, 30, 3741–3753. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, R.B.; Nachbur, U.; Yabal, M.; Wong, W.W.; Fiil, B.K.; Kastirr, M.; Rieser, E.; Rickard, J.A.; Bankovacki, A.; Peschel, C.; et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol. Cell 2012, 46, 746–758. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Castejon, G. Control of the inflammasome by the ubiquitin system. FEBS J. 2020, 287, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, M.A.; Bowman, J.W.; Fujita, H.; Orazio, N.; Shi, M.; Liang, Q.; Amatya, R.; Kelly, T.J.; Iwai, K.; Ting, J.; et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J. Exp. Med. 2014, 211, 1333–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walczak, H. TNF and ubiquitin at the crossroads of gene activation, cell death, inflammation, and cancer. Immunol. Rev. 2011, 244, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Joo, D.; Tang, Y.; Blonska, M.; Jin, J.; Zhao, X.; Lin, X. Regulation of Linear Ubiquitin Chain Assembly Complex by Caspase-Mediated Cleavage of RNF31. Mol. Cell. Biol. 2016, 36, 3010–3018. [Google Scholar] [CrossRef] [Green Version]
- Peltzer, N.; Rieser, E.; Taraborrelli, L.; Draber, P.; Darding, M.; Pernaute, B.; Shimizu, Y.; Sarr, A.; Draberova, H.; Montinaro, A.; et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. 2014, 9, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Gijbels, M.J.; Zurcher, C.; Kraal, G.; Elliott, G.R.; HogenEsch, H.; Schijff, G.; Savelkoul, H.F.; Bruijnzeel, P.L. Pathogenesis of skin lesions in mice with chronic proliferative dermatitis (cpdm/cpdm). Am. J. Pathol. 1996, 148, 941–950. [Google Scholar]
- HogenEsch, H.; Janke, S.; Boggess, D.; Sundberg, J.P. Absence of Peyer’s patches and abnormal lymphoid architecture in chronic proliferative dermatitis (cpdm/cpdm) mice. J. Immunol. 1999, 162, 3890–3896. [Google Scholar]
- Seymour, R.E.; Hasham, M.G.; Cox, G.A.; Shultz, L.D.; Hogenesch, H.; Roopenian, D.C.; Sundberg, J.P. Spontaneous mutations in the mouse Sharpin gene result in multiorgan inflammation, immune system dysregulation and dermatitis. Genes Immun. 2007, 8, 416–421. [Google Scholar] [CrossRef] [Green Version]
- Kumari, S.; Redouane, Y.; Lopez-Mosqueda, J.; Shiraishi, R.; Romanowska, M.; Lutzmayer, S.; Kuiper, J.; Martinez, C.; Dikic, I.; Pasparakis, M.; et al. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. eLife 2014, 3, e03422. [Google Scholar] [CrossRef]
- Rickard, J.A.; Anderton, H.; Etemadi, N.; Nachbur, U.; Darding, M.; Peltzer, N.; Lalaoui, N.; Lawlor, K.E.; Vanyai, H.; Hall, C.; et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. eLife 2014, 3, e03464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, B.R.; Karki, R.; Lee, E.; Zhu, Q.; Gurung, P.; Kanneganti, T.D. Innate immune adaptor MyD88 deficiency prevents skin inflammation in SHARPIN-deficient mice. Cell Death Differ. 2019, 26, 741–750. [Google Scholar] [CrossRef]
- Lee, M.S.; Kim, Y.J. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu. Rev. Biochem. 2007, 76, 447–480. [Google Scholar] [CrossRef]
