Functional Characterization of ABCA4 Missense Variants Linked to Stargardt Macular Degeneration

Abstract
:1. Introduction
2. Results
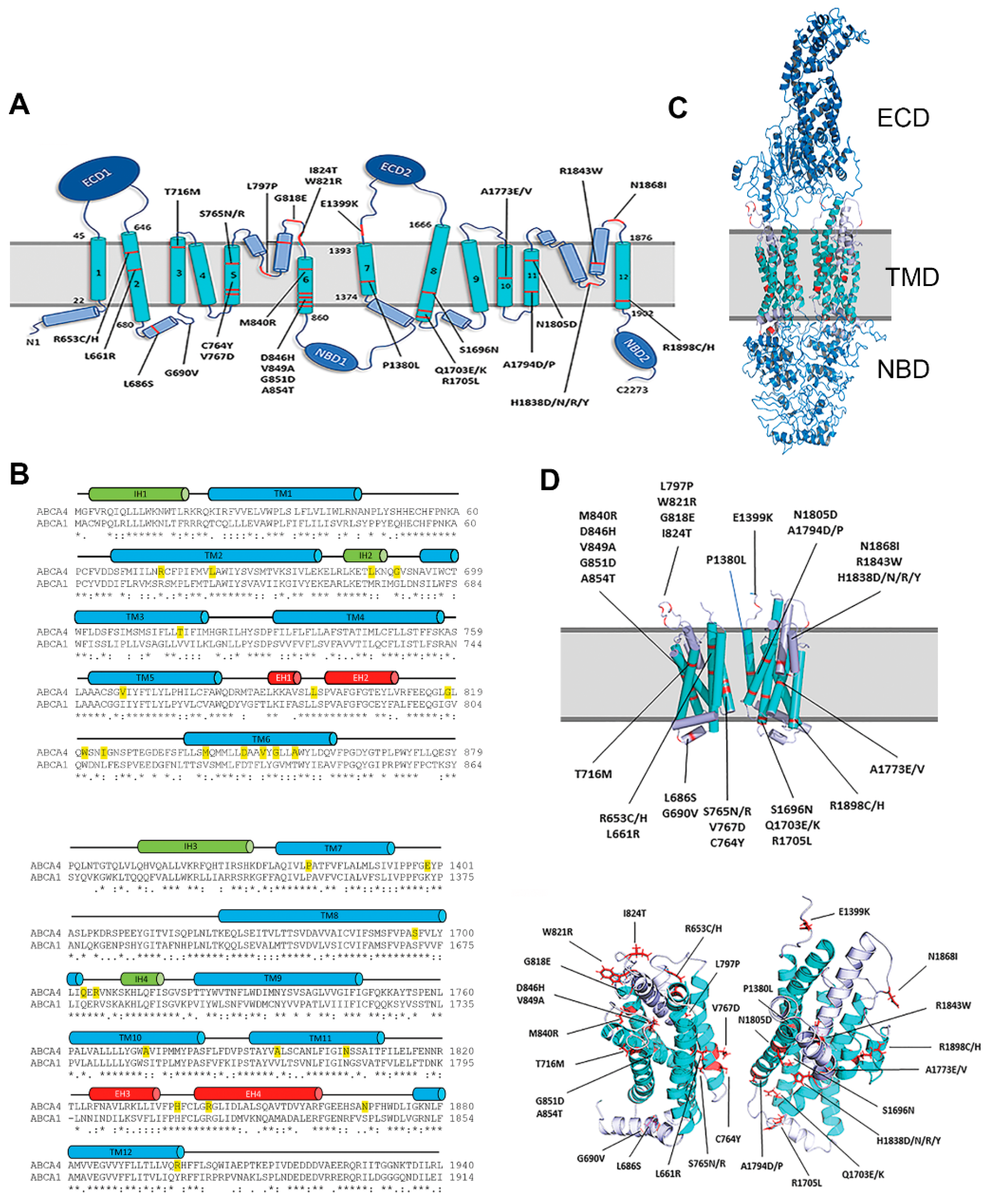
2.1. Transmembrane Domains (TMDs) of ABCA4
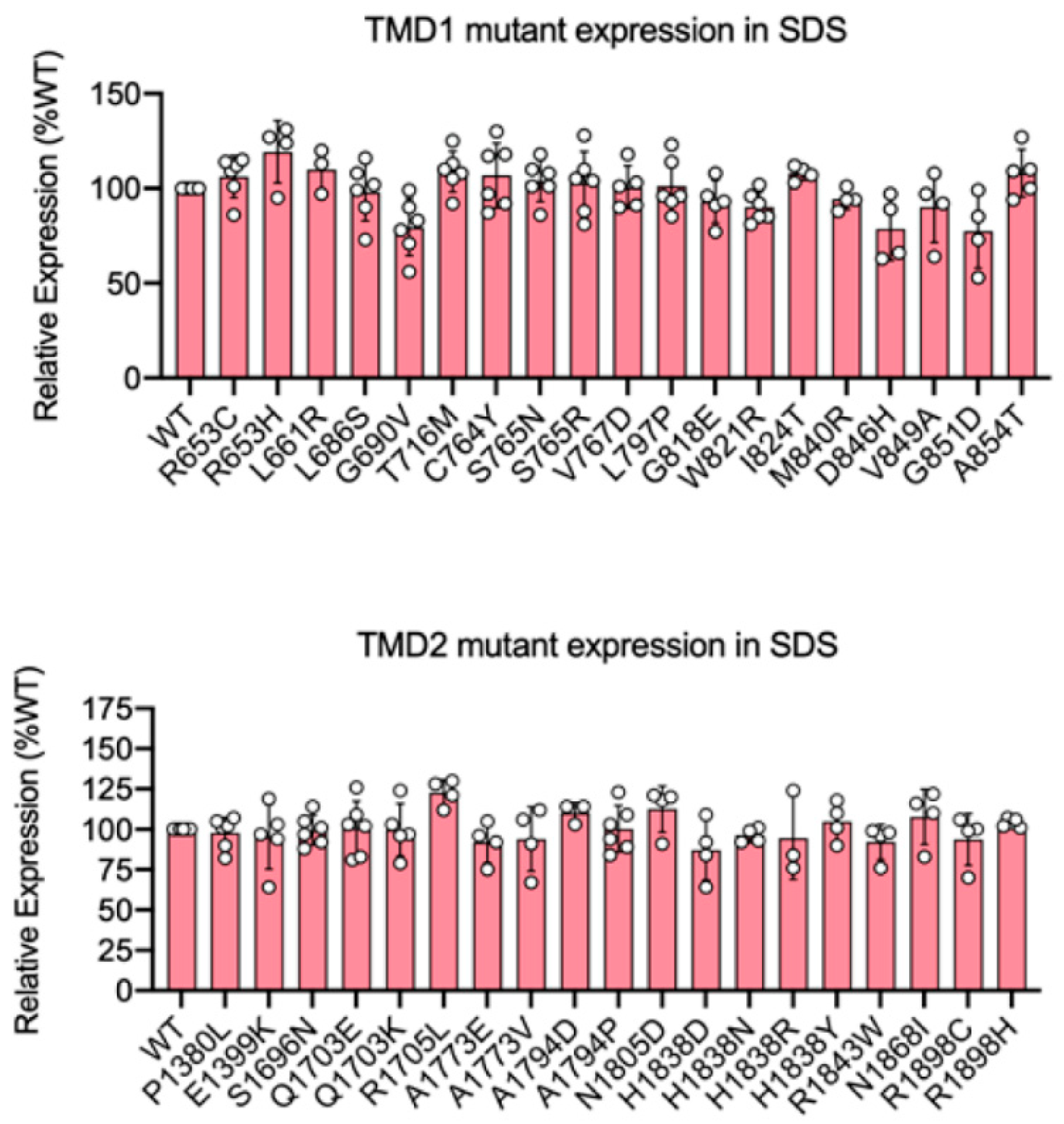
2.2. Expression of TMD Disease Variants
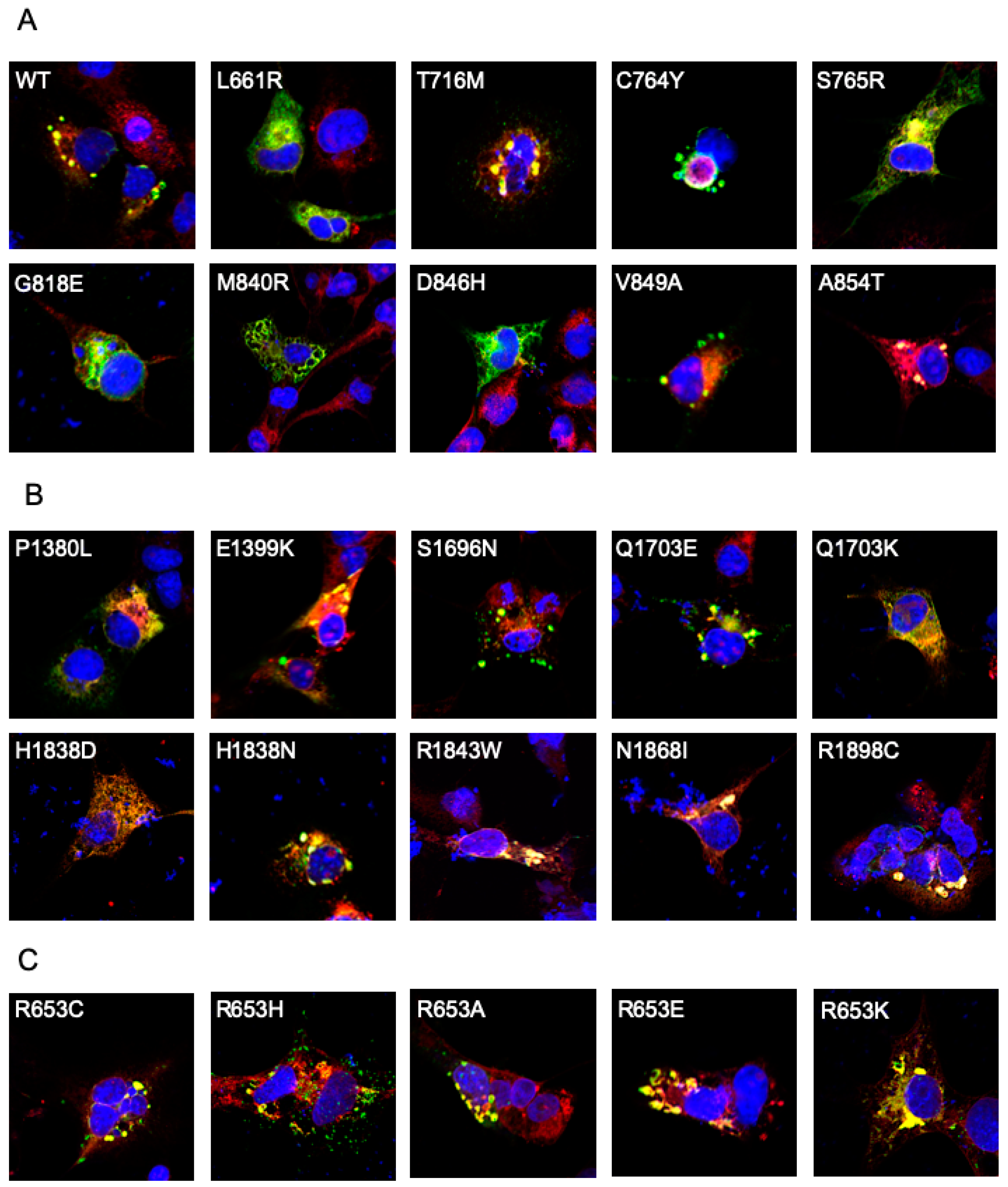
2.3. Localization of ABCA Disease Variants in COS-7 Cells
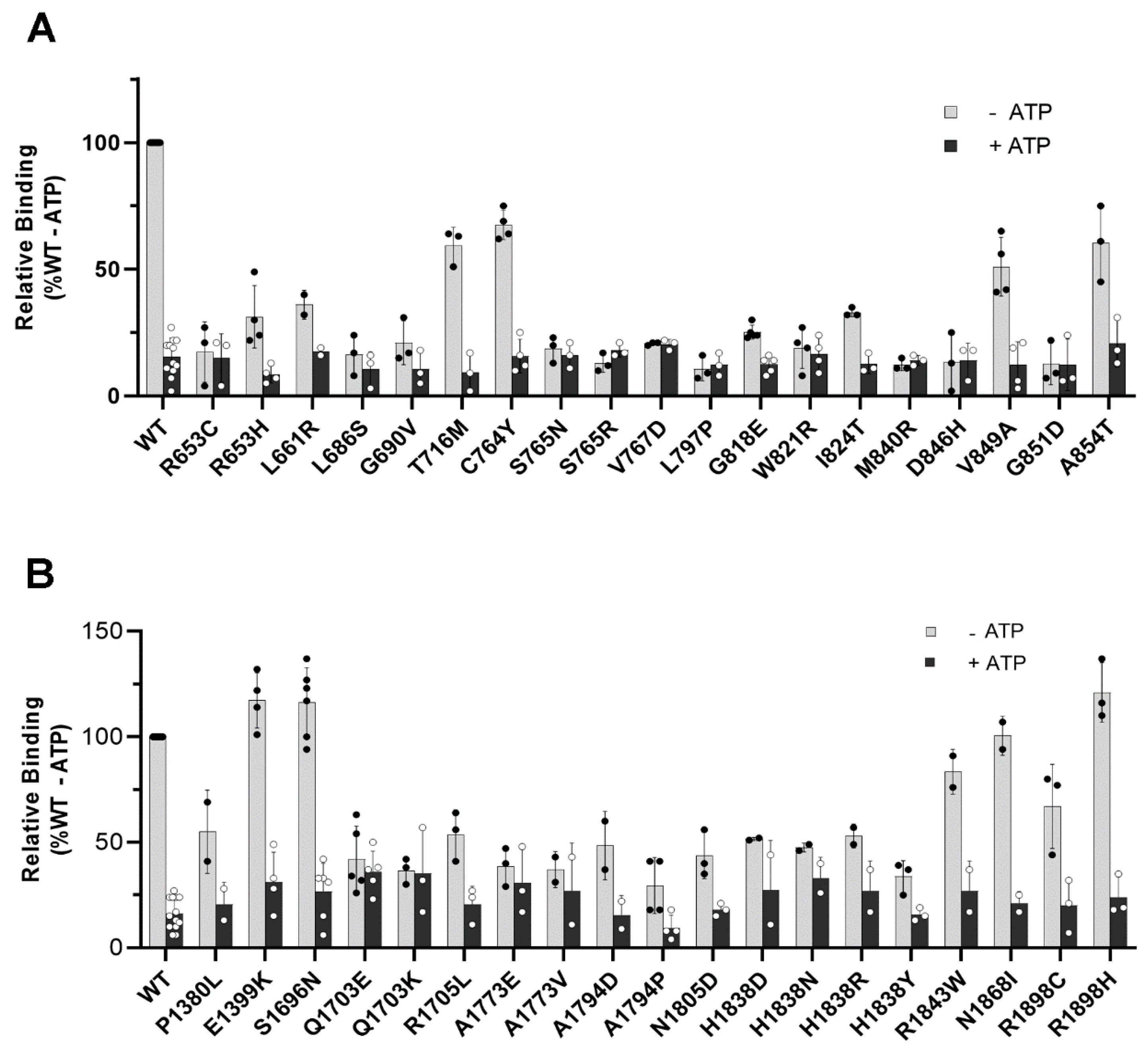
2.4. Functional Analysis of TMD Disease-Associated Variants: ATPase Assays
2.5. Functional Analysis of Disease-Associated TMD Variants: N-Ret-PE Binding Assays
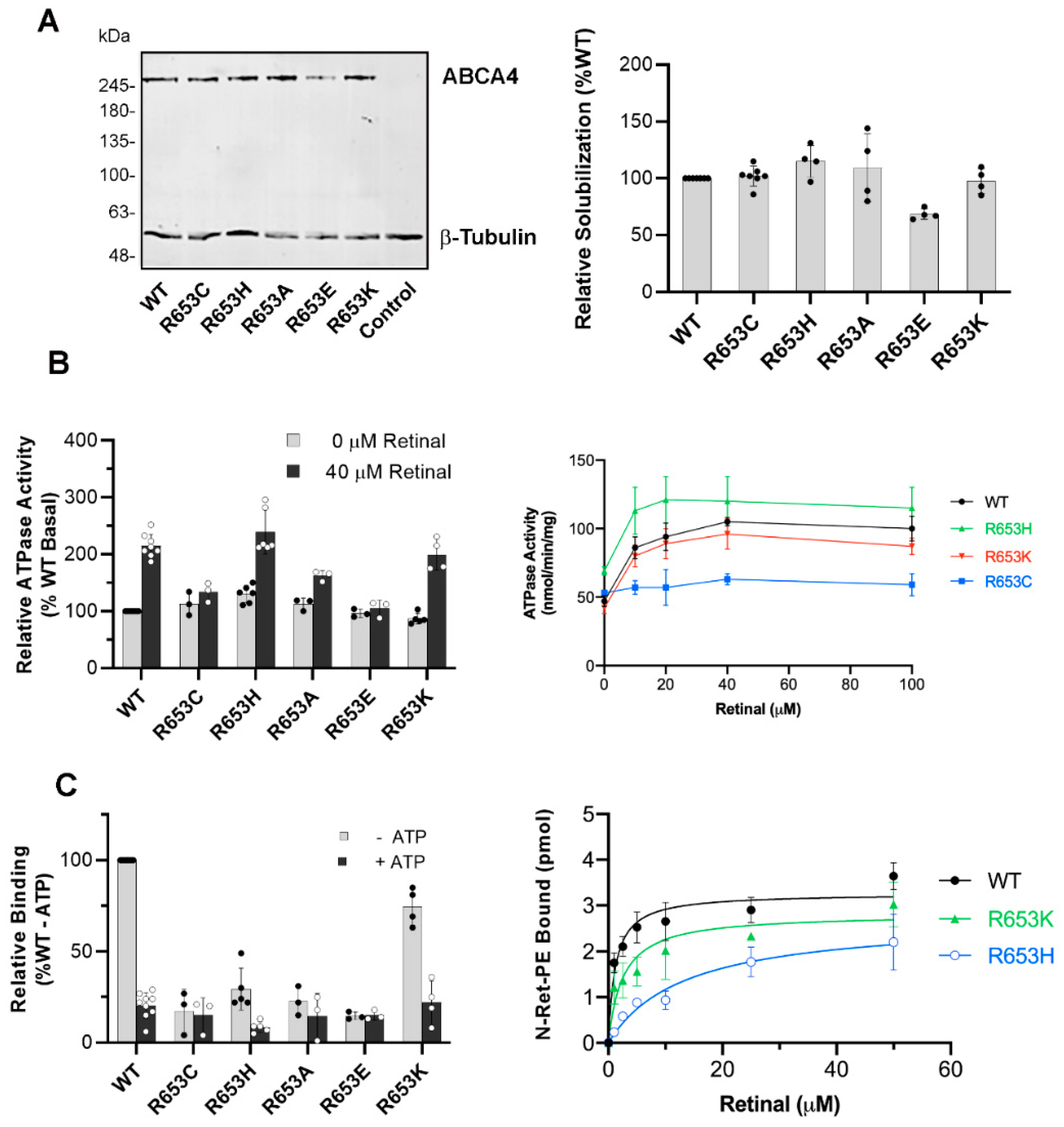
2.6. Biochemical Characterization of ABCA4 Arg653 Variants
3. Discussion
4. Materials and Methods
4.1. Prediction of Transmembrane Helices and Homology Model of ABCA4
4.2. Cloning of ABCA4 Transmembrane Variants
4.3. Antibodies
4.4. Heterologous Expression of ABCA4 Variants in HEK293T Cells
4.5. Immunofluorescence Microscopy of ABCA4 Variants in Transfected COS-7 Cells
4.6. ATPase Assay
4.7. N-Ret-PE Binding Assay
4.8. Statistical Analysis
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TMD | transmembrane domain |
| NBD | nucleotide binding domain |
| ECD | exocytoplasmic domain |
| TM | membrane spanning segment |
| ATR | all-trans retinal |
| STGD1 | Stargardt disease |
| N-Ret-PE | N-retinylidene phosphatidylethanolamine |
| PE | phosphatidylethanolamine |
| RPE | retinal pigment epithelium |
| TM | transmembrane segment |
| IH | intracellular transverse coupling helix |
| EH | exoplasmic V-shaped α-helical hairpin helix |
| BPL | brain polar lipid |
| PBS | phosphate buffered saline |
| DAPI | 4′,6-Diamidino-2-phenylindole |
| A | alanine |
| F | phenylalanine |
| C | cysteine |
| D | aspartic acid |
| N | asparagine |
| E | glutamic acid |
| Q | glutamine |
| G | glycine |
| H | histidine |
| L | leucine |
| I | isoleucine |
| K | lysine |
| M | methionine |
| P | proline |
| R | arginine |
| S | serine |
| T | threonine |
| V | valine |
| W | tryptophan |
| Y | tyrosine |
Appendix A

References
