Insights into the Interaction of LVV-Hemorphin-7 with Angiotensin II Type 1 Receptor


 , , and
, , and
Abstract
:1. Introduction
2. Results
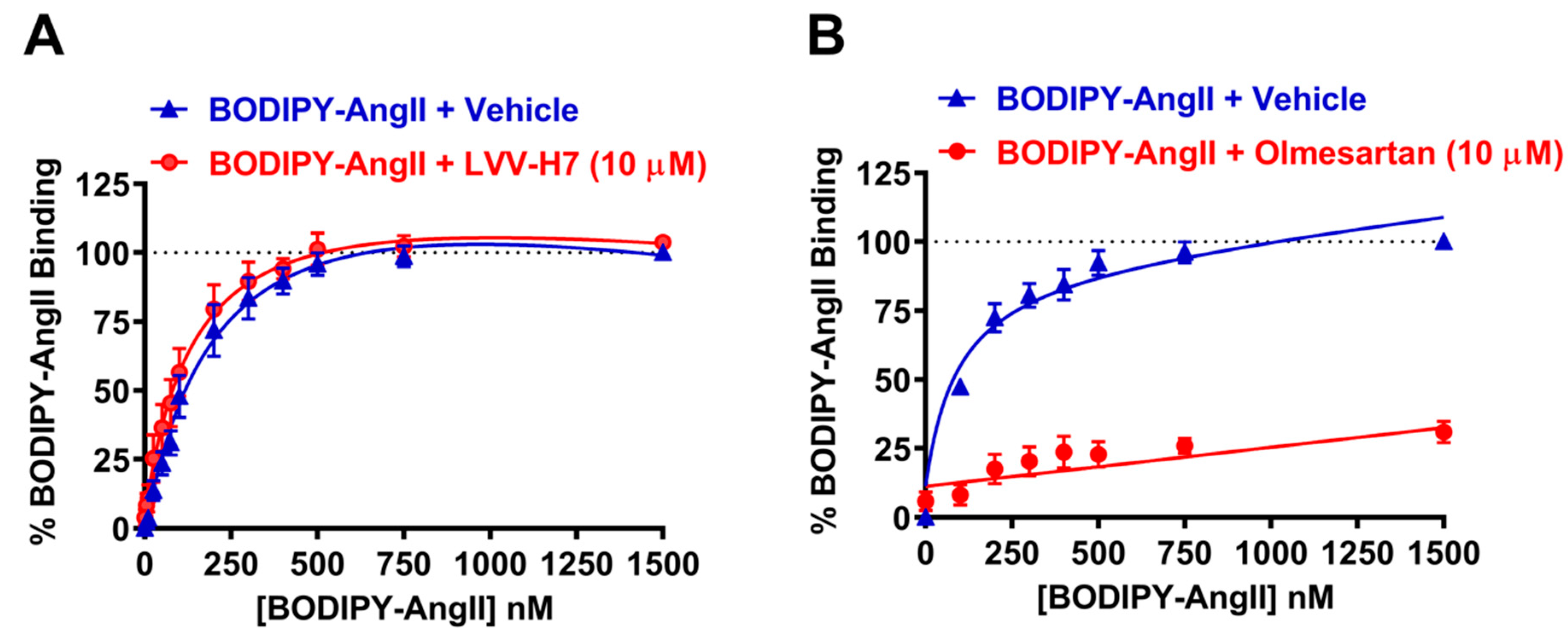
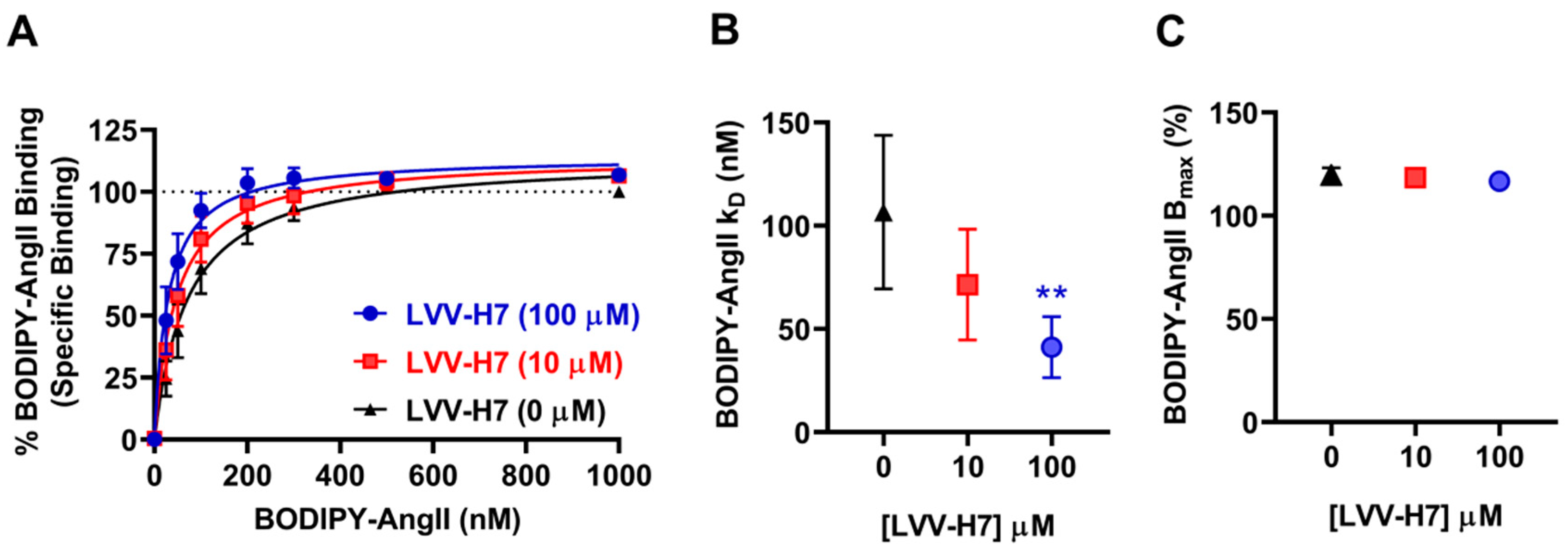
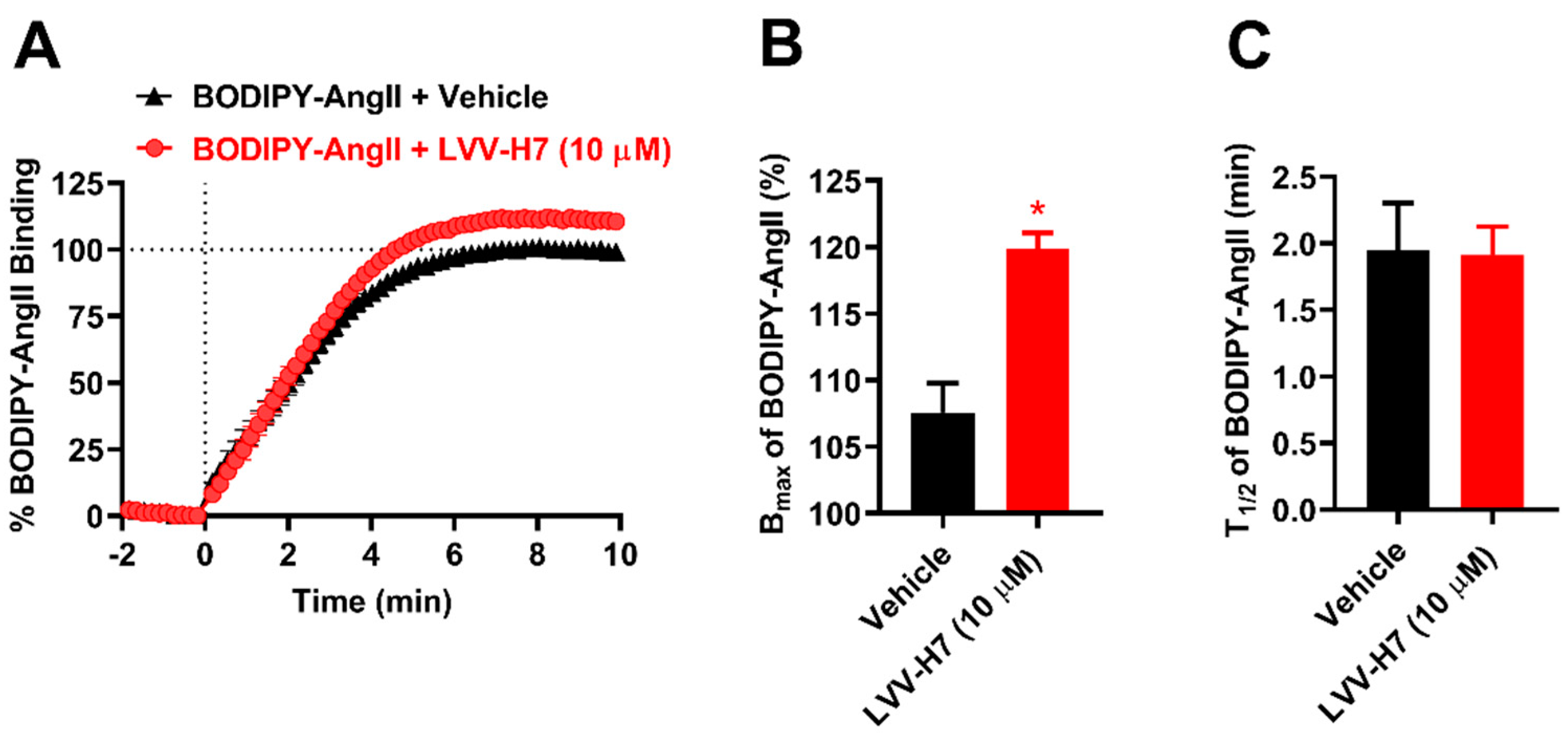
2.1. LVV-H7 Positively Affected AngII Binding on AT1R in Live HEK293 Cells
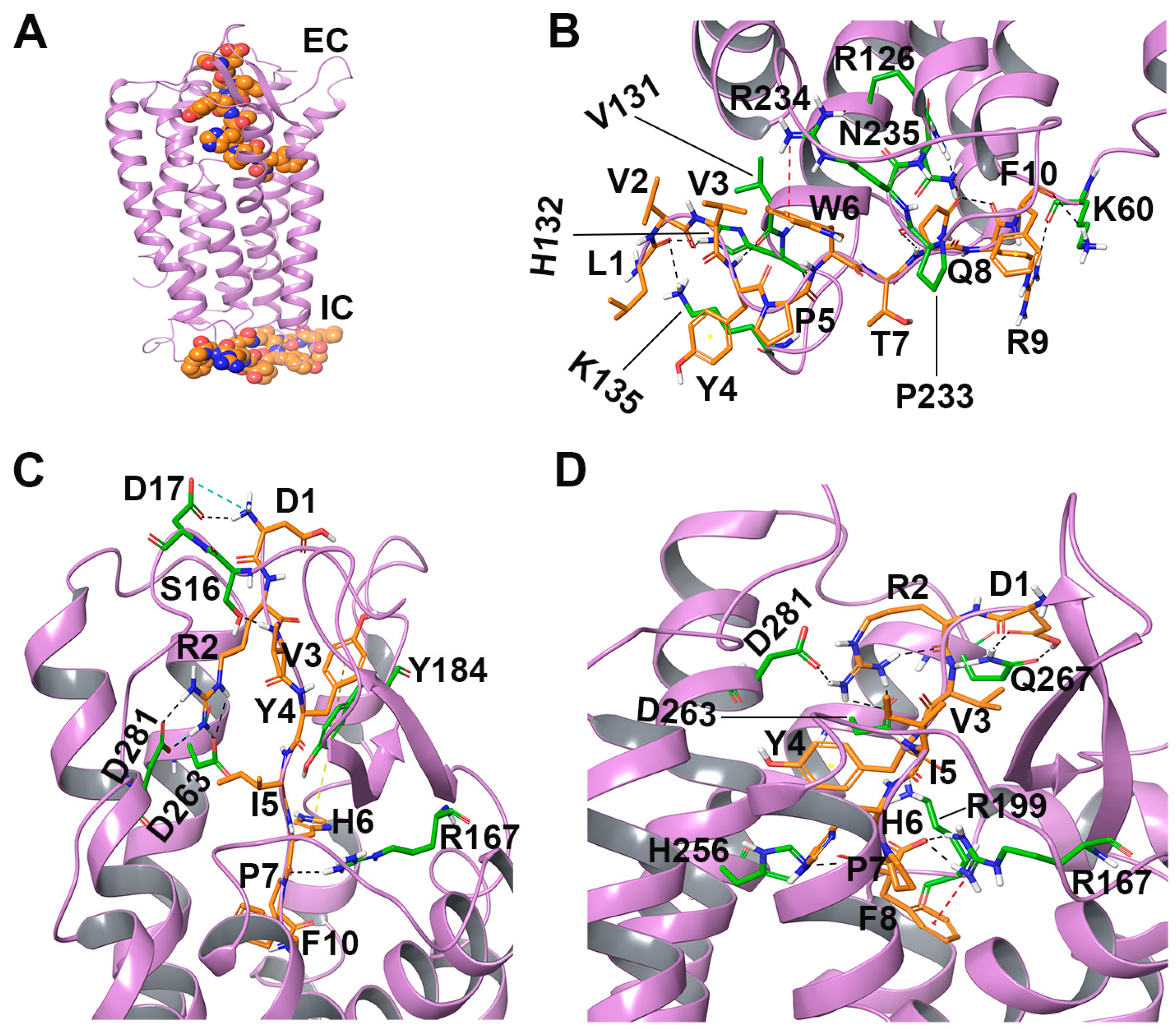
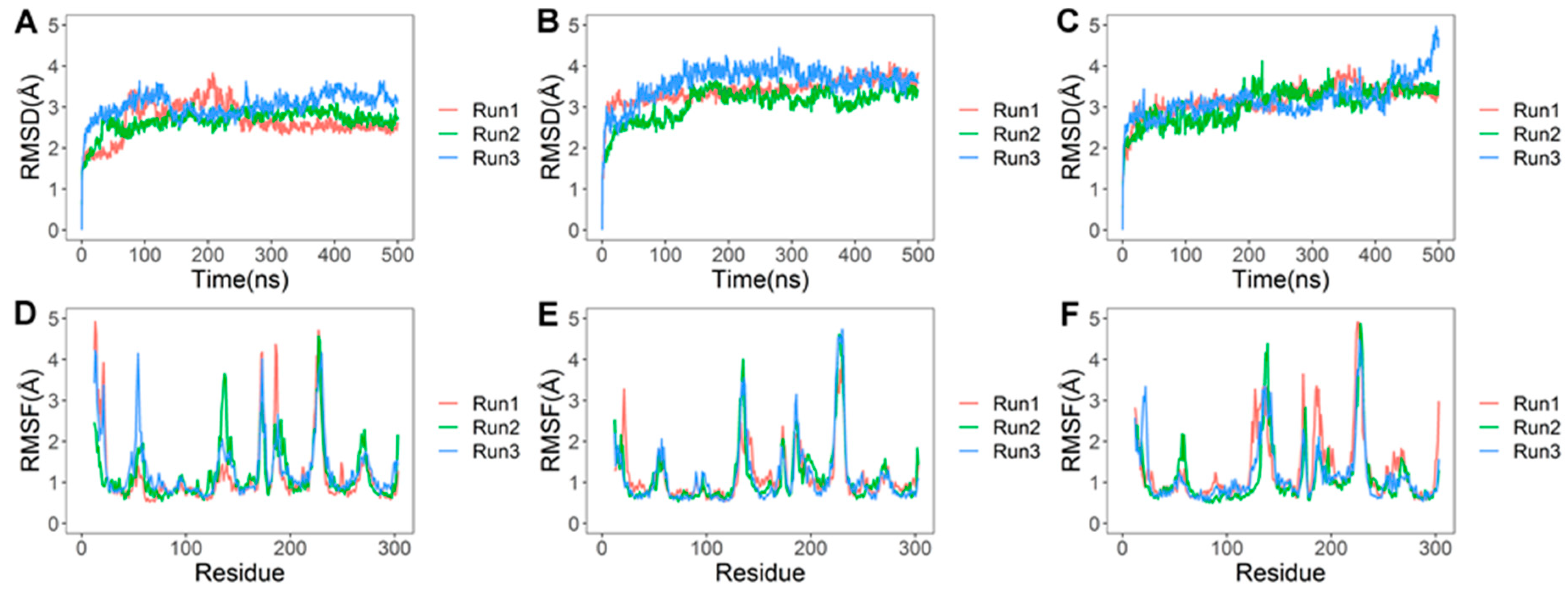
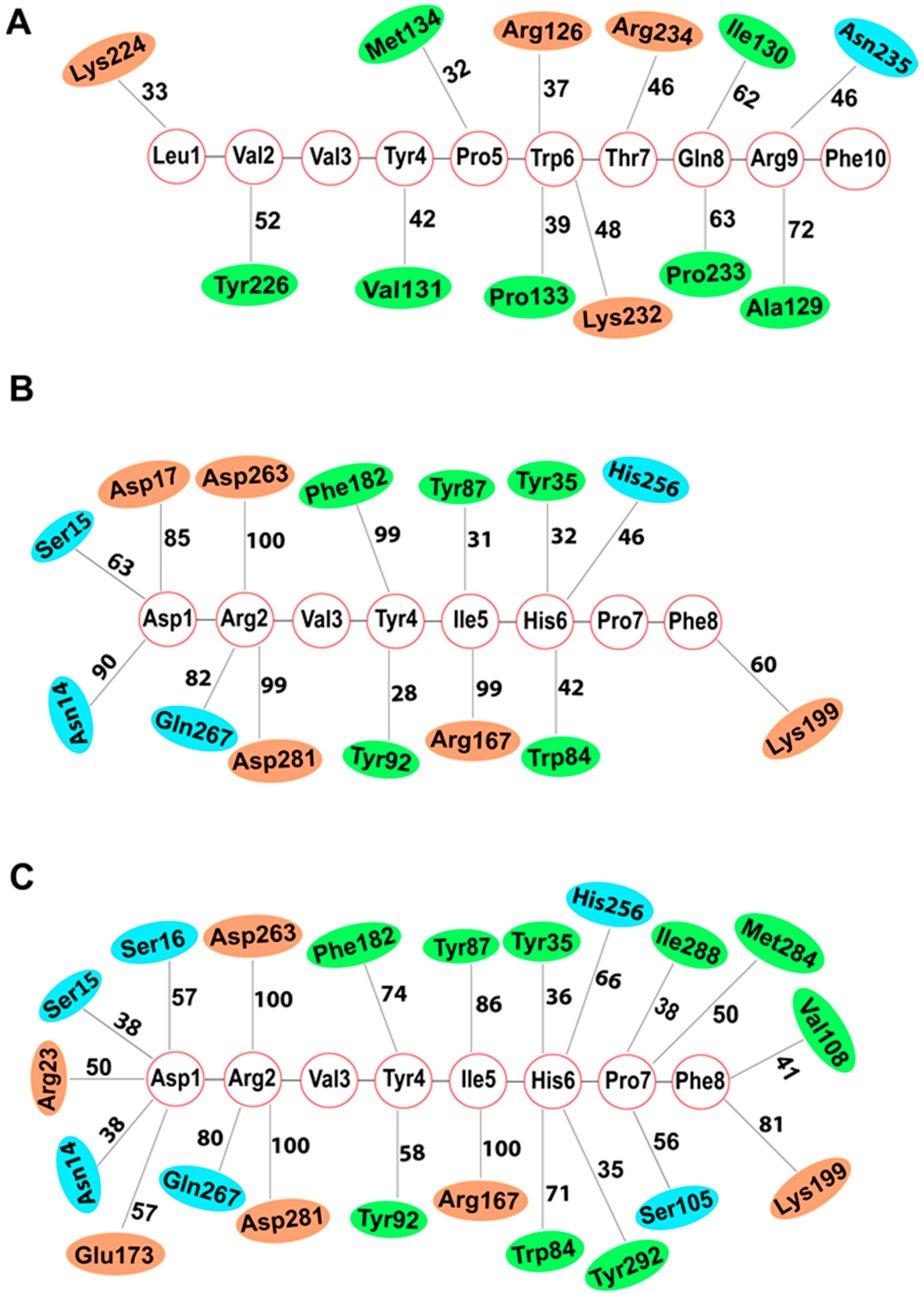
2.2. Molecular Docking and Molecular Dynamics Studies
2.2.1. LVV-H7 Binds to an Intracellular Site of AT1R
2.2.2. Intracellular Binding of LVV-H7 to AT1R Potentiated the Binding of AngII
3. Discussion
4. Materials and Methods
4.1. cDNA Constructs and Ligands
4.2. Cell Culture and Transfection
4.3. Saturation NanoBRET Assay
4.4. Real-Time NanoBRET Kinetics
4.5. Molecular Docking
4.6. Molecular Dynamics Simulations
4.7. Data Presentation and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hughes, J.; Smith, T.W.; Kosterlitz, H.W.; Fothergill, L.A.; Morgan, B.A.; Morris, H.R. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature 1975, 258, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Moisan, S.; Harvey, N.; Beaudry, G.; Forzani, P.; Burhop, K.E.; Drapeau, G.; Rioux, F. Structural requirements and mechanism of the pressor activity of Leu-Val-Val-hemorphin-7, a fragment of hemoglobin beta-chain in rats. Peptides 1998, 19, 119–131. [Google Scholar] [CrossRef]
- Albiston, A.L.; Pederson, E.S.; Burns, P.; Purcell, B.; Wright, J.W.; Harding, J.W.; Mendelsohn, F.A.; Weisinger, R.S.; Chai, S.Y. Attenuation of scopolamine-induced learning deficits by LVV-hemorphin-7 in rats in the passive avoidance and water maze paradigms. Behav. Brain Res. 2004, 154, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Alzeyoudi, S.A.R.; Almutawa, S.A.; Alnajjar, A.N.; Vijayan, R. Molecular basis of the therapeutic properties of hemorphins. Pharmacol. Res. 2020, 158, 104855. [Google Scholar] [CrossRef] [PubMed]