- Davis, M.E.; Gack, M.U. Ubiquitination in the antiviral immune response. Virology 2015, 479–480, 52–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inn, K.S.; Gack, M.U.; Tokunaga, F.; Shi, M.; Wong, L.Y.; Iwai, K.; Jung, J.U. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol. Cell 2011, 41, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belgnaoui, S.M.; Paz, S.; Samuel, S.; Goulet, M.L.; Sun, Q.; Kikkert, M.; Iwai, K.; Dikic, I.; Hiscott, J.; Lin, R. Linear ubiquitination of NEMO negatively regulates the interferon antiviral response through disruption of the MAVS-TRAF3 complex. Cell Host Microbe 2012, 12, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Y.; Feng, Q.; Jin, L.; Huang, F.; Miao, Y.; Liu, J.; Xu, Y.; Chen, X.; Zhang, H.; Guo, T.; et al. Regulation of the linear ubiquitination of STAT1 controls antiviral interferon signaling. Nat. Commun. 2020, 11, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinngrebe, J.; Rieser, E.; Taraborrelli, L.; Peltzer, N.; Hartwig, T.; Ren, H.; Kovacs, I.; Endres, C.; Draber, P.; Darding, M.; et al. LUBAC deficiency perturbs TLR3 signaling to cause immunodeficiency and autoinflammation. J. Exp. Med. 2016, 213, 2671–2689. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Hepatitis B Virus-Induced Parkin-Dependent Recruitment of Linear Ubiquitin Assembly Complex (LUBAC) to Mitochondria and Attenuation of Innate Immunity. PLoS Pathog. 2016, 12, e1005693. [Google Scholar] [CrossRef]
- MacDuff, D.A.; Baldridge, M.T.; Qaqish, A.M.; Nice, T.J.; Darbandi, A.D.; Hartley, V.L.; Peterson, S.T.; Miner, J.J.; Iwai, K.; Virgin, H.W. HOIL1 Is Essential for the Induction of Type I and III Interferons by MDA5 and Regulates Persistent Murine Norovirus Infection. J. Virol. 2018, 92, e01368-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafont, E.; Draber, P.; Rieser, E.; Reichert, M.; Kupka, S.; de Miguel, D.; Draberova, H.; von Massenhausen, A.; Bhamra, A.; Henderson, S.; et al. TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol. 2018, 20, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, D.; Sato, Y.; Ohtake, F.; Komakura, K.; Hanada, K.; Sugawara, K.; Terawaki, S.; Mizukami, Y.; Phuong, H.T.; Iio, K.; et al. Molecular bases for HOIPINs-mediated inhibition of LUBAC and innate immune responses. Commun. Biol. 2020, 3, 163. [Google Scholar] [CrossRef] [PubMed]
- Morishita, H.; Mizushima, N. Diverse Cellular Roles of Autophagy. Annu. Rev. Cell Dev. Biol. 2019, 35, 453–475. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Morita, E.; Itoh, T.; Tanaka, A.; Nakaoka, M.; Osada, Y.; Umemoto, T.; Saitoh, T.; Nakatogawa, H.; Kobayashi, S.; et al. Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J. Cell Biol. 2013, 203, 115–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiskin, E.; Bionda, T.; Dikic, I.; Behrends, C. Global Analysis of Host and Bacterial Ubiquitinome in Response to Salmonella Typhimurium Infection. Mol. Cell 2016, 62, 967–981. [Google Scholar] [CrossRef] [Green Version]
- Noad, J.; von der Malsburg, A.; Pathe, C.; Michel, M.A.; Komander, D.; Randow, F. LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-κB. Nat. Microbiol. 2017, 2, 17063. [Google Scholar] [CrossRef] [Green Version]
- Polajnar, M.; Dietz, M.S.; Heilemann, M.; Behrends, C. Expanding the host cell ubiquitylation machinery targeting cytosolic Salmonella. EMBO Rep. 2017, 18, 1572–1585. [Google Scholar] [CrossRef]
- van Wijk, S.J.L.; Fricke, F.; Herhaus, L.; Gupta, J.; Hotte, K.; Pampaloni, F.; Grumati, P.; Kaulich, M.; Sou, Y.S.; Komatsu, M.; et al. Linear ubiquitination of cytosolic Salmonella Typhimurium activates NF-κB and restricts bacterial proliferation. Nat. Microbiol. 2017, 2, 17066. [Google Scholar] [CrossRef]