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Illing, M.; Molday, L.L.; Molday, R.S. The 220-kDa Rim Protein of Retinal Rod Outer Segments Is a Member of the ABC Transporter Superfamily. J. Biol. Chem. 1997, 272, 10303–10310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molday, L.L.; Rabin, A.R.; Molday, R.S. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat. Genet. 2000, 25, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Bungert, S.; Molday, L.L.; Molday, R.S. Membrane topology of the ATP binding cassette transporter ABCR and its relationship to ABC1 and related ABCA transporters: Identification of N-linked glycosylation sites. J. Biol. Chem. 2001, 276, 23539–23546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beharry, S.; Zhong, M.; Molday, R.S. N-Retinylidene-phosphatidylethanolamine Is the Preferred Retinoid Substrate for the Photoreceptor-specific ABC Transporter ABCA4 (ABCR). J. Biol. Chem. 2004, 279, 53972–53979. [Google Scholar] [CrossRef] [Green Version]
- Quazi, F.; Lenevich, S.; Molday, R.S. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat. Commun. 2012, 3, 925. [Google Scholar] [CrossRef] [Green Version]
- Cremers, F.P.; Lee, W.; Collin, R.W.; Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef]
- Cornelis, S.S.; Bax, N.M.; Zernant, J.; Allikmets, R.; Fritsche, L.G.; Dunnen, J.T.D.; Ajmal, M.; Hoyng, C.B.; Cremers, F.P. In SilicoFunctional Meta-Analysis of 5,962ABCA4Variants in 3,928 Retinal Dystrophy Cases. Hum. Mutat. 2017, 38, 400–408. [Google Scholar] [CrossRef]
- Tanna, P.; Strauss, R.W.; Fujinami, K.; Michaelides, M. Stargardt disease: Clinical features, molecular genetics, animal models and therapeutic options. Br. J. Ophthalmol. 2016, 101, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Klevering, B.J.; Blankenagel, A.; Maugeri, A.; Cremers, F.P.; Hoyng, C.B.; Rohrschneider, K. Phenotypic spectrum of autosomal recessive cone-rod dystrophies caused by mutations in the ABCA4 (ABCR) gene. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1980–1985. [Google Scholar]