- Cejka, J.; Zelezna, B.; Velek, J.; Zicha, J.; Kunes, J. LVV-hemorphin-7 lowers blood pressure in spontaneously hypertensive rats: Radiotelemetry study. Pharmacol. Res. 2004, 53, 603–607. [Google Scholar]
- Ali, A.; Alzeyoudi, S.A.R.; Almutawa, S.A.; Alnajjar, A.N.; Al Dhaheri, Y.; Vijayan, R. Camel Hemorphins Exhibit a More Potent Angiotensin-I Converting Enzyme Inhibitory Activity than Other Mammalian Hemorphins: An In Silico and In Vitro Study. Biomolecules 2020, 10, 486. [Google Scholar] [CrossRef] [Green Version]
- Cheng, B.C.; Tao, P.L.; Cheng, Y.Y.; Huang, E.Y. LVV-hemorphin 7 and angiotensin IV in correlation with antinociception and anti-thermal hyperalgesia in rats. Peptides 2012, 36, 9–16. [Google Scholar] [CrossRef]
- Ali, A.; Baby, B.; Soman, S.S.; Vijayan, R. Molecular insights into the interaction of hemorphin and its targets. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Davis, T.P.; Gillespie, T.J.; Porreca, F. Peptide fragments derived from the beta-chain of hemoglobin (hemorphins) are centrally active in vivo. Peptides 1989, 10, 747–751. [Google Scholar] [CrossRef]
- Liebmann, C.; Schrader, U.; Brantl, V. Opioid receptor affinities of the blood-derived tetrapeptides hemorphin and cytochrophin. Eur. J. Pharmacol. 1989, 166, 523–526. [Google Scholar] [CrossRef]