- Komander, D.; Clague, M.J.; Urbe, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul Rehman, S.A.; Kristariyanto, Y.A.; Choi, S.Y.; Nkosi, P.J.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol. Cell 2016, 63, 146–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haahr, P.; Borgermann, N.; Guo, X.; Typas, D.; Achuthankutty, D.; Hoffmann, S.; Shearer, R.; Sixma, T.K.; Mailand, N. ZUFSP Deubiquitylates K63-Linked Polyubiquitin Chains to Promote Genome Stability. Mol. Cell 2018, 70, 165–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lork, M.; Verhelst, K.; Beyaert, R. CYLD, A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: So similar, yet so different. Cell Death Differ. 2017, 24, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Keusekotten, K.; Elliott, P.R.; Glockner, L.; Fiil, B.K.; Damgaard, R.B.; Kulathu, Y.; Wauer, T.; Hospenthal, M.K.; Gyrd-Hansen, M.; Krappmann, D.; et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 2013, 153, 1312–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivkin, E.; Almeida, S.M.; Ceccarelli, D.F.; Juang, Y.C.; MacLean, T.A.; Srikumar, T.; Huang, H.; Dunham, W.H.; Fukumura, R.; Xie, G.; et al. The linear ubiquitin-specific deubiquitinase gumby regulates angiogenesis. Nature 2013, 498, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Fiil, B.K.; Damgaard, R.B.; Wagner, S.A.; Keusekotten, K.; Fritsch, M.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C.; Komander, D.; Gyrd-Hansen, M. OTULIN restricts Met1-linked ubiquitination to control innate immune signaling. Mol. Cell 2013, 50, 818–830. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.R.; Nielsen, S.V.; Marco-Casanova, P.; Fiil, B.K.; Keusekotten, K.; Mailand, N.; Freund, S.M.; Gyrd-Hansen, M.; Komander, D. Molecular basis and regulation of OTULIN-LUBAC interaction. Mol. Cell 2014, 54, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Takiuchi, T.; Nakagawa, T.; Tamiya, H.; Fujita, H.; Sasaki, Y.; Saeki, Y.; Takeda, H.; Sawasaki, T.; Buchberger, A.; Kimura, T.; et al. Suppression of LUBAC-mediated linear ubiquitination by a specific interaction between LUBAC and the deubiquitinases CYLD and OTULIN. Genes Cells 2014, 19, 254–272. [Google Scholar] [CrossRef] [Green Version]
- Damgaard, R.B.; Walker, J.A.; Marco-Casanova, P.; Morgan, N.V.; Titheradge, H.L.; Elliott, P.R.; McHale, D.; Maher, E.R.; McKenzie, A.N.J.; Komander, D. The Deubiquitinase OTULIN Is an Essential Negative Regulator of Inflammation and Autoimmunity. Cell 2016, 166, 1215–1230. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Yu, X.; Demirkaya, E.; Deuitch, N.; Stone, D.; Tsai, W.L.; Kuehn, H.S.; Wang, H.; Yang, D.; Park, Y.H.; et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl. Acad. Sci. USA 2016, 113, 10127–10132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabavi, M.; Shahrooei, M.; Rokni-Zadeh, H.; Vrancken, J.; Changi-Ashtiani, M.; Darabi, K.; Manian, M.; Seif, F.; Meyts, I.; Voet, A.; et al. Auto-inflammation in a Patient with a Novel Homozygous OTULIN Mutation. J. Clin. Immunol. 2019, 39, 138–141. [Google Scholar] [CrossRef]
- Damgaard, R.B.; Elliott, P.R.; Swatek, K.N.; Maher, E.R.; Stepensky, P.; Elpeleg, O.; Komander, D.; Berkun, Y. OTULIN deficiency in ORAS causes cell type-specific LUBAC degradation, dysregulated TNF signalling and cell death. EMBO Mol. Med. 2019, 11, e9324. [Google Scholar] [CrossRef] [PubMed]