- Issa, P.C.; Barnard, A.R.; Singh, M.S.; Carter, E.; Jiang, Z.; Radu, R.A.; Schraermeyer, U.; MacLaren, R.E. Fundus autofluorescence in the Abca4(-/-) mouse model of Stargardt disease—correlation with accumulation of A2E, retinal function, and histology. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5602–5612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasonkin, I.; Illing, M.; Koehler, M.R.; Schmid, M.; Molday, R.S.; Weber, B.H.F. Mapping of the rod photoreceptor ABC transporter (ABCR) to 1p21-p22.1 and identification of novel mutations in Stargardt’s disease. Hum. Genet. 1998, 102, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Smallwood, P.M.; Nathans, J. Biochemical defects in ABCR protein variants associated with human retinopathies. Nat. Genet. 2000, 26, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Garces, F.; Jiang, K.; Molday, L.L.; Stöhr, H.; Weber, B.H.; Lyons, C.J.; Maberley, D.; Molday, R.S. Correlating the Expression and Functional Activity of ABCA4 Disease Variants with the Phenotype of Patients With Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2305–2315. [Google Scholar] [CrossRef] [Green Version]
- Quazi, F.; Molday, R.S. Differential Phospholipid Substrates and Directional Transport by ATP-binding Cassette Proteins ABCA1, ABCA7, and ABCA4 and Disease-causing Mutants. J. Biol. Chem. 2013, 288, 34414–34426. [Google Scholar] [CrossRef] [Green Version]
- Zhong, M.; Molday, L.L.; Molday, R.S. Role of the C Terminus of the Photoreceptor ABCA4 Transporter in Protein Folding, Function, and Retinal Degenerative Diseases. J. Biol. Chem. 2009, 284, 3640–3649. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Tsybovsky, Y.; Kolesnikov, A.V.; Rozanowska, M.; Swider, M.; Schwartz, S.B.; Stone, E.M.; Palczewska, G.; Maeda, A.; Kefalov, V.J.; et al. Protein misfolding and the pathogenesis of ABCA4-associated retinal degenerations. Hum. Mol. Genet. 2015, 24, 3220–3237. [Google Scholar] [CrossRef]
- Wiszniewski, W.; Zaremba, C.M.; Yatsenko, A.N.; Jamrich, M.; Wensel, T.G.; Mann, J.; Lupski, J.R. ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies. Hum. Mol. Genet. 2005, 14, 2769–2778. [Google Scholar] [CrossRef] [Green Version]