- Szikra, J.; Benyhe, S.; Orosz, G.; Darula, Z.; Piot, J.M.; Fruitier, I.; Monory, K.; Hanoune, J.; Borsodi, A. Radioligand binding properties of VV-hemorphin 7, an atypical opioid peptide. Biochem. Biophys. Res. Commun. 2001, 281, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Piot, J.M. Investigation of inhibition angiotensin-converting enzyme (ACE) activity and opioid activity of two hemorphins, LVV-hemorphin-5 and VV-hemorphin-5, isolated from a defined peptic hydrolysate of bovine hemoglobin. Neuropeptides 1997, 31, 147–153. [Google Scholar] [CrossRef]
- Fruitier-Arnaudin, I.; Cohen, M.; Bordenave, S.; Sannier, F.; Piot, J.M. Comparative effects of angiotensin IV and two hemorphins on angiotensin-converting enzyme activity. Peptides 2002, 23, 1465–1470. [Google Scholar] [CrossRef]
- Ali, A.; Palakkott, A.; Ashraf, A.; Al Zamel, I.; Baby, B.; Vijayan, R.; Ayoub, M.A. Positive modulation of angiotensin II type 1 receptor–mediated signaling by LVV–hemorphin-7. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Stoddart, L.A.; Johnstone, E.K.M.; Wheal, A.J.; Goulding, J.; Robers, M.B.; Machleidt, T.; Wood, K.V.; Hill, S.J.; Pfleger, K.D.G. Application of BRET to monitor ligand binding to GPCRs. Nat. Methods 2015, 12, 661–663. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the SC’06 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Zhang, H.; Qiao, A.; Yang, D.; Yang, L.; Dai, A.; de Graaf, C.; Reedtz-Runge, S.; Dharmarajan, V.; Han, G.W.; Grant, T.D.; et al. Structure of the full-length glucagon class B G-protein-coupled receptor. Nature 2017, 546, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Luderman, K.D.; Conroy, J.L.; Free, R.B.; Southall, N.; Ferrer, M.; Sanchez-Soto, M.; Moritz, A.E.; Willette, B.K.; Fyfe, T.J.; Jain, P.; et al. Identification of positive allosteric modulators of the D1 dopamine receptor that act at diverse binding sites. Mol. Pharmacol. 2018, 94, 1197–1209. [Google Scholar] [CrossRef]
