The Bacterial Toxin CNF1 Protects Human Neuroblastoma SH-SY5Y Cells against 6-Hydroxydopamine-Induced Cell Damage: The Hypothesis of CNF1-Promoted Autophagy as an Antioxidant Strategy

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
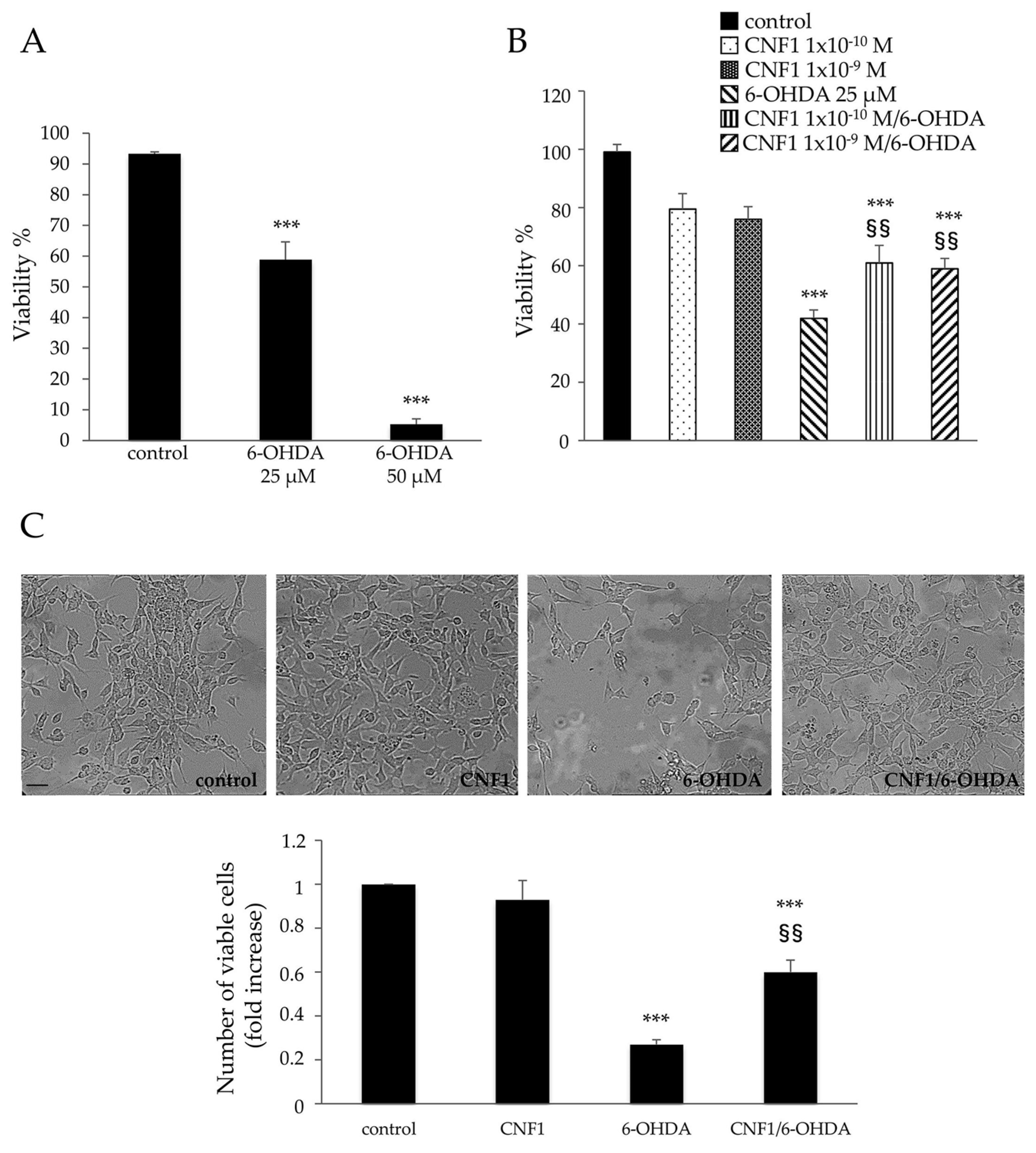
2.1. CNF1 Pre-Treatment Partially Rescues Cell Viability in SH-SY5Y Cells Exposed to the Neurotoxic 6-OHDA
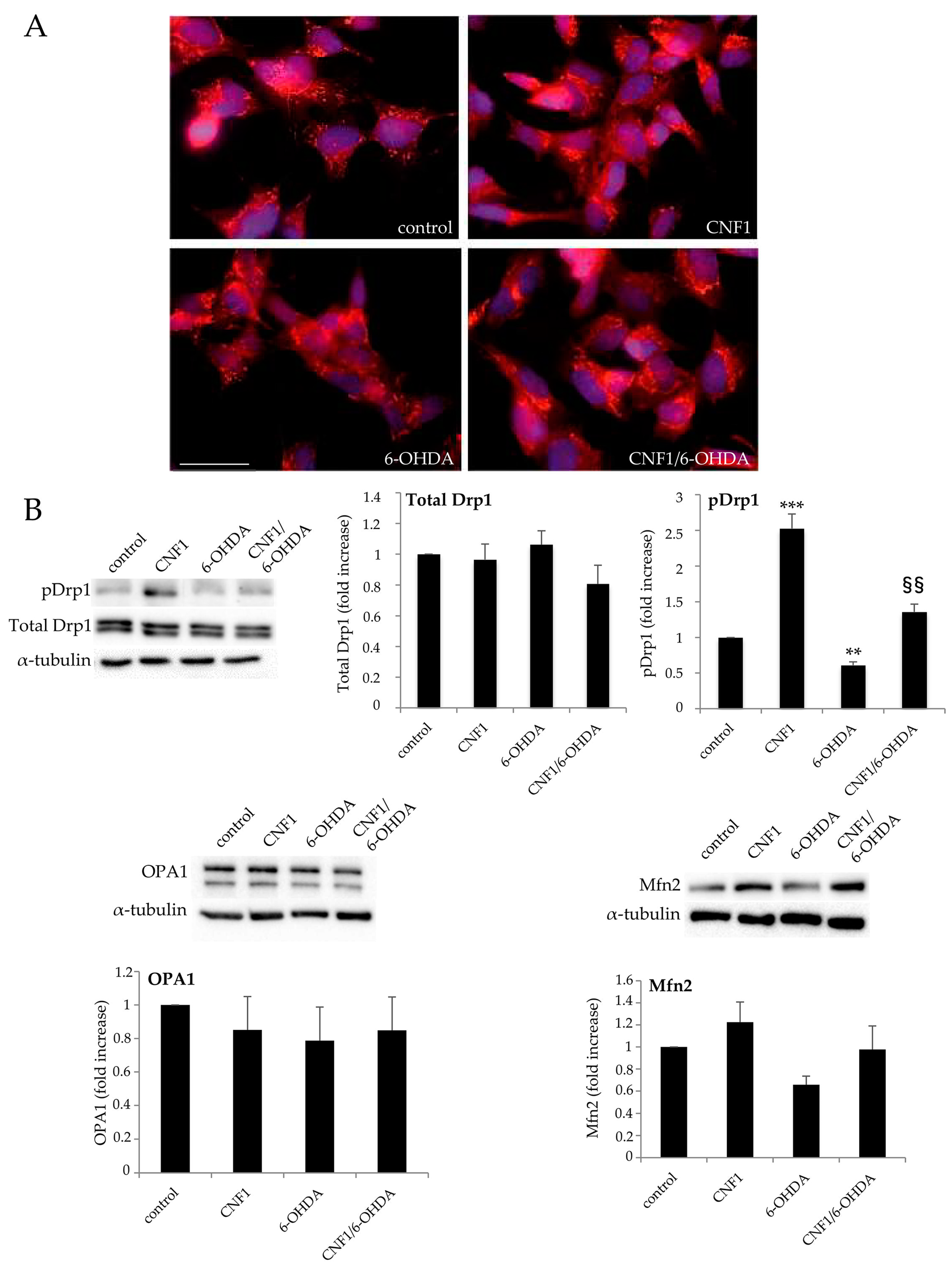
2.2. CNF1 Influences Mitochondrial Morphology and Dynamics in SH-SY5Y Cells
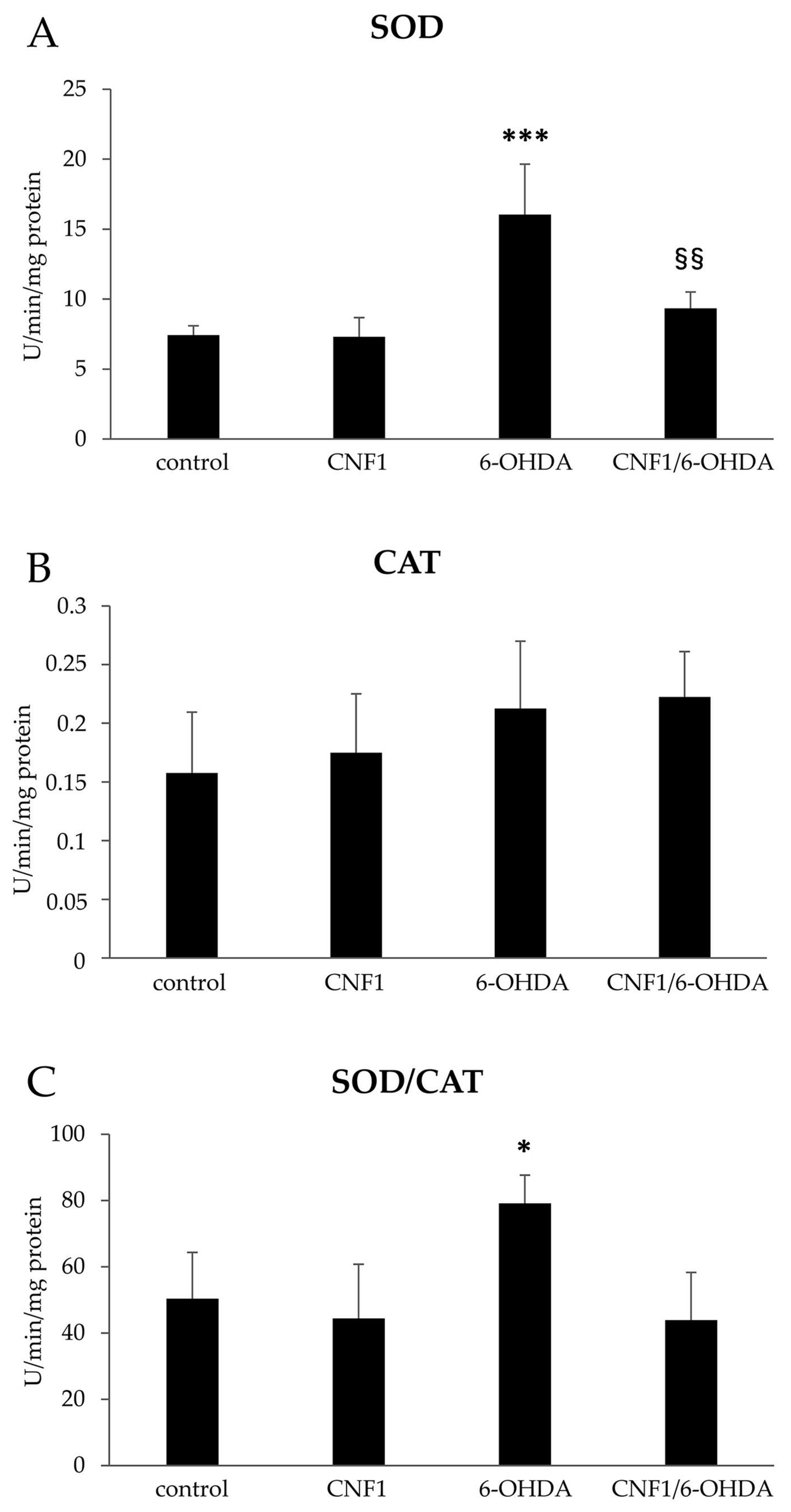
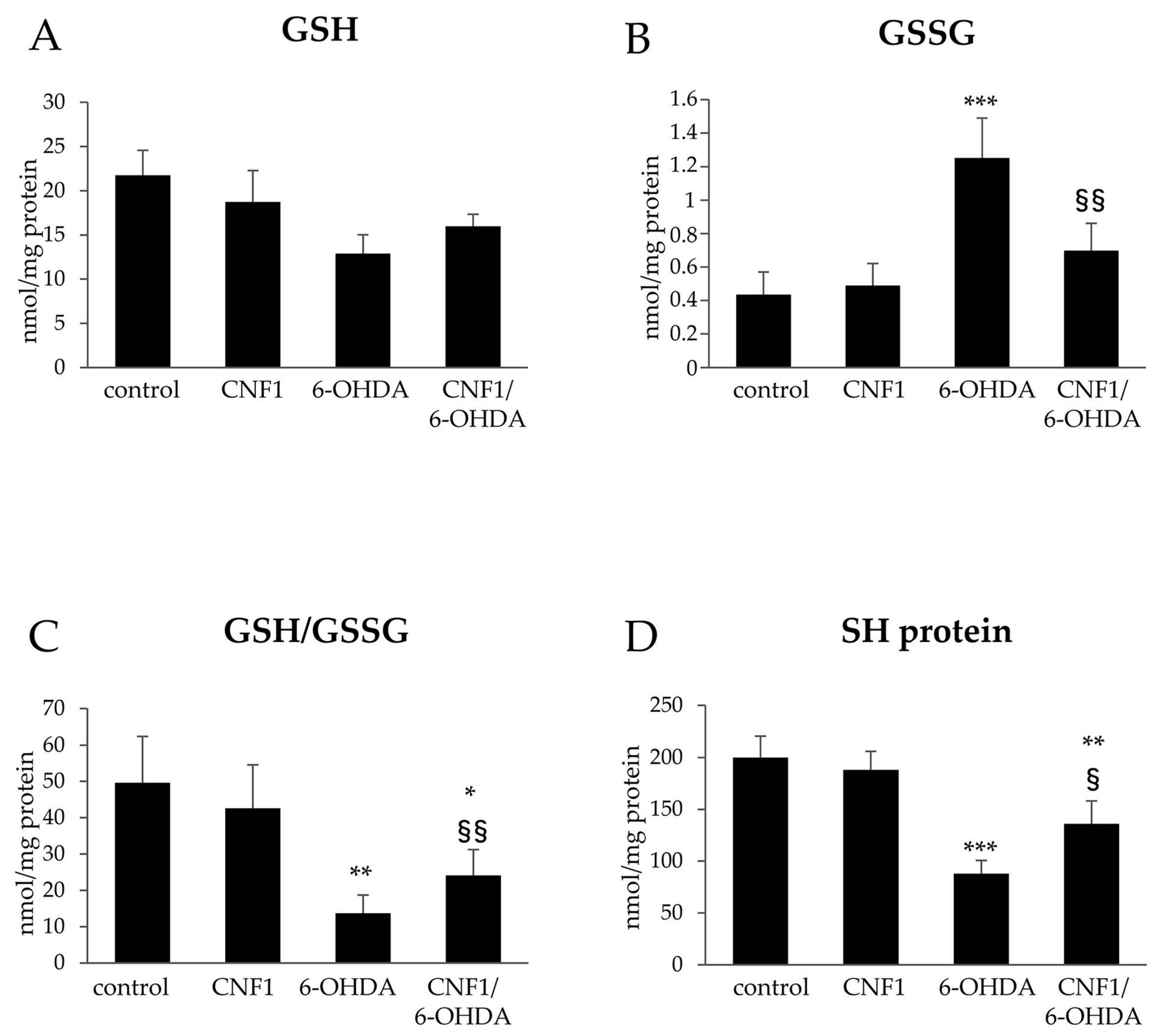
2.3. CNF1 Counteracts 6-OHDA-Induced Oxidative Stress
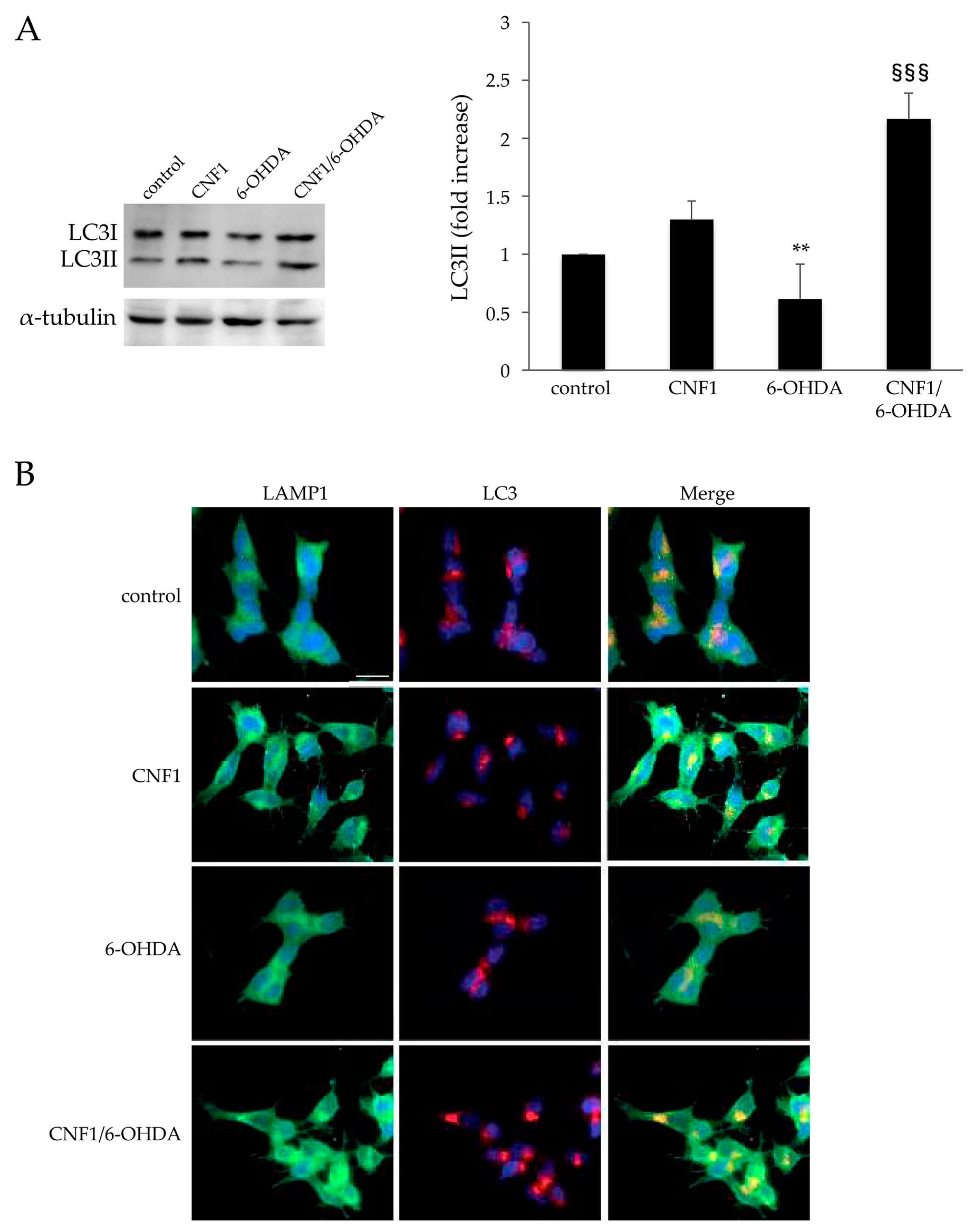
2.4. CNF1 Triggers Autophagy in SH-SY5Y Cells
3. Discussion
4. Materials and Methods
4.1. CNF1 Preparation
4.2. Cell Culture and Drug Treatment Procedures
4.3. Cell Viability
4.4. Immunofluorescence Analysis
4.5. Western Blot
4.6. Determination of SOD and CAT Activities
4.7. Determination of GSSG, Reduced GSH Glutathione
4.8. Determination of Protein Sulfhydryl Groups
4.9. Protein Quantification
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-hydroxydopamine |
| CAT | Catalase |
| CNF1 | Cytotoxic necrotizing factor 1 |
| Drp1 | Dynamin-related protein 1 |
| DTNB | 5,5’-dithiobis-2-nitrobenzoic acid |
| GSH | Reduced glutathione |
| GSSG | Oxidized glutathione |
| IVM | Intensified video microscopy |
| LAMP1 | Lysosomal-associated membrane protein 1 |
| LC3 | Microtubule-associated proteins 1A/1B light chain 3 |
| mAb | Monoclonal antibodies |
| pAb | Polyclonal antibodies |
| PD | Parkinson’s disease |
| pDrp1 | Phosphorylated dynamin related protein 1 |
| PKA | Protein kinase A |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
References
- Taylor, J.P.; McKeith, I.G.; Burn, D.J.; Boeve, B.F.; Weintraub, D.; Bamford, C.; Allan, L.M.; Thomas, A.J.; O’Brien, J.T. New evidence on the management of Lewy body dementia. Lancet Neurol. 2020, 19, 157–169. [Google Scholar] [CrossRef]