- Curtis, S.B.; Molday, L.L.; Garces, F.A.; Molday, R.S. Functional analysis and classification of homozygous and hypomorphic ABCA4 variants associated with Stargardt macular degeneration. Hum. Mutat. 2020, 41, 1944–1956. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Boulton, M. RPE lipofuscin and its role in retinal pathobiology. Exp. Eye Res. 2005, 80, 595–606. [Google Scholar] [CrossRef]
- Weng, J.; Mata, N.L.; Azarian, S.M.; Tzekov, R.T.; Birch, D.G.; Travis, G.H. Insights into the Function of Rim Protein in Photoreceptors and Etiology of Stargardt’s Disease from the Phenotype in abcr Knockout Mice. Cell 1999, 98, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Molday, L.L.; Wahl, D.; Sarunic, M.V.; Molday, R.S. Localization and functional characterization of the p.Asn965Ser (N965S) ABCA4 variant in mice reveal pathogenic mechanisms underlying Stargardt macular degeneration. Hum. Mol. Genet. 2018, 27, 295–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, N.P.; Higbee, D.; Currin, M.B.; Blakeley, L.R.; Chen, C.; Ablonczy, Z.; Crouch, R.K.; Koutalos, Y. Lipofuscin and N-retinylidene-N-retinylethanolamine (A2E) accumulate in retinal pigment epithelium in absence of light exposure: Their origin is 11-cis-retinal. J. Biol. Chem. 2012, 287, 22276–22286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, T.R.; Duncker, T.; Woods, R.L.; Greenberg, J.P.; Zernant, J.; Tsang, S.H.; Smith, R.T.; Allikmets, R.; Sparrow, J.R.; Delori, F.C. Quantitative Fundus Autofluorescence in Recessive Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2841–2852. [Google Scholar] [CrossRef] [Green Version]
- Mata, N.L.; Weng, J.; Travis, G.H. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc. Natl. Acad. Sci. USA 2000, 97, 7154–7159. [Google Scholar] [CrossRef] [Green Version]
- Mäkeläinen, S.; Gòdia, M.; Hellsand, M.; Viluma, A.; Hahn, D.; Makdoumi, K.; Zeiss, C.J.; Mellersh, C.; Ricketts, S.L.; Narfström, K.; et al. An ABCA4 loss-of-function mutation causes a canine form of Stargardt disease. PLoS Genet. 2019, 15, e1007873. [Google Scholar] [CrossRef] [Green Version]