- Wang, X.; Heinz, B.A.; Qian, Y.W.; Carter, J.H.; Gadski, R.A.; Beavers, L.S.; Little, S.P.; Yang, C.R.; Beck, J.P.; Hao, J.; et al. Intracellular Binding Site for a Positive Allosteric Modulator of the Dopamine D1 Receptor. Mol. Pharmacol. 2018, 94, 1232–1245. [Google Scholar] [CrossRef]
- Wingler, L.M.; McMahon, C.; Staus, D.P.; Lefkowitz, R.J.; Kruse, A.C. Distinctive Activation Mechanism for Angiotensin Receptor Revealed by a Synthetic Nanobody. Cell 2019, 176, 479–490.e12. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.D.; Unal, H.; Desnoyer, R.; Karnik, S.S. Mechanism of Hormone Peptide Activation of a GPCR: Angiotensin II Activated State of AT1R Initiated by van der Waals Attraction. J. Chem. Inf. Model. 2019, 59, 373–385. [Google Scholar] [CrossRef]
- Fillion, D.; Cabana, J.; Guillemette, G.; Leduc, R.; Lavigne, P.; Escher, E. Structure of the human angiotensin II type 1 (AT1) receptor bound to angiotensin II from multiple chemoselective photoprobe contacts reveals a unique peptide binding mode. J. Biol. Chem. 2013, 288, 8187–8197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillion, D.; Lemieux, G.; Basambombo, L.L.; Lavigne, P.; Guillemette, G.; Leduc, R.; Escher, E. The amino-terminus of angiotensin II contacts several ectodomains of the angiotensin II receptor AT1. J. Med. Chem. 2010, 53, 2063–2075. [Google Scholar] [CrossRef] [PubMed]
- Noda, K.; Saad, Y.; Karnik, S.S. Interaction of Phe8 of angiotensin II with Lys199 and His256 of AT1 receptor in agonist activation. J. Biol. Chem. 1995, 270, 28511–28514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Unal, H.; Desnoyer, R.; Han, G.W.; Patel, N.; Katritch, V.; Karnik, S.S.; Cherezov, V.; Stevens, R.C. Structural basis for ligand recognition and functional