- Heger, K.; Wickliffe, K.E.; Ndoja, A.; Zhang, J.; Murthy, A.; Dugger, D.L.; Maltzman, A.; de Sousa, E.M.F.; Hung, J.; Zeng, Y.; et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 2018, 559, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Douglas, T.; Saleh, M. Post-translational Modification of OTULIN Regulates Ubiquitin Dynamics and Cell Death. Cell Rep. 2019, 29, 3652–3663.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verboom, L.; Martens, A.; Priem, D.; Hoste, E.; Sze, M.; Vikkula, H.; Van Hove, L.; Voet, S.; Roels, J.; Maelfait, J.; et al. OTULIN Prevents Liver Inflammation and Hepatocellular Carcinoma by Inhibiting FADD- and RIPK1 Kinase-Mediated Hepatocyte Apoptosis. Cell Rep. 2020, 30, 2237–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Song, K.; Hao, W.; Wang, L.; Patil, G.; Li, Q.; Xu, L.; Hua, F.; Fu, B.; Schwamborn, J.C.; et al. Non-proteolytic ubiquitination of OTULIN regulates NF-κB signaling pathway. J. Mol. Cell Biol. 2020, 12, 163–175. [Google Scholar] [CrossRef]
- Stangl, A.; Elliott, P.R.; Pinto-Fernandez, A.; Bonham, S.; Harrison, L.; Schaub, A.; Kutzner, K.; Keusekotten, K.; Pfluger, P.T.; El Oualid, F.; et al. Regulation of the endosomal SNX27-retromer by OTULIN. Nat. Commun. 2019, 10, 4320. [Google Scholar] [CrossRef]
- Bignell, G.R.; Warren, W.; Seal, S.; Takahashi, M.; Rapley, E.; Barfoot, R.; Green, H.; Brown, C.; Biggs, P.J.; Lakhani, S.R.; et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 2000, 25, 160–165. [Google Scholar] [CrossRef]
- Regamey, A.; Hohl, D.; Liu, J.W.; Roger, T.; Kogerman, P.; Toftgard, R.; Huber, M. The tumor suppressor CYLD interacts with TRIP and regulates negatively nuclear factor kappaB activation by tumor necrosis factor. J. Exp. Med. 2003, 198, 1959–1964. [Google Scholar] [CrossRef] [Green Version]
- Trompouki, E.; Hatzivassiliou, E.; Tsichritzis, T.; Farmer, H.; Ashworth, A.; Mosialos, G. CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature 2003, 424, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Reyes-Turcu, F.; Licchesi, J.D.; Odenwaelder, P.; Wilkinson, K.D.; Barford, D. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 2009, 10, 466–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrdinka, M.; Fiil, B.K.; Zucca, M.; Leske, D.; Bagola, K.; Yabal, M.; Elliott, P.R.; Damgaard, R.B.; Komander, D.; Jost, P.J.; et al. CYLD Limits Lys63- and Met1-Linked Ubiquitin at Receptor Complexes to Regulate Innate Immune Signaling. Cell Rep. 2016, 14, 2846–2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokunaga, F.; Nishimasu, H.; Ishitani, R.; Goto, E.; Noguchi, T.; Mio, K.; Kamei, K.; Ma, A.; Iwai, K.; Nureki, O. Specific recognition of linear polyubiquitin by A20 zinc finger 7 is involved in NF-κB regulation. Embo J. 2012, 31, 3856–3870. [Google Scholar] [CrossRef]
- Draber, P.; Kupka, S.; Reichert, M.; Draberova, H.; Lafont, E.; de Miguel, D.; Spilgies, L.; Surinova, S.; Taraborrelli, L.; Hartwig, T.; et al. LUBAC-Recruited CYLD and A20 Regulate Gene Activation and Cell Death by Exerting Opposing Effects on Linear Ubiquitin in Signaling Complexes. Cell Rep. 2015, 13, 2258–2272. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Goto, E.; Shibata, Y.; Kubota, Y.; Yamagata, A.; Goto-Ito, S.; Kubota, K.; Inoue, J.; Takekawa, M.; Tokunaga, F.; et al. Structures of CYLD USP with Met1- or Lys63-linked diubiquitin reveal mechanisms for dual specificity. Nat. Struct. Mol. Biol. 2015, 22, 222–229. [Google Scholar] [CrossRef]
- Yamanaka, S.; Sato, Y.; Oikawa, D.; Goto, E.; Fukai, S.; Tokunaga, F.; Takahashi, H.; Sawasaki, T. Subquinocin, a small molecule inhibitor of CYLD and USP-family deubiquitinating enzymes, promotes NF-κB signaling. Biochem. Biophys. Res. Commun. 2020, 524, 1–7. [Google Scholar] [CrossRef]
- Uematsu, A.; Kido, K.; Takahashi, H.; Takahashi, C.; Yanagihara, Y.; Saeki, N.; Yoshida, S.; Maekawa, M.; Honda, M.; Kai, T.; et al. The E3 ubiquitin ligase MIB2 enhances inflammation by degrading the deubiquitinating enzyme CYLD. J. Biol. Chem. 2019, 294, 14135–14148. [Google Scholar] [CrossRef]