- Peña-Oyarzun, D.; Bravo-Sagua, R.; Diaz-Vega, A.; Aleman, L.; Chiong, M.; Garcia, L.; Bambs, C.; Troncoso, R.; Cifuentes, M.; Morselli, E.; et al. Autophagy and oxidative stress in non-communicable diseases: A matter of the inflammatory state? Free Radic. Biol. Med. 2018, 124, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Leadsham, J.E.; Sanders, G.; Giannaki, S.; Bastow, E.L.; Hutton, R.; Naeimi, W.R.; Breitenbach, M.; Gourlay, C.W. Loss of cytochrome c oxidase promotes RAS-dependent ROS production from the ER resident NADPH oxidase, Yno1p, in yeast. Cell Metab. 2013, 18, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Reddy, P.H. Mitochondrial medicine for aging and neurodegenerative diseases. Neuromol. Med. 2008, 10, 291–315. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Reddy, T.P. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr. Alzheimer Res. 2011, 8, 393–409. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 2010, 51, 440–450. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Does mitochondrial DNA play a role in Parkinson’s disease? A review of cybrid and other supportive evidence. Antioxidants Redox Signal. 2012, 16, 950–964. [Google Scholar] [CrossRef]
- Steinhubl, S.R. Why Have Antioxidants Failed in Clinical Trials? Am. J. Cardiol. 2008, 101, 14D–19D. [Google Scholar] [CrossRef]
- Chan, E.D.; Riches, D.W.H.; White, C.W. Redox Paradox: Effect of N-Acetylcysteine and Serum on Oxidation Reduction–Sensitive Mitogen-Activated Protein Kinase Signaling Pathways. Am. J. Respir. Cell Mol. Biol. 2001, 24, 627–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, B.D.; Guo, D.F.; Kulkarni, C.A.; Rahmouni, K.; Kerns, R.J.; Sivitz, W.I. Metabolic effects of a mitochondrial-targeted coenzyme Q analog in high fat fed obese mice. Pharmacol. Res. Perspect. 2017, 5, e00301. [Google Scholar] [CrossRef] [PubMed]
- Sakellariou, G.K.; Pearson, T.; Lightfoot, A.P.; Nye, G.A.; Wells, N.; Giakoumaki, I.I.; Vasilaki, A.; Griffiths, R.D.; Jackson, M.J.; McArdle, A. Mitochondrial ROS regulate oxidative damage and mitophagy but not age-related muscle fiber atrophy. Sci. Rep. 2016, 6, 33944. [Google Scholar] [CrossRef] [Green Version]