selectivity at angiotensin receptor. J. Biol. Chem. 2015, 290, 29127–29139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Leger, A.J.; Baleja, J.D.; Rana, R.; Corlin, T.; Nguyen, N.; Koukos, G.; Bohm, A.; Covic, L.; Kuliopulos, A. Allosteric Activation of a G Protein-coupled Receptor with Cell-penetrating Receptor Mimetics. J. Biol. Chem. 2015, 290, 15785–15798. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, K.; Kuliopulos, A.; Covic, L. Turning receptors on and off with intracellular pepducins: New insights into G-protein-coupled receptor drug development. J. Biol. Chem. 2012, 287, 12787–12796. [Google Scholar] [CrossRef] [Green Version]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J. Structure of the CCR5 chemokine receptor–HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef] [Green Version]
- Covic, L.; Gresser, A.L.; Talavera, J.; Swift, S.; Kuliopulos, A. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc. Natl. Acad. Sci. USA 2002, 99, 643–648. [Google Scholar] [CrossRef] [Green Version]
- Bruns, R.F.; Fergus, J.H. Allosteric enhancement of adenosine A1 receptor binding and function by 2-amino-3-benzoylthiophenes. Mol. Pharmacol. 1990, 38, 939–949. [Google Scholar]
- Kruse, A.C.; Ring, A.M.; Manglik, A.; Hu, J.; Hu, K.; Eitel, K.; Hubner, H.; Pardon, E.; Valant, C.; Sexton, P.M.; et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 2013, 504, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Congreve, M.; Oswald, C.; Marshall, F.H. Applying Structure-Based Drug Design Approaches to Allosteric Modulators of GPCRs. Trends Pharmacol. Sci. 2017, 38, 837–847. [Google Scholar] [CrossRef] [PubMed]