- Qian, H.; Zhao, X.; Cao, P.; Lei, J.; Yan, N.; Gong, X. Structure of the Human Lipid Exporter ABCA1. Cell 2017, 169, 1228–1239.e10. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Molday, R.S.; Nathans, J. Retinal Stimulates ATP Hydrolysis by Purified and Reconstituted ABCR, the Photoreceptor-specific ATP-binding Cassette Transporter Responsible for Stargardt Disease. J. Biol. Chem. 1999, 274, 8269–8281. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Molday, R.S. Purification and characterization of ABCR from bovine rod outer segments. Methods Enzymol. 2000, 315, 864–879. [Google Scholar] [CrossRef]
- Molday, R.S. Insights into the Molecular Properties of ABCA4 and Its Role in the Visual Cycle and Stargardt Disease. Prog. Mol. Biol. Transl. Sci. 2015, 134, 415–431. [Google Scholar] [CrossRef]
- Quazi, F.; Molday, R.S. ATP-binding cassette transporter ABCA4 and chemical isomerization protect photoreceptor cells from the toxic accumulation of excess 11-cis-retinal. Proc. Natl. Acad. Sci. USA 2014, 111, 5024–5029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fingert, J.H.; Eliason, D.A.; Phillips, N.C.; Lotery, A.J.; Sheffield, V.C.; Stone, E.M. Case of Stargardt Disease Caused by Uniparental Isodisomy. Arch. Ophthalmol. 2006, 124, 744–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.C.; Zernant, J.; Allikmets, R.; Barile, G.R.; Chang, S.; Smith, R.T. Peripapillary Atrophy in Stargardt Disease. Retina 2009, 29, 181–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cideciyan, A.V.; Swider, M.; Aleman, T.S.; Tsybovsky, Y.; Schwartz, S.B.; Windsor, E.A.; Roman, A.J.; Sumaroka, A.; Steinberg, J.D.; Jacobson, S.G.; et al. ABCA4 disease progression and a proposed strategy for gene therapy. Hum. Mol. Genet. 2008, 18, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Fakin, A.; Robson, A.G.; Fujinami, K.; Moore, A.T.; Michaelides, M.; Holder, G.E.; Webster, A.R. The Effect on Retinal Structure and Function of 15 Specific ABCA4 Mutations: A Detailed Examination of 82 Hemizygous Patients. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5963–5973. [Google Scholar] [CrossRef] [PubMed]
- Zernant, J.; Lee, W.; Collison, F.T.; Fishman, G.A.; Sergeev, Y.V.; Schuerch, K.; Sparrow, J.R.; Tsang, S.H.; Allikmets, R. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J. Med. Genet. 2017, 54, 404–412. [Google Scholar] [CrossRef]