- Sheu, S.-S.; Nauduri, D.; Anders, M.W. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim. Biophys. Acta 2006, 1762, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.J.; Murphy, M.P.; Taylor, K.M.; Protect Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef]
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Travaglione, S.; Loizzo, S.; Rizza, T.; Del Brocco, A.; Ballan, G.; Guidotti, M.; Vona, R.; Di Nottia, M.; Torraco, A.; Carrozzo, R.; et al. Enhancement of mitochondrial ATP production by the Escherichia coli cytotoxic necrotizing factor 1. FEBS J. 2014, 281, 3473–3488. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, A.; Travaglione, S.; Fiorentini, C. Escherichia coli cytotoxic necrotizing factor 1 (CNF1): Toxin biology, in vivo applications and therapeutic potential. Toxins 2010, 2, 283–296. [Google Scholar] [CrossRef]
- Travaglione, S.; Ballan, G.; Fortuna, A.; Ferri, A.; Guidotti, M.; Campana, G.; Fiorentini, C.; Loizzo, S. CNF1 Enhances Brain Energy Content and Counteracts Spontaneous Epileptiform Phenomena in Aged DBA/2J Mice. PLoS ONE 2015, 10, e0140495. [Google Scholar] [CrossRef] [Green Version]
- De Filippis, B.; Fabbri, A.; Simone, D.; Canese, R.; Ricceri, L.; Malchiodi-Albedi, F.; Laviola, G.; Fiorentini, C. Modulation of RhoGTPases Improves the Behavioral Phenotype and Reverses Astrocytic Deficits in a Mouse Model of Rett Syndrome. Neuropsychopharmacology 2012, 37, 1152–1163. [Google Scholar] [CrossRef] [PubMed]
- Musilli, M.; Ciotti, M.T.; Pieri, M.; Martino, A.; Borrelli, S.; Dinallo, V.; Diana, G. Therapeutic effects of the Rho GTPase modulator CNF1 in a model of Parkinson’s disease. Neuropharmacology 2016, 109, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Lerm, M.; Selzer, J.; Hoffmeyer, A.; Rapp, U.R.; Aktories, K.; Schmidt, G. Deamidation of Cdc42 and Rac by Escherichia coli cytotoxic necrotizing factor 1: Activation of c-Jun N-terminal kinase in HeLa cells. Infect. Immun. 