- Dobson-Stone, C.; Hallupp, M.; Shahheydari, H.; Ragagnin, A.M.G.; Chatterton, Z.; Carew-Jones, F.; Shepherd, C.E.; Stefen, H.; Paric, E.; Fath, T.; et al. CYLD is a causative gene for frontotemporal dementia—Amyotrophic lateral sclerosis. Brain 2020, 143, 783–799. [Google Scholar] [CrossRef]
- Wagner, S.; Carpentier, I.; Rogov, V.; Kreike, M.; Ikeda, F.; Lohr, F.; Wu, C.J.; Ashwell, J.D.; Dotsch, V.; Dikic, I.; et al. Ubiquitin binding mediates the NF-κB inhibitory potential of ABIN proteins. Oncogene 2008, 27, 3739–3745. [Google Scholar] [CrossRef] [Green Version]
- Verhelst, K.; Carpentier, I.; Kreike, M.; Meloni, L.; Verstrepen, L.; Kensche, T.; Dikic, I.; Beyaert, R. A20 inhibits LUBAC-mediated NF-κB activation by binding linear polyubiquitin chains via its zinc finger 7. Embo J. 2012, 31, 3845–3855. [Google Scholar] [CrossRef] [PubMed]
- Fennell, L.M.; Rahighi, S.; Ikeda, F. Linear ubiquitin chain-binding domains. FEBS J. 2018, 285, 2746–2761. [Google Scholar] [CrossRef] [Green Version]
- Rahighi, S.; Ikeda, F.; Kawasaki, M.; Akutsu, M.; Suzuki, N.; Kato, R.; Kensche, T.; Uejima, T.; Bloor, S.; Komander, D.; et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell 2009, 136, 1098–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, Y.C.; Lin, S.C.; Rospigliosi, C.C.; Conze, D.B.; Wu, C.J.; Ashwell, J.D.; Eliezer, D.; Wu, H. Structural basis for recognition of diubiquitins by NEMO. Mol. Cell 2009, 33, 602–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doffinger, R.; Smahi, A.; Bessia, C.; Geissmann, F.; Feinberg, J.; Durandy, A.; Bodemer, C.; Kenwrick, S.; Dupuis-Girod, S.; Blanche, S.; et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-κB signaling. Nat. Genet. 2001, 27, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Hubeau, M.; Ngadjeua, F.; Puel, A.; Israel, L.; Feinberg, J.; Chrabieh, M.; Belani, K.; Bodemer, C.; Fabre, I.; Plebani, A.; et al. New mechanism of X-linked anhidrotic ectodermal dysplasia with immunodeficiency: Impairment of ubiquitin binding despite normal folding of NEMO protein. Blood 2011, 118, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Rezaie, T.; Child, A.; Hitchings, R.; Brice, G.; Miller, L.; Coca-Prados, M.; Heon, E.; Krupin, T.; Ritch, R.; Kreutzer, D.; et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science 2002, 295, 1077–1079. [Google Scholar] [CrossRef]
- Ying, H.; Yue, B.Y. Cellular and molecular biology of optineurin. Int. Rev. Cell Mol. Biol. 2012, 294, 223–258. [Google Scholar]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Ito, H.; Nakamura, M.; Komure, O.; Ayaki, T.; Wate, R.; Maruyama, H.; Nakamura, Y.; Fujita, K.; Kaneko, S.; Okamoto, Y.; et al. Clinicopathologic study on an ALS family with a heterozygous E478G optineurin mutation. Acta Neuropathol. 2011, 122, 223–229. [Google Scholar] [CrossRef]
- Nakazawa, S.; Oikawa, D.; Ishii, R.; Ayaki, T.; Takahashi, H.; Takeda, H.; Ishitani, R.; Kamei, K.; Takeyoshi, I.; Kawakami, H.; et al. Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat. Commun. 2016, 7, 12547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstrepen, L.; Carpentier, I.; Beyaert, R. The biology of A20-binding inhibitors of NF-κB activation (ABINs). Adv. Exp. Med. Biol. 2014, 809, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.M.; Lin, S.C.; Hong, J.Y.; Su, T.W.; Kuo, B.J.; Chang, W.H.; Tu, Y.F.; Lo, Y.C. Structural Insights into Linear Tri-ubiquitin Recognition by A20-Binding Inhibitor of NF-κB, ABIN-2. Structure 2017, 25, 66–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herhaus, L.; van den Bedem, H.; Tang, S.; Maslennikov, I.; Wakatsuki, S.; Dikic, I.; Rahighi, S. Molecular Recognition of M1-Linked Ubiquitin Chains by Native and Phosphorylated UBAN Domains. J. Mol. Biol. 2019, 431, 3146–3156. [Google Scholar] [CrossRef] [Green Version]