- De Amici, M.; Dallanoce, C.; Holzgrabe, U.; Trankle, C.; Mohr, K. Allosteric ligands for G protein-coupled receptors: A novel strategy with attractive therapeutic opportunities. Med. Res. Rev. 2010, 30, 463–549. [Google Scholar] [CrossRef] [PubMed]
- Bock, A.; Merten, N.; Schrage, R.; Dallanoce, C.; Batz, J.; Klockner, J.; Schmitz, J.; Matera, C.; Simon, K.; Kebig, A.; et al. The allosteric vestibule of a seven transmembrane helical receptor controls G-protein coupling. Nat. Commun. 2012, 3, 1044. [Google Scholar] [CrossRef] [PubMed]
- Proska, J.; Tucek, S. Mechanisms of steric and cooperative actions of alcuronium on cardiac muscarinic acetylcholine receptors. Mol. Pharmacol. 1994, 45, 709–717. [Google Scholar] [PubMed]
- Carlson, K.E.; McMurry, T.J.; Hunt III, S.W. Pepducins: Lipopeptide allosteric modulators of GPCR signaling. Drug Discov. Today Technol. 2012, 9, e33–e39. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Elgeti, M.; Roman, K.; Martin, H.; Takefumi, M.; Eglof, R.; Patrick, S.; Oliver, P.; Friedrich, S.; Klaus, P.; Franz, J.; et al. Conserved Tyr223(5.58) plays different roles in the activation and G-protein interaction of rhodopsin. J. Am. Chem. Soc. 2011, 133, 7159–7165. [Google Scholar] [CrossRef]
- Matsoukas, M.-T.; Potamitis, C.; Plotas, P.; Androutsou, M.-E.; Agelis, G.; Matsoukas, J.; Zoumpoulakis, P. Insights into AT1 receptor activation through AngII binding studies. J. Chem. Inf. Model. 2013, 53, 2798–2811. [Google Scholar] [CrossRef]
- Rasmussen, S.G.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D. Crystal structure of the β 2 adrenergic receptor–Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Maraninchi, M.; Feron, D.; Fruitier-Arnaudin, I.; Bégu-Le Corroller, A.; Nogueira, P.; Mancini, J.; Valéro, R.; Piot, J.M.; Vialettes, B. Serum hemorphin-7 levels are decreased in obesity. Obesity 