- Runhart, E.H.; Sangermano, R.; Cornelis, S.S.; Verheij, J.B.; Plomp, A.S.; Boon, C.J.; Lugtenberg, D.; Roosing, S.; Bax, N.M.; Blokland, E.A.; et al. The Common ABCA4 Variant p.Asn1868Ile Shows Nonpenetrance and Variable Expression of Stargardt Disease When Present in trans with Severe Variants. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3220–3231. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Zhang, Q.; Li, S.; Guan, L.; Xiao, X.; Zhang, J.; Jia, X.; Sun, W.; Zhu, Z.; Gao, Y.; et al. Exome Sequencing of 47 Chinese Families with Cone-Rod Dystrophy: Mutations in 25 Known Causative Genes. PLoS ONE 2013, 8, e65546. [Google Scholar] [CrossRef] [Green Version]
- Fujinami, K.; Zernant, J.; Chana, R.K.; Wright, G.A.; Tsunoda, K.; Ozawa, Y.; Tsubota, K.; Robson, A.G.; Holder, G.E.; Allikmets, R.; et al. Clinical and Molecular Characteristics of Childhood-Onset Stargardt Disease. Ophthalmology 2015, 122, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Molday, R.S.; MacKenzie, D. Monoclonal antibodies to rhodopsin: Characterization, cross-reactivity, and application as structural probes. Biochemistry 1983, 22, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Garwin, G.G.; Saari, J.C. High-performance liquid chromatography analysis of visual cycle retinoids. Methods Enzymol. 2000, 316, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Molday, R.S. Binding of Retinoids to ABCA4, the Photoreceptor ABC Transporter Associated with Stargardt Macular Degeneration. Methods Mol. Biol. 2010, 652, 163–176. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Localization | Relative Solubilization | Basal ATPase Activity | N-Ret-PE-Induced ATPase Activity | N-ret-PE Binding no ATP | N-Ret-PE Binding with ATP | Class * | Predicted Severity |
|---|---|---|---|---|---|---|---|---|
| WT | Vesicles/ER | 100 | 100 | 222 ± 22 | 100 | 15 ± 7 | -- | Normal |
| R653C | Vesicles/ER | 102 ± 9 | 113 ± 21 | 134 ± 17 | 17 ± 12 | 15 ± 10 | 1 | Severe |
| R653H | Vesicles/ER | 115 ± 14 | 130 ± 15 | 239 ± 39 | 31 ± 12 | 9 ± 3 | 2 | Moderate/ Mild * |