- Ma, A.; Malynn, B.A. A20: Linking a complex regulator of ubiquitylation to immunity and human disease. Nat. Rev. Immunol. 2012, 12, 774–785. [Google Scholar] [CrossRef] [Green Version]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef]
- Garcia-Carbonell, R.; Wong, J.; Kim, J.Y.; Close, L.A.; Boland, B.S.; Wong, T.L.; Harris, P.A.; Ho, S.B.; Das, S.; Ernst, P.B.; et al. Elevated A20 promotes TNF-induced and RIPK1-dependent intestinal epithelial cell death. Proc. Natl. Acad. Sci. USA 2018, 115, E9192–E9200. [Google Scholar] [CrossRef] [Green Version]
- Martens, A.; Priem, D.; Hoste, E.; Vetters, J.; Rennen, S.; Catrysse, L.; Voet, S.; Deelen, L.; Sze, M.; Vikkula, H.; et al. Two distinct ubiquitin-binding motifs in A20 mediate its anti-inflammatory and cell-protective activities. Nat. Immunol. 2020, 21, 381–387. [Google Scholar] [CrossRef]
- Razani, B.; Whang, M.I.; Kim, F.S.; Nakamura, M.C.; Sun, X.; Advincula, R.; Turnbaugh, J.A.; Pendse, M.; Tanbun, P.; Achacoso, P.; et al. Non-catalytic ubiquitin binding by A20 prevents psoriatic arthritis-like disease and inflammation. Nat. Immunol. 2020, 21, 422–433. [Google Scholar] [CrossRef]
- Aksentijevich, I.; Zhou, Q. NF-κB Pathway in Autoinflammatory Diseases: Dysregulation of Protein Modifications by Ubiquitin Defines a New Category of Autoinflammatory Diseases. Front. Immunol. 2017, 8, 399. [Google Scholar] [CrossRef] [Green Version]
- Peltzer, N.; Darding, M.; Montinaro, A.; Draber, P.; Draberova, H.; Kupka, S.; Rieser, E.; Fisher, A.; Hutchinson, C.; Taraborrelli, L.; et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature 2018, 557, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Boisson, B.; Laplantine, E.; Prando, C.; Giliani, S.; Israelsson, E.; Xu, Z.; Abhyankar, A.; Israel, L.; Trevejo-Nunez, G.; Bogunovic, D.; et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 2012, 13, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Kim, C.; Bradfield, J.; Guo, Y.; Toskala, E.; Otieno, F.G.; Hou, C.; Thomas, K.; Cardinale, C.; Lyon, G.J.; et al. Whole-genome DNA/RNA sequencing identifies truncating mutations in RBCK1 in a novel Mendelian disease with neuromuscular and cardiac involvement. Genome Med. 2013, 5, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, J.; Schoser, B.; Laforet, P.; Kalev, O.; Lindberg, C.; Romero, N.B.; Davila Lopez, M.; Akman, H.O.; Wahbi, K.; Iglseder, S.; et al. Polyglucosan body myopathy caused by defective ubiquitin ligase RBCK1. Ann. Neurol. 2013, 74, 914–919. [Google Scholar] [CrossRef]
- Boisson, B.; Laplantine, E.; Dobbs, K.; Cobat, A.; Tarantino, N.; Hazen, M.; Lidov, H.G.; Hopkins, G.; Du, L.; Belkadi, A.; et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med. 2015, 212, 939–951. [Google Scholar] [CrossRef] [Green Version]
- Oda, H.; Beck, D.B.; Kuehn, H.S.; Sampaio Moura, N.; Hoffmann, P.; Ibarra, M.; Stoddard, J.; Tsai, W.L.; Gutierrez-Cruz, G.; Gadina, M.; et al. Second Case of HOIP Deficiency Expands Clinical Features and Defines Inflammatory Transcriptome Regulated by LUBAC. Front. Immunol. 2019, 10, 479. [Google Scholar] [CrossRef] [Green Version]
- Pasqualucci, L.; Zhang, B. Genetic drivers of NF-κB deregulation in diffuse large B-cell lymphoma. Semin. Cancer Biol. 2016, 39, 26–31. [Google Scholar] [CrossRef]