2013, 21, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Ingrid Fruitier-Arnaudin, S.; Daniel, B.; Jean-Marie, P. Serum levels of Hemorphin-7 peptides in patients with breast cancer. Clin. Chim. Acta 2002, 337, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Glämsta, L.; Mørkrid, L.; Lantz, I.; Nyberg, F. Concomitant increase in blood plasma levels of immunoreactive hemorphin-7 and beta-endorphin following long distance running. Regul. Pept. 1993, 49, 9–18. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Watts, K.S.; Dalal, P.; Murphy, R.B.; Sherman, W.; Friesner, R.A.; Shelley, J.C. ConfGen: A conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Model. 2010, 50, 534–546. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [Green Version]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Matyszewska, D.; Bilewicz, R. DPPC monolayers as simple models of biological membranes for studies of interactions with perfluorinated compounds. Ann. UMCS Chem. 2008, 63, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Tuckerman, M.; Berne, B.J.; Martyna, G.J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992, 97, 1990–2001. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | AT1R PBD ID | GlideScore Docking Score (kcal/mol) | MM-GBSA Binding Energy (kcal/mol) | Residues Forming Hydrogen Bonds | Residues Forming Hydrophobic Interactions | Residues Forming π-π Stacking or Cation-π Interactions |
|---|---|---|---|---|---|---|
| LVV-H7 | 4ZUD | −10.51 | −115.53 | Lys60, Arg126, Val131, His132, Lys135, Pro233, Asn235 | Val62, Ala129, Ile130, Pro133, Met134, Leu138, Met142, Leu217, Ala221, Ala225, Ile238 | Arg234 |