| L661R | ER | 20 ± 11 | 13 ± 4 | 16 ± 3 | 36 ± 6 | 18 ± 2 | 1 | Severe |
| L686S | ER | 27 ± 8 | 18 ± 9 | 30 ± 16 | 16 ± 8 | 11 ± 7 | 1 | Severe |
| G690V | ER | 33 ± 12 | 41 ± 12 | 59 ± 14 | 21 ± 9 | 11 ± 7 | 1 | Severe |
| T716M | Vesicles/ER | 98 ± 16 | 94 ± 4 | 228 ± 13 | 59 ± 7 | 9 ± 8 | 3 | Mild |
| C764Y | Vesicles/ER | 100 ± 12 | 133 ± 5 | 270 ± 39 | 68 ± 6 | 16 ± 7 | 3 | Mild |
| S765N | ER | 30 ± 9 | 21 ± 9 | 45 ± 7 | 19 ± 5 | 16 ± 5 | 1 | Severe |
| S765R | ER | 29 ± 11 | 18 ± 6 | 29 ± 4 | 13 ± 4 | 18 ± 3 | 1 | Severe |
| V767D | ER | 24 ± 11 | 40 ± 14 | 63 ± 17 | 21 ± 1 | 20 ± 2 | 1 | Severe |
| L797P | ER | 25 ± 5 | 31 ± 5 | 37 ± 2 | 11 ± 5 | 12 ± 5 | 1 | Severe |
| G818E | ER | 51 ± 13 | 63 ± 5 | 111 ± 8 | 25 ± 3 | 12 ± 3 | 2 | Moderate /Severe |
| W821R | ER | 51 ± 6 | 58 ± 7 | 125 ± 7 | 19 ± 8 | 17 ± 6 | 2 | Moderate /Severe |
| I824T | ER | 41 ± 17 | 64 ± 3 | 145 ± 6 | 33 ± 2 | 13 ± 4 | 2 | Moderate /Severe |
| M840R | ER | 26 ± 4 | 37 ± 12 | 47 ± 14 | 12 ± 2 | 14 ± 2 | 1 | Severe |
| D846H | ER | 30 ± 9 | 42 ± 22 | 50 ± 18 | 13 ± 12 | 14 ± 7 | 1 | Severe |
| V849A | Vesicles/ER | 88 ± 20 | 86 ± 5 | 216 ± 13 | 51 ± 12 | 12 ± 9 | 3 | Mild |
| G851D | ER | 27 ± 11 | 31 ± 11 | 46 ± 12 | 13 ± 8 | 12 ± 10 | 1 | Severe |
| A854T | Vesicles/ER | 82 ± 6 | 70 +/−10 | 148 ± 26 | 60 ± 5 | 21 ± 9 | 2 | Moderate |
| Variant | Localization | Relative Solubilization | Basal ATPase Activity | N-Ret-PE-Induced ATPase Activity | N-Ret-PE Binding no ATP | N-Ret-PE Binding with ATP | Class | Predicted Severity |
|---|---|---|---|---|---|---|---|---|
| WT | Vesicles/ER | 100 | 100 | 222 ± 16 | 100 | 16 ± 7 | -- | Normal |
| p.P1380L | ER | 53 ± 12 | 61 ± 19 | 144 ± 31 | 55 ± 20 | 21 ± 11 | 2 | Moderate |
| p.E1399K | Vesicles/ER | 102 ± 15 | 94 ± 11 | 202 ± 34 | 117 ± 13 | 31 ± 14 | 3 | Mild |
| p.S1696N | Vesicles/ER | 96 ± 17 | 120 ± 8 | 255 ± 7 | 116 ± 16 | 27 ± 13 | 3 | Mild |
| p.Q1703E | Vesicles/ER | 89 ± 13 | 36 ± 9 | 20 ± 9 | 42 ± 16 | 36 ± 10 | 1 | Severe |
| p.Q1703K | ER | 52 ± 11 | 17 ± 2 | 12 ± 1 | 37 ± 6 | 35 ± 20 | 1 | Severe |
| p.R1705L | ER | 40 ± 13 | 76 ± 5 | 113 ± 9 | 54 ± 12 | 21 ± 9 | 2 | Moderate /Severe |