- Yang, Y.; Schmitz, R.; Mitala, J.; Whiting, A.; Xiao, W.; Ceribelli, M.; Wright, G.W.; Zhao, H.; Yang, Y.; Xu, W.; et al. Essential role of the linear ubiquitin chain assembly complex in lymphoma revealed by rare germline polymorphisms. Cancer Discov. 2014, 4, 480–493. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Kelly, P.; Shaffer, A.L., III; Schmitz, R.; Yoo, H.M.; Liu, X.; Huang, D.W.; Webster, D.; Young, R.M.; Nakagawa, M.; et al. Targeting Non-proteolytic Protein Ubiquitination for the Treatment of Diffuse Large B Cell Lymphoma. Cancer Cell 2016, 29, 494–507. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, E.J.; Diefenbacher, M.E.; Nelson, J.K.; Sancho, R.; Pucci, F.; Chakraborty, A.; Moreno, P.; Annibaldi, A.; Liccardi, G.; Encheva, V.; et al. LUBAC determines chemotherapy resistance in squamous cell lung cancer. J. Exp. Med. 2019, 216, 450–465. [Google Scholar] [CrossRef] [Green Version]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar] [CrossRef] [PubMed]
- Winklhofer, K.F.; Tatzelt, J.; Haass, C. The two faces of protein misfolding: Gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008, 27, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, Y.; Tsuji, K.; Ayaki, T.; Mori, M.; Tokunaga, F.; Ito, H. Linear Polyubiquitin Chain Modification of TDP-43-Positive Neuronal Cytoplasmic Inclusions in Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2020, 79, 256–265. [Google Scholar] [CrossRef]
- Nakayama, Y.; Sakamoto, S.; Tsuji, K.; Ayaki, T.; Tokunaga, F.; Ito, H. Identification of linear polyubiquitin chain immunoreactivity in tau pathology of Alzheimer’s disease. Neurosci. Lett. 2019, 703, 53–57. [Google Scholar] [CrossRef]
- van Well, E.M.; Bader, V.; Patra, M.; Sanchez-Vicente, A.; Meschede, J.; Furthmann, N.; Schnack, C.; Blusch, A.; Longworth, J.; Petrasch-Parwez, E.; et al. A protein quality control pathway regulated by linear ubiquitination. EMBO J. 2019, 38, e100730. [Google Scholar] [CrossRef]
- Strickson, S.; Campbell, D.G.; Emmerich, C.H.; Knebel, A.; Plater, L.; Ritorto, M.S.; Shpiro, N.; Cohen, P. The anti-inflammatory drug BAY 11-7082 suppresses the MyD88-dependent signalling network by targeting the ubiquitin system. Biochem. J. 2013, 451, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, H.; Egashira, S.; Saito, N.; Kirisako, T.; Miller, S.; Sasaki, Y.; Matsumoto, T.; Shimonishi, M.; Komatsu, T.; Terai, T.; et al. Gliotoxin suppresses NF-κB activation by selectively inhibiting linear ubiquitin chain assembly complex (LUBAC). ACS Chem. Biol. 2015, 10, 675–681. [Google Scholar] [CrossRef]
- De Cesare, V.; Johnson, C.; Barlow, V.; Hastie, J.; Knebel, A.; Trost, M. The MALDI-TOF E2/E3 Ligase Assay as Universal Tool for Drug Discovery in the Ubiquitin Pathway. Cell Chem. Biol. 2018, 25, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Johansson, H.; Isabella Tsai, Y.C.; Fantom, K.; Chung, C.W.; Kumper, S.; Martino, L.; Thomas, D.A.; Eberl, H.C.; Muelbaier, M.; House, D.; et al. Fragment-Based Covalent Ligand Screening Enables Rapid Discovery of Inhibitors for the RBR E3 Ubiquitin Ligase HOIP. J. Am. Chem. Soc. 2019, 141, 2703–2712. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.I.; Johansson, H.; Dixon, D.; Martin, S.; Chung, C.W.; Clarkson, J.; House, D.; Rittinger, K. Single-Domain Antibodies as Crystallization Chaperones to Enable Structure-Based Inhibitor Development for RBR E3 Ubiquitin Ligases. Cell Chem. Biol. 2020, 27, 83–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuya, K.; Hori, Y.; Oikawa, D.; Yamamoto, T.; Umetani, K.; Urashima, T.; Kinoshita, T.; Ayukawa, K.; Tokunaga, F.; Tamaru, M. High-Throughput Screening for Linear Ubiquitin Chain Assembly Complex (LUBAC) Selective Inhibitors Using Homogenous Time-Resolved Fluorescence (HTRF)-Based Assay System. SLAS Discov. 