1999, 67, 496–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, A. Rho family GTPases. Biochem. Soc. Trans. 2012, 40, 1378–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorentini, C.; Donelli, G.; Matarrese, P.; Fabbri, A.; Paradisi, S.; Boquet, P. Escherichia coli cytotoxic necrotizing factor 1: Evidence for induction of actin assembly by constitutive activation of the p21 Rho GTPase. Infect. Immun. 1995, 63, 3936–3944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minin, A.A.; Kulik, A.V.; Gyoeva, F.K.; Li, Y.; Goshima, G.; Gelfand, V.I. Regulation of mitochondria distribution by RhoA and formins. J. Cell Sci. 2006, 119, 659–670. [Google Scholar] [CrossRef] [Green Version]
- Matveeva, E.A.; Venkova, L.S.; Chernoivanenko, I.S.; Minin, A.A. Vimentin is involved in regulation of mitochondrial motility and membrane potential by Rac1. Biol. Open 2015, 4, 1290–1297. [Google Scholar] [CrossRef] [Green Version]
- Loizzo, S.; Rimondini, R.; Travaglione, S.; Fabbri, A.; Guidotti, M.; Ferri, A.; Campana, G.; Fiorentini, C. CNF1 Increases Brain Energy Level, Counteracts Neuroinflammatory Markers and Rescues Cognitive Deficits in a Murine Model of Alzheimer’s Disease. PLoS ONE 2013, 8, e65898. [Google Scholar] [CrossRef]
- De Filippis, B.; Valenti, D.; de Bari, L.; De Rasmo, D.; Musto, M.; Fabbri, A.; Ricceri, L.; Fiorentini, C.; Laviola, G.; Vacca, R.A. Mitochondrial free radical overproduction due to respiratory chain impairment in the brain of a mouse model of Rett syndrome: Protective effect of CNF1. Free Radic. Biol. Med. 2015, 83, 167–177. [Google Scholar] [CrossRef]
- De Filippis, B.; Valenti, D.; Chiodi, V.; Ferrante, A.; de Bari, L.; Fiorentini, C.; Domenici, M.R.; Ricceri, L.; Vacca, R.A.; Fabbri, A.; et al. Modulation of Rho GTPases rescues brain mitochondrial dysfunction, cognitive deficits and aberrant synaptic plasticity in female mice modeling Rett syndrome. Eur. Neuropsychopharmacol. 2015, 25, 889–901. [Google Scholar] [CrossRef]