| p.A1773E | ER | 21 ± 4 | 38 ± 9 | 43 ± 1 | 39 ± 9 | 31 ± 16 | 1 | Severe |
| p.A1773V | ER | 36 ± 20 | 25 ± 4 | 46 ± 9 | 37 ± 8 | 27 ± 23 | 1 | Severe |
| p.A1794D | ER | 76 ± 4 | 46 ± 13 | 57 ± 14 | 49 ± 16 | 16 ± 9 | 1 | Severe |
| p.A1794P | ER | 55 ± 15 | 74 ± 11 | 137 ± 27 | 30 ± 13 | 10 ± 6 | 2 | Moderate /Severe |
| p.N1805D | Vesicles/ER | 82 ± 18 | 74 ± 15 | 154 ± 19 | 44 ± 11 | 18 ± 3 | 2 | Moderate |
| p.H1838D | ER | 56 ± 16 | 48 ± 13 | 96 ± 40 | 52 ± 1 | 28 ± 23 | 2 | Moderate /Severe |
| p.H1838N | Vesicles/ER | 107 ± 14 | 60 ± 6 | 132 ± 13 | 48 ± 2 | 33 ± 10 | 2 | Moderate |
| p.H1838R | ER | 27 ± 6 | 23 ± 8 | 28 ± 11 | 53 ± 6 | 27 ± 14 | 1 | Severe |
| p.H1838Y | ER | 48 ± 11 | 48 ± 18 | 75 ± 20 | 34 ± 8 | 16 ± 3 | 1 | Severe |
| p.R1843W | Vesicles/ER | 99 ± 8 | 68 ± 3 | 131 ± 14 | 84 ± 11 | 27 ± 14 | 3 | Moderate/Mild |
| p.N1868I | Vesicles/ER | 111 ± 25 | 134 ± 14 | 259 ± 29 | 101 ± 9 | 21 ± 6 | 3 | Mild |
| p.R1898C | Vesicles/ER | 84 ± 11 | 114 ± 8 | 238 ± 13 | 67 ± 20 | 20 ± 13 | 3 | Mild |
| p.R1898H | Vesicles/ER | 102 ± 28 | 112 ± 17 | 270 ± 35 | 121 ± 14 | 24 ± 10 | 3 | Mild |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garces, F.A.; Scortecci, J.F.; Molday, R.S. Functional Characterization of ABCA4 Missense Variants Linked to Stargardt Macular Degeneration. Int. J. Mol. Sci. 2021, 22, 185. https://doi.org/10.3390/ijms22010185
Garces FA, Scortecci JF, Molday RS. Functional Characterization of ABCA4 Missense Variants Linked to Stargardt Macular Degeneration. International Journal of Molecular Sciences. 2021; 22(1):185. https://doi.org/10.3390/ijms22010185
Chicago/Turabian StyleGarces, Fabian A., Jessica F. Scortecci, and Robert S. Molday. 2021. "Functional Characterization of ABCA4 Missense Variants Linked to Stargardt Macular Degeneration" International Journal of Molecular Sciences 22, no. 1: 185. https://doi.org/10.3390/ijms22010185
APA StyleGarces, F. A., Scortecci, J. F., & Molday, R. S. (2021). Functional Characterization of ABCA4 Missense Variants Linked to Stargardt Macular Degeneration. International Journal of Molecular Sciences, 22(1), 185. https://doi.org/10.3390/ijms22010185