2018, 23, 1018–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuya, K.; Oikawa, D.; Iio, K.; Obika, S.; Hori, Y.; Urashima, T.; Ayukawa, K.; Tokunaga, F. Small-molecule inhibitors of linear ubiquitin chain assembly complex (LUBAC), HOIPINs, suppress NF-κB signaling. Biochem. Biophys. Res. Commun. 2019, 509, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Harden, J.L.; Krueger, J.G.; Bowcock, A.M. The immunogenetics of Psoriasis: A comprehensive review. J. Autoimmun. 2015, 64, 66–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greb, J.E.; Goldminz, A.M.; Elder, J.T.; Lebwohl, M.G.; Gladman, D.D.; Wu, J.J.; Mehta, N.N.; Finlay, A.Y.; Gottlieb, A.B. Psoriasis. Nat. Rev. Dis. Primers 2016, 2, 16082. [Google Scholar] [CrossRef]
- Yu, K.; Phu, L.; Varfolomeev, E.; Bustos, D.; Vucic, D.; Kirkpatrick, D.S. Immunoaffinity enrichment coupled to quantitative mass spectrometry reveals ubiquitin-mediated signaling events. J. Mol. Biol. 2015, 427, 2121–2134. [Google Scholar] [CrossRef]
- Morimoto, D.; Walinda, E.; Fukada, H.; Sou, Y.S.; Kageyama, S.; Hoshino, M.; Fujii, T.; Tsuchiya, H.; Saeki, Y.; Arita, K.; et al. The unexpected role of polyubiquitin chains in the formation of fibrillar aggregates. Nat. Commun. 2015, 6, 6116. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Mutations | Symptoms | References |
|---|---|---|---|
| HOIL-1L | p.Q185X (c.553C > T), p.L41fsX7 (c.121_122delCT), c.ex1_ex4del | polyglucosan myopathy with immunodeficiency, sepsis, chronic autoinflammation | [132] |
| p.Q222X (c.90C > T) p.E190fs (c.68_69insAGGAGCG) c.456+1G > C | polyglucosan body myopathy, cardiomyopathy, muscle atrophy | [133] | |
| p.E243X (c.727G > T), p.N387S (c.1160A > G), p.E299VfsX18 (c.896_899delAGTG), p.A241GfsX34 (c.722delC), p.A18P (c.52G > C), p.E243GfsX114 (c.727_728ins GGCG), c.ex1_ex4del p.R352X (c.1054C > T) p.R298RfsX40 (c.917+3_917+4insG), p.R165RfsX111 (c.494delG) | polyglucosan myopathy without immunodeficiency | [134] | |
| HOIP | p.L72P (c.215T > C) | multiorgan autoinflammation with immunodeficiency, amylopectinosis, systemic lymphangiectasia | [135] |
| p.Q399H (c.1197G > C) c.1737+3A > G | eczematous dermatitis, splenomegaly, clubbing of fingers | [136] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oikawa, D.; Sato, Y.; Ito, H.; Tokunaga, F. Linear Ubiquitin Code: Its Writer, Erasers, Decoders, Inhibitors, and Implications in Disorders. Int. J. Mol. Sci. 2020, 21, 3381. https://doi.org/10.3390/ijms21093381
Oikawa D, Sato Y, Ito H, Tokunaga F. Linear Ubiquitin Code: Its Writer, Erasers, Decoders, Inhibitors, and Implications in Disorders. International Journal of Molecular Sciences. 2020; 21(9):3381. https://doi.org/10.3390/ijms21093381
Chicago/Turabian StyleOikawa, Daisuke, Yusuke Sato, Hidefumi Ito, and Fuminori Tokunaga. 2020. "Linear Ubiquitin Code: Its Writer, Erasers, Decoders, Inhibitors, and Implications in Disorders" International Journal of Molecular Sciences 21, no. 9: 3381. https://doi.org/10.3390/ijms21093381
APA StyleOikawa, D., Sato, Y., Ito, H., & Tokunaga, F. (2020). Linear Ubiquitin Code: Its Writer, Erasers, Decoders, Inhibitors, and Implications in Disorders. International Journal of Molecular Sciences, 21(9), 3381. https://doi.org/10.3390/ijms21093381