- Bové, J.; Prou, D.; Perier, C.; Przedborski, S. Toxin-induced models of Parkinson’s disease. NeuroRX 2005, 2, 484–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Lazaro, M.; Bonekamp, N.A.; Galindo, M.F.; Jordán, J.; Schrader, M. 6-Hydroxydopamine (6-OHDA) induces Drp1-dependent mitochondrial fragmentation in SH-SY5Y cells. Free Radic. Biol. Med. 2008, 44, 1960–1969. [Google Scholar] [CrossRef]
- Solesio, M.E.; Saez-atienzar, S.; Jordán, J.; Galindo, M.F. Characterization of mitophagy in the 6-hydoxydopamine Parkinson’s disease model. Toxicol. Sci. 2012, 129, 411–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Rosenwald, A. Small GTPase proteins in macroautophagy. Small GTPases 2018, 9, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. 1), 216–231. [Google Scholar] [CrossRef]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Cereghetti, G.M.; Stangherlin, A.; Martins De Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [Green Version]
- Storch, A.; Kaftan, A.; Burkhardt, K.; Schwarz, J. 6-Hydroxydopamine toxicity towards human SH-SY5Y dopaminergic neuroblastoma cells: Independent of mitochondrial energy metabolism. J. Neural Transm. 2000, 107, 281–293. [Google Scholar] [CrossRef]
- Aguilera, M.O.; Berón, W.; Colombo, M.I. The actin cytoskeleton participates in the early events of autophagosome formation upon starvation induced autophagy. Autophagy 2012, 8, 1590–1603. [Google Scholar] [CrossRef] [Green Version]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, T.R.; Linseman, D.A. Rho family GTPases: Key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell. Neurosci. 2014, 8, 314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falzano, L.; Quaranta, M.G.; Travaglione, S.; Filippini, P.; Fabbri, A.; Viora, M.; Donelli, G.; Fiorentini, C. Cytotoxic necrotizing factor 1 enhances reactive oxygen species-dependent transcription and secretion of proinflammatory cytokines in human uroepithelial cells. Infect. Immun. 2003, 71, 4178–4181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Flint Beal, M. Mitochondrial Diseases of the Brain. Free Radic. Biol. Med. 2013, 63, 1–29. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Coskun, P.; Wyrembak, J.; Schriner, S.E.; Chen, H.W.; Marciniack, C.; Laferla, F.; Wallace, D.C. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Van Laar, V.S.; Berman, S.B. Mitochondrial dynamics in Parkinson’s disease. Exp. Neurol. 2009, 218, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Mhatrea, M.; Floyd, R.A.; Hensley, K. Oxidative stress and neuroinflammation in Alzheimer’s disease and amyotrophic lateral sclerosis: Common links and potential therapeutic targets. J. Alzheimer’s Dis. 2004, 6, 147–157. [Google Scholar] [CrossRef]
- Miraglia, A.G.; Travaglione, S.; Meschini, S.; Falzano, L.; Matarrese, P.; Quaranta, M.G.; Viora, M.; Fiorentini, C.; Fabbri, A. Cytotoxic necrotizing factor 1 prevents apoptosis via the Akt/IkappaB kinase pathway: Role of nuclear factor-kappaB and Bcl-2. Mol. Biol. Cell 2007, 18, 2735–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahani-Asl, A.; Slack, R.S. The phosphorylation state of Drp1 determines cell fate. EMBO Rep. 2007, 8, 912–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpentieri, A.; Cozzoli, E.; Scimeca, M.; Bonanno, E.; Sardanelli, A.M.; Gambacurta, A. Differentiation of human neuroblastoma cells toward the osteogenic lineage by mTOR inhibitor. Cell Death Dis. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Autophagy in Human Health and Disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Xiong, F.; Zhang, L. Role of the hypothalamic-pituitary-adrenal axis in developmental programming of health and disease. Front. Neuroendocrinol. 2013, 34, 27–46. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Gu, L.; Smerin, D.; Mao, S.; Xiong, X. The Interrelation between Reactive Oxygen Species and Autophagy in Neurological Disorders. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Huang, M.; Chiang, S.; Kalinowski, D.; Bae, D.; Sahni, S.; Richardson, D. The Role of the Antioxidant Response in Mitochondrial Dysfunction in Degenerative Diseases: Cross-Talk Between Antioxidant Defense, Autophagy, and Apoptosis. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Denton, D.; Nicolson, S.; Kumar, S. Cell death by autophagy: Facts and apparent artefacts. Cell Death Differ. 2012, 19, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.M.; Codogno, P. Autophagy is a survival force via suppression of necrotic cell death. Exp. Cell Res. 2012, 318, 1304–1308. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T. Diversity in the Regulation of Autophagy and Mitophagy: Lessons from Parkinson’s Disease. Parkinsons. Dis. 2011, 2011, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morán, M.; Delmiro, A.; Blázquez, A.; Ugalde, C.; Arenas, J.; Martín, M.A. Bulk autophagy, but not mitophagy, is increased in cellular model of mitochondrial disease. Biochim. Biophys. Acta 2014, 1842, 1059–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, L.C.; Benedetto, G.D.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Solitro, A.R.; MacKeigan, J.P. Leaving the lysosome behind: Novel developments in autophagy inhibition. Future Med. Chem. 2016, 8, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Kast, D.J.; Dominguez, R. The Cytoskeleton-Autophagy Connection. Curr. Biol. 2017, 27, R318–R326. [Google Scholar] [CrossRef] [Green Version]
- Falzano, L.; Fiorentini, C.; Donelli, G.; Michel, E.; Kocks, C.; Cossart, P.; Cabanié, L.; Oswald, E.; Boquet, P. Induction of phagocytic behaviour in human epithelial cells by Escherichia coli cytotoxic necrotizing factor type 1. Mol. Microbiol. 1993, 9, 1247–1254. [Google Scholar] [CrossRef]
- Marklund, S.; Marklund, G. Involvement of the Superoxide Anion Radical in the Autoxidation of Pyrogallol and a Convenient Assay for Superoxide Dismutase. Eur. J. Biochem. 1974, 47, 469–474. [Google Scholar] [CrossRef]
- Aebi, H. [13] Catalase in vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar]
- Anderson, M.E. [70] Determination of glutathione and glutathione disulfide in biological samples. Methods Enzymol. 1985, 113, 548–555. [Google Scholar]