| AngII | 4ZUD | −13.03 | −125.03 | Ser16, Asp17, Arg167, Asp263, Asp281 | Leu13, Cys18, Pro19, Ala21, Ile31, Tyr35, Trp84, Tyr87, Tyr92, Val108, Ile172, Ala181, Phe182, Trp253, Ile266, Ala277, Val280, Met284, Pro285, ILe288, Tyr292 | Tyr184 |
| AngII with LVV-H7 | 4ZUD | −14.67 | −142.81 | Tyr87, Arg167, Phe182, Lys199, His256, Asp263, Gln267, Val280, Asp281 | Ile12, Leu13, Tyr35, Trp84, Tyr87, Tyr92, Val108, Ala106, Leu112, Ala163, Val179, Cys180, Ala181, Tyr184, Leu202, Trp253, Phe259, Ile266, Ala277, Ala283, Met284, Pro285, Ile288, Tyr292 | His166, Phe204 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Johnstone, E.K.M.; Baby, B.; See, H.B.; Song, A.; Rosengren, K.J.; Pfleger, K.D.G.; Ayoub, M.A.; Vijayan, R. Insights into the Interaction of LVV-Hemorphin-7 with Angiotensin II Type 1 Receptor. Int. J. Mol. Sci. 2021, 22, 209. https://doi.org/10.3390/ijms22010209
Ali A, Johnstone EKM, Baby B, See HB, Song A, Rosengren KJ, Pfleger KDG, Ayoub MA, Vijayan R. Insights into the Interaction of LVV-Hemorphin-7 with Angiotensin II Type 1 Receptor. International Journal of Molecular Sciences. 2021; 22(1):209. https://doi.org/10.3390/ijms22010209
Chicago/Turabian StyleAli, Amanat, Elizabeth K. M. Johnstone, Bincy Baby, Heng B. See, Angela Song, K. Johan Rosengren, Kevin D. G. Pfleger, Mohammed Akli Ayoub, and Ranjit Vijayan. 2021. "Insights into the Interaction of LVV-Hemorphin-7 with Angiotensin II Type 1 Receptor" International Journal of Molecular Sciences 22, no. 1: 209. https://doi.org/10.3390/ijms22010209
APA StyleAli, A., Johnstone, E. K. M., Baby, B., See, H. B., Song, A., Rosengren, K. J., Pfleger, K. D. G., Ayoub, M. A., & Vijayan, R. (2021). Insights into the Interaction of LVV-Hemorphin-7 with Angiotensin II Type 1 Receptor. International Journal of Molecular Sciences, 22(1), 209. https://doi.org/10.3390/ijms22010209