- Di Monte, D.; Ross, D.; Bellomo, G.; Eklöw, L.; Orrenius, S. Alterations in intracellular thiol homeostasis during the metabolism of menadione by isolated rat hepatocytes. Arch. Biochem. Biophys. 1984, 235, 334–342. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Travaglione, S.; Loizzo, S.; Vona, R.; Ballan, G.; Rivabene, R.; Giordani, D.; Guidotti, M.; Dupuis, M.L.; Maroccia, Z.; Baiula, M.; et al. The Bacterial Toxin CNF1 Protects Human Neuroblastoma SH-SY5Y Cells against 6-Hydroxydopamine-Induced Cell Damage: The Hypothesis of CNF1-Promoted Autophagy as an Antioxidant Strategy. Int. J. Mol. Sci. 2020, 21, 3390. https://doi.org/10.3390/ijms21093390
Travaglione S, Loizzo S, Vona R, Ballan G, Rivabene R, Giordani D, Guidotti M, Dupuis ML, Maroccia Z, Baiula M, et al. The Bacterial Toxin CNF1 Protects Human Neuroblastoma SH-SY5Y Cells against 6-Hydroxydopamine-Induced Cell Damage: The Hypothesis of CNF1-Promoted Autophagy as an Antioxidant Strategy. International Journal of Molecular Sciences. 2020; 21(9):3390. https://doi.org/10.3390/ijms21093390
Chicago/Turabian StyleTravaglione, Sara, Stefano Loizzo, Rosa Vona, Giulia Ballan, Roberto Rivabene, Danila Giordani, Marco Guidotti, Maria Luisa Dupuis, Zaira Maroccia, Monica Baiula, and et al. 2020. "The Bacterial Toxin CNF1 Protects Human Neuroblastoma SH-SY5Y Cells against 6-Hydroxydopamine-Induced Cell Damage: The Hypothesis of CNF1-Promoted Autophagy as an Antioxidant Strategy" International Journal of Molecular Sciences 21, no. 9: 3390. https://doi.org/10.3390/ijms21093390
APA StyleTravaglione, S., Loizzo, S., Vona, R., Ballan, G., Rivabene, R., Giordani, D., Guidotti, M., Dupuis, M. L., Maroccia, Z., Baiula, M., Rimondini, R., Campana, G., & Fiorentini, C. (2020). The Bacterial Toxin CNF1 Protects Human Neuroblastoma SH-SY5Y Cells against 6-Hydroxydopamine-Induced Cell Damage: The Hypothesis of CNF1-Promoted Autophagy as an Antioxidant Strategy. International Journal of Molecular Sciences, 21(9), 3390. https://doi.org/10.3390/ijms21093390